

Abstract

More cycling–fewer steps
The first enantioselective total synthesis of (−)-minovincine has been accomplished in nine chemical steps and 13% overall yield. A novel, one-step Diels–Alder/β-elimination/conjugate addition organocascade sequence allowed rapid access to the central tetracyclic core in an asymmetric manner.
Keywords: organocatalysis, total synthesis, cascade catalysis, enantioselective, alkaloids
Since its isolation in 1962, minovincine[1] (1) has garnered considerable interest within the chemical synthesis community due to its characteristic spiroindoline framework, a common structural feature found among a number of high-profile natural products derived from the Aspidosperma, Kopsia, and Catharanthus genre.[2] Indeed, this structural core has led to minovincine being described as a “biogenetic turntable” between the vindolinine and kopsinine classes of isolates,[3a,b] and has provided the impetus over several decades for a number of racemic total syntheses of this biosynthetic linchpin. At the present time, however, no asymmetric total synthesis of minovincine has been reported,[4] a surprising fact given the range of catalytic technologies that have been developed to forge this broadly represented spiroindoline framework. In this communication, we detail the first enantioselective total synthesis of (−)-minovincine in only nine chemical steps, using a novel organocascade[5] catalysis transformation that incorporates an enantioselective Diels–Alder cycloaddition/β-elimination/conjugate addition sequence.[6] Central to the development of this new catalysis cascade has been the identification of ketone substrates and amine catalysts which combine to provide direct access to the key functional and architectural elements found in minovincine.
We were initially drawn to minovincine on the basis of its (i) benchmark spiroindoline framework, which is broadly represented across a large range of Aspidosperma, Kopsia, and Catharanthus isolates; and (ii) the exocyclic ketone substituent that is not common within these alkaloid families.[2] Previous studies conducted by our group have revealed that similar tetracycles are readily constructed via iminium-activated organocascade catalysis protocols that involve enantioselective [4 + 2] cycloaddition of 2-vinyl indoles with an unsaturated aldehyde partner.[6] We questioned whether this strategy might be extended to ketone dienophiles, such as 3-butyn-2-one, thereby providing direct access to the exocyclic ketone present in minovincine and several other natural alkaloids. Despite their apparent similarities on paper, aldehydes and ketones exhibit markedly different behavior in organocatalytic processes, and the latter have historically posed a significant challenge in amine-catalyzed Diels–Alder and cascade catalysis reactions (Figure 2).[7] More specifically, ketones typically exhibit (i) attenuated reactivity towards condensation with secondary amines in comparison to their formyl counterparts – a limitation that can dramatically impact overall reaction efficiency; and (ii) they are prone to non-selective iminium geometry formation with amine catalysts, a loss of organizational control that often leads to diminished enantioselectivity in the critical bond-forming step. As such, we were intrigued by the possibility of expanding the scope of organocascade catalysis to include this more challenging family of carbonyls while developing a concise route to minovincine.
Figure 2.

Issues with ketone substrates in organocatalytic cascades.
The pathway for the proposed organocascade sequence involves a series of catalytic cycles outlined in Scheme 1. In the first cycle, condensation between 3-butyn-2-one (3, R = Me) and amine catalyst 4 would transiently generate a LUMO-lowered unsaturated iminium ion poised to undergo [4 + 2] cycloaddition with vinyl indole 2.[8] We hypothesized that incorporation of a suitable leaving group on the terminus of the diene would result in facile βelimination, delivering indolinium ion 6. At this stage, 5-exo-heterocyclization of the pendent amine onto the α,β-unsaturated iminium moiety would directly yield tetracycle 11 (Path A), which, after hydrolysis, would deliver spiroindoline 13 and regenerate the amine catalyst. Based on previous studies, we recognized the possibility of an alternative second cycle in which indolinium trapping by the pendent amine would furnish pyrroloindoline 8 (Path B), an isolable intermediate when propynal is utilized as the dienophile (R = H). Importantly, we have demonstrated that this aldehyde undergoes facile conversion to the desired spiroindoline 13 (R = H) in the presence of the tribromoacetic acid salt of N-methyl 4 (incapable of undergoing iminium ion formation), illustrating the feasibility of Brønsted acid catalysis as an operative pathway in this organocascade sequence.[6] As a key stereocontrol element, we hypothesized that condensation of 3-butyn-2-one with the imidazolidinone catalyst would lead to an iminium ion geometry wherein the acetylenic functionality of the conjugated system is projected towards the benzylic unit on the catalyst scaffold (enabling stabilizing cation-π interactions between the pendent arene and the terminal alkyne). In this geometry, the catalyst substituents effectively shield one π-face of the iminium ion, thereby ensuring that endo-selective Diels–Alder cycloaddition[8,9] occurs from the opposing trajectory (TS-A, Figure 3), a feature that should induce high levels of enantiocontrol.
Scheme 1.

