

Abstract
The direct β-coupling of cyclic ketones with aryl ketones has been achieved via the synergistic combination of photoredox catalysis and organocatalysis. Diaryl oxymethyl or aryl–alkyl oxymethyl radicals, transiently generated via single-electron reduction of ketone precursors, readily merge with β-enaminyl radical species – generated by photon-induced enamine oxidation – to produce γ-hydroxyketone adducts. Experimental evidence indicates that two discrete reaction pathways can be operable in this process depending upon the nature of the ketyl radical precursor and the photocatalyst.
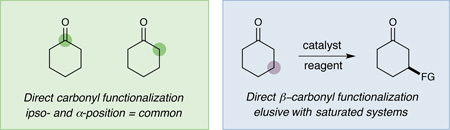
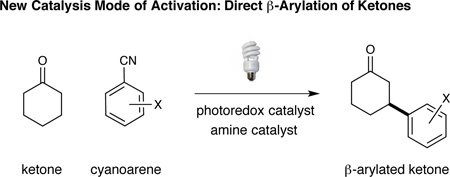
The direct β-functionalization of saturated ketones and aldehydes is an important yet elusive goal in organic chemistry.1 While carbonyl groups are readily amenable to ipso- and α-carbon substitution with a range of nucleophiles and electrophiles respectively,2,3 activation at the β-methylene position poses a significant synthetic challenge. With a few notable exceptions,1,4 carbonyl β-functionalization has traditionally been restricted to the conjugate addition of soft nucleophiles into α,β-unsaturated carbonyl systems. As such, the development of a general catalytic platform5 for the direct β- functionalization of saturated ketones or aldehydes would represent a conceptual and practical advance for the field. In this context, our lab has recently introduced an unprecedented 5πe− carbonyl activation mode that capitalizes on the synergistic merger of photoredox catalysis and amine organocatalysis to accomplish the direct β-arylation of saturated aldehydes and ketones (Eq 1).1a This strategy relies on the coupling of two catalytically generated radical species: a β-enaminyl radical formed via oxidation and deprotonation of a ketone-derived enamine and a radical anion generated by photocatalytic reduction of an aryl nitrile.6 In this communication, we further advance this activation concept to describe the first β-functionalization of saturated cyclic ketones with aryl ketones to deliver γ-hydroxyketone motifs, a protocol that formally represents a homo-enolate aldol reaction using simple carbonyl substrates, a household light source, and two commercial catalysts (Eq 2).
 |
(Eq 1) |
 |
(Eq 2) |
Design Plan
Among the most fundamental carbonyl α-functionalization reactions in organic chemistry is the aldol coupling of nucleophilic enolates with electrophilic ketones or aldehydes to deliver valuable β-hydroxycarbonyl motifs.7 Although the aldol reaction has been widely exploited for the α-functionalization of carbonyl substrates for over 140 years,8 analogous “homo-aldol” transformations that allow for the direct β-functionalization of carbonyls remain elusive. Typically, homoaldol-type synthons are accessed via carbene catalysis,9,10 nucleophilic addition of acetal-protected Grignard reagents,11 or stoichiometric metal-activated homoenolate equivalents.12–16
Drawing from the mechanistic insights gained in the course of our β-arylation program,1a we envisioned a direct β-coupling of saturated ketones with aryl–alkyl and diaryl ketone precursors. Specifically, we postulated that a transiently formed nucleophilic β-enaminyl 5πe− species (1) would be intercepted by a ketyl radical (2) to directly form a γ-hydroxyketone adduct (Eq 2).17 Notably, both radical species would be generated in catalytic quantities through the operation of two concurrent activation pathways: a photoredox cycle (en route to 2) and an organocatalytic cycle (en route to 1).