Proposed mechanism for [4 + 2] cycloadddition/β-elimination/hetero-conjugate addition organocascade sequence.
Figure 3.

Proposed stereochemical model for cascade Diels–Alder.
To evaluate the key organocascade sequence, a series of halide-or selenium-bearing vinyl indoles were prepared via a straightforward, three-step protocol (Scheme 2). More specifically, N-indole protection of commercially available tetrahydro-β-carboline derivative 18 was followed by selenium dioxide-mediated oxidation[10] and subsequent olefination to afford diene 16 as the vinyl bromide, iodide, or selenide.
Scheme 2.
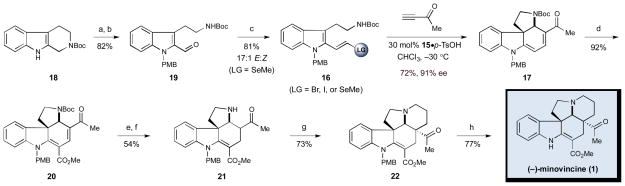
Total synthesis of (−)-minovincine: a) NaH, PMB-Cl, DMF, 0 °C, 94%; b) SeO2, 95:5 dioxane:H2O, 100 °C, 87%; c) LG = Br: [(PPh3)CH2Br]Br, t-BuOK, THF, −78 °C, 21%; LG = I: CrCl2, CHI3, THF, 0 °C, 50%; LG = SeMe: (EtO)2P(O)CH2SeMe, KHMDS, 18-crown-6, −78 °C to rt, 81%; d) N-iodosuccinimide, Pd(PhCN)2Cl2 (10 mol%), Et3N, MeOH, MeCN, CO (1 atm), 60 °C, 92%; e) L-Selectride, THF, −78 to 0 °C, 75%; f) (Me)3Si-I, Et3N, CH2Cl2, 0 °C, 72%; g) 1,3-diiodopropane, NaHCO3, DMF, 35 °C; then t-BuOK, t-BuOH, rt, 73%; h) 1:1 TFA:CH2Cl2, 0 °C to rt, 77%.
As illustrated in Table 1, our initial attempts to construct spiroindoline 17 via our proposed organocascade sequence using the vinyl bromide derivative of 16 were unsuccessful (entry 1). However, employing the indole vinyl iodide derivative in conjunction with the camphorsulfonic acid salt of catalyst 14 (known to efficiently condense with cyclic ketones to form enamines)[11] produced the desired tetracycle, albeit with low yield and enantioselectivity (entry 2, 15% yield, 24% ee). To our delight, further investigation revealed that use of the vinyl selenide system afforded cascade product 17 in a promising 45% yield and 36% ee (entry 3). Reaction medium was also critical with respect to obtaining high levels of asymmetric induction, as demonstrated by the implementation of chloroform (entry 4, 79% ee). Further improvements in enantioselectivity were achieved by increasing the size of the pendent aromatic unit on the catalyst framework, presumably allowing for greater facial shielding of the transient iminium ion (entry 5, catalyst 15, 95% ee). Finally, a pronounced acid co-catalyst effect was identified, wherein changing from camphorsulfonic acid[12] to p-toluenesulfonic acid provided the desired spiroindoline in 72% yield while maintaining excellent levels of enantioselectivity (entry 6, 91% ee).
Table 1.
Evaluation of key organocascade step.