The specific mechanistic details of our proposed synergistic merger of visible light-mediated photoredox catalysis and organocatalysis18,19 are outlined in Scheme 1. Irradiation of tris(2-phenylpyridinato-C2, N)iridium(III) [Ir(ppy)3] (8) with visible light produces a long-lived (1.9 µs) photoexcited state,20*Ir(ppy)3 (9), which can be readily oxidized or reduced by an appropriate substrate quencher. While*Ir(ppy)3 (9) is a strong reductant (E1/2red [Ir(ppy)3+/*Ir(ppy)3] = −1.73 V vs. SCE),21 its capacity for single electron transfer (SET) with diarylketones such as benzophenone would be endergonic (E1/2red = −1.83 V vs. SCE).22 However, in acidic medium, the standard reduction potential of the ketone is elevated and therefore it is easier to reduce, rendering this step experimentally feasible.23 As such, electron transfer between an aryl ketone and excited state 9 would provide the oxidized IrIV(ppy)3 (10) system along with the corresponding ketyl radical 2. Concurrent with this photoredox cycle, we envisioned a second organocatalytic cycle, commencing with condensation of amine catalyst 3 with the ketone coupling partner (i.e. cyclohexanone) to generate an electron-rich enamine, 4. The facile oxidation of this intermediate by IrIV(ppy)3 (E1/2red [Ir(ppy)3+/Ir(ppy)3] = +0.77 V vs. SCE; E1/2red 4 = +0.38 V vs. SCE)24 serves to reduce the photocatalyst to its ground state, thereby completing the photocatalytic cycle. Formation of the desired enaminyl radical cation 5 would then induce an increase in the acidity of the allylic C–H bond, facilitating deprotonation at the β-position.1a The transiently formed 5pe− species 1 (an intermediate which represents the desired activation mode) should then readily couple with ketyl radical 2 to form the γ-hydroxyketone enamine 6. Finally, enamine hydrolysis would serve to release the β-union product 7 and regenerate the amine catalyst 3, thereby completing the organocatalytic cycle. With respect to achieving chemoselective reduction of benzophenone in the presence of cyclohexanone, it was expected that the significantly lower standard reduction potential of cyclohexanone (E1/2red = −2.79 V vs. SCE)25 would render this substrate thermodynamically indisposed toward adventitious reduction.
Scheme 1.

Proposed Mechanism for the β-Ketone Coupling.
We first explored the proposed direct β-carbonyl coupling reaction in the context of cyclohexanone and benzophenone (Table 1). Examination of a range of photocatalysts, amines, bases, and solvents (entries 1–6) revealed the combination of Ir(ppy)3 (8) and 1,4-diazabicyclo[2.2.2]octane (DABCO) as base to be most effective. For example, when a DMPU solution26 containing the ketone substrates, Ir(ppy)3 (8), DABCO, and azepane organocatalyst (3)27 was irradiated with 26 W fluorescent light, the γ-alkyloxy adduct was formed in 67% yield (entry 3). The desired product was accompanied by significant amounts (12%) of benzophenone dimer, which we assume arises from the combination of two molecules of ketyl radical 2. Notably, further improvements in reaction efficiency were achieved through addition of an equivalent of LiAsF6,28 which suppresses formation of this undesired diol, presumably via the production of a lithium alkoxide ketyl radical that is inert to dimerization (entry 9, 81% yield). The critical roles of the photocatalyst, organocatalyst, and light were demonstrated through control experiments, wherein no desired product was detected in the absence of any of these components (entries 10– 12).
Table 1.
Initial Studies towards the β-Coupling of Ketones.
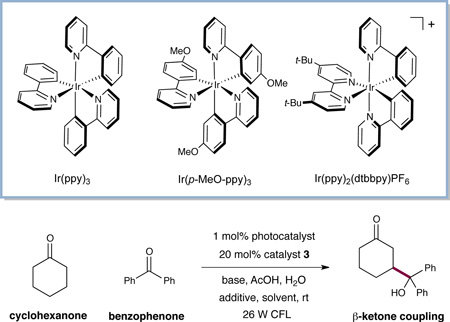 | |||||
|---|---|---|---|---|---|
| entry | photocatalyst | base | additive | solvent | yielda |
| 1 | Ir(ppy)3 | NaOAc | -- | DMPU | 52% |
| 2 | Ir(ppy)3 | 2,6-lutidine | -- | DMPU | 34% |
| 3 | Ir(ppy)3 | DABCO | -- | DMPU | 67% |
| 4 | Ir(p-MeO ppy)3 | DABCO | -- | DMPU | 65% |
| 5 | Ir(ppy)2(dtbbpy)PF6 | DABCO | -- | DMPU | 59% |
| 6 | Ir(ppy)3 | DABCO | -- | DMF | 59% |
| 7 | Ir(ppy)3 | DABCO | CsClO4 | DMPU | 72% |
| 8 | Ir(ppy)3 | DABCO | KAsF6 | DMPU | 75% |
| 9 | Ir(ppy)3 | DABCO | LiAsF6 | DMPU | 81% |
| 10 | none | DABCO | LiAsF6 | DMPU | 0% |
| 11 | Ir(ppy)3b | DABCO | LiAsF6 | DMPU | 0% |
| 12 | Ir(ppy)3c | DABCO | LiAsF6 | DMPU | 0% |
Yield determined by 1H NMR using 1,3-benzodioxole as an internal standard.
Reaction performed in the absence of catalyst 3.
Reaction performed in the absence of light. CFL = compact fluorescent light.
Reaction Scope
Having identified optimal conditions for this photocatalytic, direct β-ketone-ketone coupling reaction, we aimed to define the scope of the enaminyl radical precursor. As shown in Table 2, a series of differentially substituted cyclohexanone-derived substrates was readily coupled with benzophenone. It is of note that incorporation of both alkyl and aryl substituents at positions 3 and 4 of the cyclohexanone ring is well-tolerated (entries 2–5, 43–79% yield). As expected, the presence of groups at the ring 4-position induces higher levels of diastereoselectivity than substituents at the cyclohexanone 3-position (cf. entries 4 and 5). Interestingly, while cyclopentanone served as a suitable substrate for this reaction (entry 6, 65% yield), 7-membered ketones gave low yields of the desired β-alkyloxy product (10–20% yield, see Supporting Information).
Table 2.
Scope of the Ketone-Enaminyl Radical Precursor.a
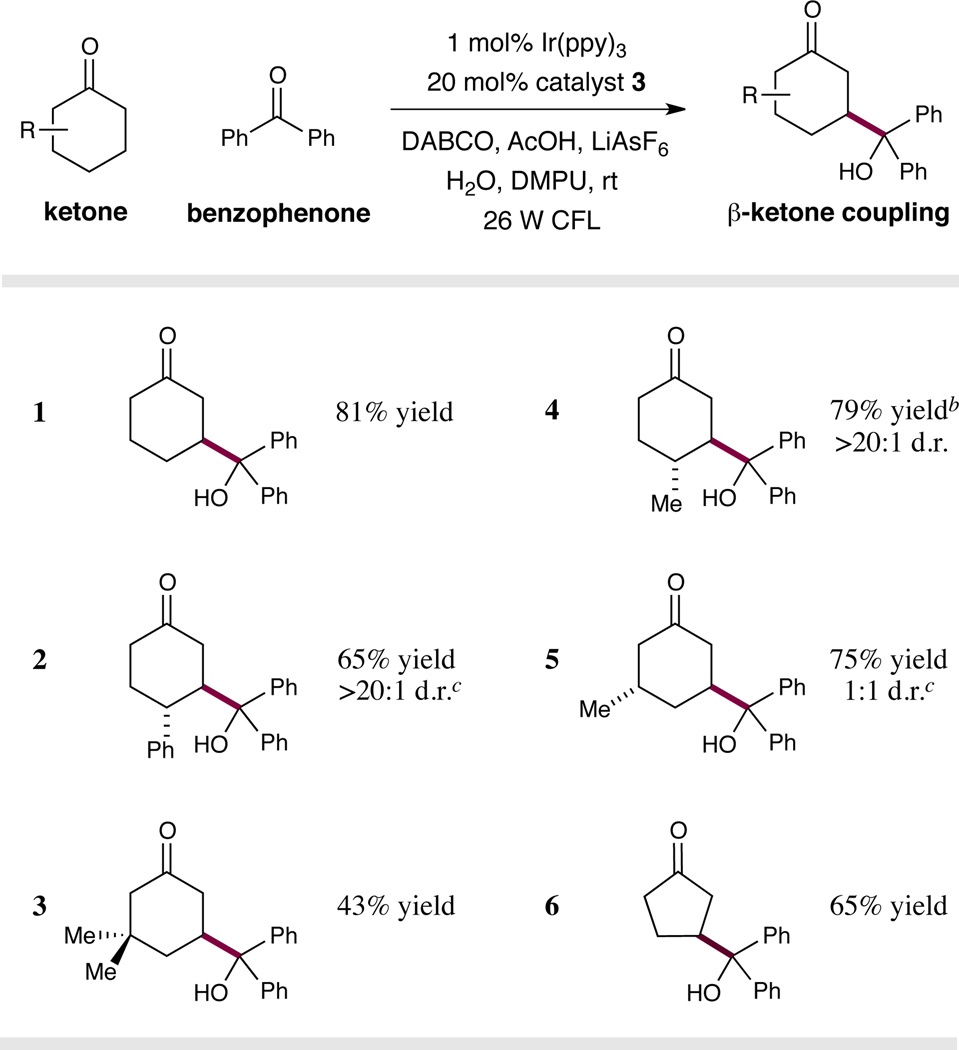 |
Reaction typically performed with 20 mol% of amine catalyst 3, 20 mol% of AcOH, 1 equiv. of LiAsF6, 2 equiv. of DABCO, 2 equiv. of water.