| |||||
|---|---|---|---|---|---|
| entry[a] | catalyst•HX | LG | solvent | % yield[b] | % ee[c] |
| 1 | 14•(+)-CSA | Br | CH2Cl2 | 0 | -- |
| 2 | 14•(+)-CSA | I | CH2Cl2 | 15 | 24 |
| 3 | 14•(+)-CSA | SeMe | CH2Cl2 | 45 | 36 |
| 4 | 14•(+)-CSA | SeMe | CHCl3 | 36 | 79 |
| 5 | 15•(+)-CSA | SeMe | CHCl3 | 38 | 95 |
| 6 | 15•p-TSA | SeMe | CHCl3 | 72 | 91 |
Entries 1–4 performed at −20 °C; entries 5 and 6 performed at −30 °C.
Isolated yields.
Determined by chiral HPLC analysis.
Having established our optimal ketone activation cascade catalysis sequence, and with the enantioenriched tetracyclic core 17 in hand, we next focused upon completing the natural product synthesis in an expeditious fashion (Scheme 2). With the goal of installing the requisite minovincine ester group, we exposed spiroindoline 17 to a variety of traditional acylating agents (i.e. phosgene, methyl chloroformate, etc.), however, with little success. Fortunately, the palladium-catalyzed carbomethoxylation of the dienylogous amide in the presence of carbon monoxide and methanol proceeded smoothly to afford vinylogous carbamate 20 in excellent yield (92%).[13] Regioselective 1,4-conjugate reduction of the resulting α,β,γ,δ-unsaturated ketone was accomplished with the aid of the sterically demanding reducing agent L-Selectride,[14] followed by Lewis acid-mediated Boc-deprotection to deliver the βamino ketone 21 in 54% yield over two steps.[15] Closure of the final ring was achieved through N-alkylation with 1,3-diiodopropane and treatment with potassium tert-butoxide to provide the natural product in its protected form in 73% yield as a single diastereomer. Finally, removal of the para-methoxybenzyl group from the indoline nitrogen afforded (−)-minovincine in 77% yield for the final step, and in 13% overall yield for the nine-step sequence.[16] Synthetic (−)-minovincine was found to be identical in all spectroscopic respects to the natural isolate.
In summary, the first enantioselective total synthesis of (−)-minovincine has been completed in nine chemical steps and 13% overall yield from commercial materials. A prominent feature of this synthesis involves an organocatalytic Diels–Alder/βelimination/conjugate addition cascade to rapidly construct the tetracyclic framework of minovincine in a highly enantioselective manner. This total synthesis exemplifies the capacity of ketone dienophiles to be viable substrates for iminium catalysis in the context of complex target settings.
Supplementary Material
Figure 1.