Reaction performed with 10 equiv. of water.
Diastereoselectivity determined by 1H NMR analysis.
We next sought to establish the scope of the ketyl radical substrate in this β-carbonyl functionalization reaction. As shown in Table 3, a range of substituted benzophenones can serve as viable coupling partners using our optimized conditions (entries 1–3, 56–81% yield) to furnish γ-hydroxyketone products with high levels of efficiency. Notably, a slightly diminished yield was obtained with 4-methoxybenzophenone (entry 2, 56% yield), likely due to the electron-donating group on the aromatic ring, which lowers the standard reduction potential of the ketone thereby rendering formation of the ketyl radical less thermodynamically favorable.
Table 3.
Scope of the Ketyl Radical Coupling Precursora
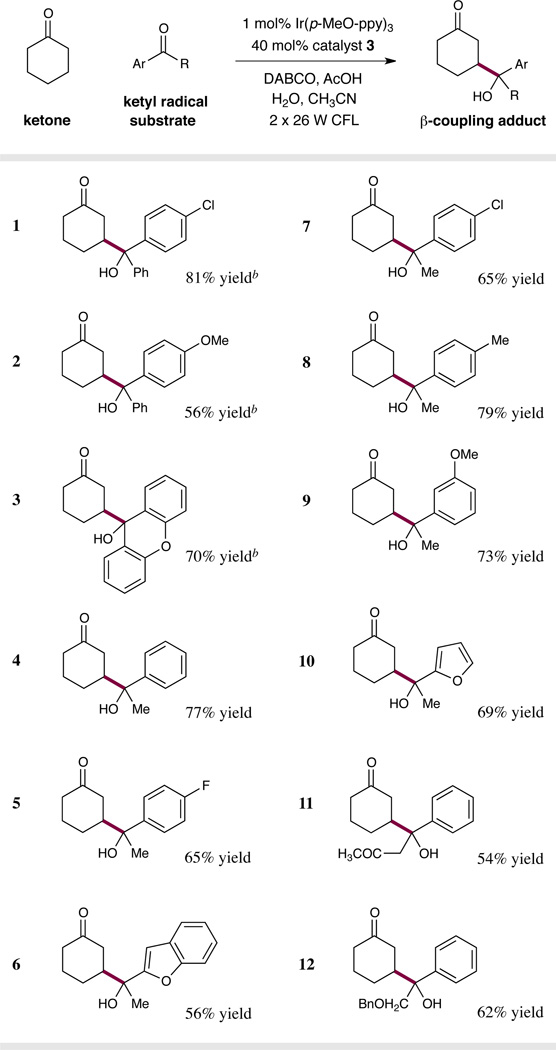 |
Diastereoselectivity, where relevant, was determined by 1H NMR analysis to be 1–1.2:1
Reaction conditions as performed in Table 2.