Apocynaceae alkaloids – common pentacyclic framework.
Acknowledgments
Financial support was provided by the NIHGMS (R01 GM078201-06), Merck and Amgen. M.P. is grateful for a Marie-Curie Postdoctoral fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org
References
- 1.Isolation of (−)- and (+)-minovincine from Vinca minor and Tabernaemontana ridelli: Plat M, LeMen J, Janot MM, Budzikiewicz H, Wilson JM, Durham LJ, Djerassi C. Bull Soc Chim Fr. 1962:2237–2241.Cava MP, Tjoa SS, Ahmed QA, Da Rocha AI. J Org Chem. 1968;33:1055–1059. doi: 10.1021/jo01267a023.
- 2.Aspidosperma, Kopsia, and Catharanthus alkaloids: Saxton JE. The Indoles: the monoterpenoid indole alkaloids. Vol. 25. Wiley-Interscience; 1983. Saxton JE. In: The Alkaloids: Chemistry and Biology. Cordell GA, editor. Vol. 51. Academic Press; San Diego: 1998. pp. 1–197.Toyota M, Ihara M. Nat Prod Rep. 1998;15:327–340.van der Heijden R, Jacobs DI, Snoeijer W, Hallard D, Verpoorte R. Curr Med Chem. 2004;11:607–628. doi: 10.2174/0929867043455846.Saxton JE. The Chemistry of Heterocyclic Compounds. Wiley; New York: 2008. pp. 331–437.
- 3.For racemic total syntheses of minovincine, see: Kuehne ME, Earley WG. Tetrahedron. 1983;39:3707–3714.Kuehne ME, Earley WG. Tetrahedron. 1983;39:3715–3717.Kalaus G, Juhász I, Kajtár-Peredy M, Brlik J, Szabó L, Szántay C. J Org Chem. 1997;62:9188–9191.Kalaus G, Léder L, Greiner I, Kajtár-Peredy M, Vékey K, Szabó L, Szántay C. Tetrahedron. 2003;59:5661–5666.
- 4.For a semisynthesis of (−)-minovincine beginning from vindoline, see: Langlois N, Andriamialiosa RZ. J Org Chem. 1979;44:2468–2471. doi: 10.1021/jo01336a006.
- 5.a) Enders D, Grondal C, Hüttl MRM. Angew Chem. 2007;119:1590–1601. [Google Scholar]; Angew Chem Int Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]; b) Walji AM, MacMillan DWC. Synlett. 2007:1477–1489. [Google Scholar]; c) Pellissier H. Adv Synth Catal. 2012;354:237–294. [Google Scholar]
- 6.Jones SB, Simmons B, Mastracchio A, MacMillan DWC. Nature. 2011;475:183–188. doi: 10.1038/nature10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Northrup AB, MacMillan DWC. J Am Chem Soc. 2002;124:2458–2460. doi: 10.1021/ja017641u. [DOI] [PubMed] [Google Scholar]; b) Wu LY, Bencivenni G, Mancinelli M, Mazzanti A, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2009;48:7196–7199. doi: 10.1002/anie.200903280. [DOI] [PubMed] [Google Scholar]
- 8.Jones SB, Simmons B, MacMillan DWC. J Am Chem Soc. 2009;131:13606–13607. doi: 10.1021/ja906472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Acetylenic [4 + 2] cycloadditions are typically exo-selective: Corey EJ, Lee TW. Tetrahedron Lett. 1997;38:5755–5758.Ishihara K, Kondo S, Kurihara H, Yamamoto H. J Org Chem. 1997;62:3026–3027. doi: 10.1021/jo970171v.
- 10.Gatta F, Misiti D. J Heterocycl Chem. 1987;24:1183–1187. [Google Scholar]
- 11.The installation of aromatic groups on the catalyst scaffold has been shown to be useful in ketone SOMO-arylations. See: Mastracchio A, Warkentin AA, Walji AM, MacMillan DWC. Proc Natl Acad Sci. 2010;107:20648–20651. doi: 10.1073/pnas.1002845107.
- 12.Both enantiomers of camphorsulfonic acid produced similar results with regard to both yield and enantioselectivity.
- 13.For a similar, two-step alkoxycarbonylation of uridine derivatives, see: Ito T, Ueno Y, Komatsu Y, Matsuda A. Nucleic Acids Res. 2003;31:2514–2523. doi: 10.1093/nar/gkg374.
- 14.For an example of L-Selectride 1,4-conjugate reduction of a vinylogous amide, see: Donohoe TJ, Connolly MJ, Walton L. Org Lett. 2009;11:5562–5565. doi: 10.1021/ol902402v.
- 15.Variable temperature NMR studies suggested a rotameric mixture of the reduced product as a single diastereomer (see Supporting Information). Amine 21 was obtained as a single diastereomer.
- 16.A slight decrease in enantiomeric excess between intermediate 17 (91% ee) and (−)-minovincine (86% ee) was observed upon completion of the synthesis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.