At this stage we turned our attention to aryl–alkyl ketone reaction partners, a more challenging substrate class given that ketyl radical formation would be thermodynamically disfavored with respect to the analogous benzophenone system. Indeed, initial efforts to achieve this photocatalytic β-heterocoupling reaction using acetophenone met with little success using our previously optimized conditions. We recognized that the excited state of the photocatalyst (9) is not sufficiently reducing to induce ketyl radical formation from acetophenone via electron transfer (E1/2red = −2.14 V vs. SCE)20 a reduction potential that is considerably lower than that of benzophenone. Fortunately, we identified Ir(p-MeO-ppy)3 as an effective photocatalyst for the reduction of aryl–alkyl ketones, and indeed, this system enabled a large increase in the ketyl-radical partner scope (Table 3, entries 4–12). We initially speculated that incorporation of electron-donating substituents on the photocatalyst aryl ligand would enhance the reduction potential of the IrL3 excited state, thereby allowing ketyl radical formation to become facile with aryl–alkyl ketones.29 However, subsequent studies suggest that the use of Ir(p-MeO-ppy)3 leads to a change in the order of electron transfer events with enamine oxidation becoming the primary interaction for the IrL3 excited state (vide infra). Using the Ir(p-MeO-ppy)3 catalyst, both electron-deficient (entries 5 and 7, 65% yield) and electron-rich (entries 8 and 9, 73–79% yield) acetophenone derivatives readily coupled with cyclohexanone to generate γ-hydroxyketone adducts. Moreover, heteroaryl–methyl ketones were also suitable reaction partners in this transformation (entries 6 and 10, 56–69% yield). Although higher alkyl homologs of acetophenone failed to participate in this β-functionalization protocol, reaction efficiency was recovered via the introduction of electron-withdrawing groups onto the carbonyl alkyl substituent (entries 11 and 12, 54–62% yield).
To further probe the mechanistic course of this transformation and the specific utility of each photocatalyst as a function of ketyl radical subclass, we performed a series of Stern–Volmer quenching studies. As expected, these experiments revealed that benzophenone quenches the excited state of Ir(ppy)3 (9) (Figure 1), lending support to our initial hypothesis presented in Scheme 1. However, similar experiments performed with acetophenone (or acetophenone in the presence of acetic acid)30 and Ir(p-MeO-ppy)3 revealed that no excited state quenching was observed with this ketone class, thereby demonstrating that an alternative mechanism is operative using aryl–alkyl ketone substrates (Figure 2). Indeed, pregenerated enamine 4 was found to quench*Ir(p-MeO-ppy)3, providing evidence that oxidation of the enamine occurs prior to reduction of acetophenone when aryl–alkyl ketones are employed. This change in the sequence of the oxidation and reduction steps in the photoredox cycle is consistent with the observed requirement for a different photocatalyst depending on the ketone acceptor employed (e.g. benzophenone = Ir(ppy)3 preffered, acetophenone = Ir(p-MeO-ppy)3 preffered).
Figure 1.

Ir(ppy)3 Emission Quenching with Benzophenone and Enamine 4.
Figure 2.

Ir(p-MeO-ppy)3 Emission Quenching with Acetophenone and Enamine 4.
In summary, through the synergistic merger of photoredox catalysis and organocatalysis, we have successfully achieved the direct β-coupling of cyclic ketones with diaryl or aryl–alkyl ketones to form γ-hydroxyketones. This approach features the concurrent in situ formation of two different radical species, generated via: (1) single-electron reduction of a diaryl or aryl–alkyl ketone and (2) single-electron oxidation of a transiently formed ketone-derived enamine. These catalytically formed radical intermediates are selectively coupled to provide direct access to synthetically useful γ-hydroxyketone motifs. Notably, we have found that the order of these single electron transfer events is dependent on the nature of the photocatalyst employed. To our knowledge, this approach represents the first example of selective functionalization of two distinct ketones in a predictable β-functionalization mechanism. Moreover, this transformation further provides a ketone–ketone selective heterocoupling in an intermolecular process without need for preactivation or masking of either of the carbonyl coupling partners.
Supplementary Material
Acknowledgement
Financial support was provided by the NIGMS (R01 GM103558-01) and kind gifts from Merck, Amgen, and AbbVie.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593. doi: 10.1126/science.1232993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang S-L, Xie H-X, Zhu J, Li H, Zhang X-S, Li J, Wang W. Nat. Commun. 2011;2:211. doi: 10.1038/ncomms1214. [DOI] [PubMed] [Google Scholar]; (c) Leskinen MV, Yip K-T, Valkonen A, Pihko PM. J. Am. Chem. Soc. 2012;134:5750. doi: 10.1021/ja300684r. [DOI] [PubMed] [Google Scholar]
- 2.Carey FA, Sundberg RJ. Advanced Organic Chemistry: Part B: Reactions and Synthesis. New York: Springer; 2001. [Google Scholar]
- 3.(a) Mukherjee S, Yang JW, Hoffmann S, List B. Chem. Rev. 2007;107:5471. doi: 10.1021/cr0684016. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Helmchen G, Rüping M. Asymmetric Synthesis–The Essentials. Wiley-VCH: Weinheim; 2006. [Google Scholar]
- 4.For direct β-functionalizations of amides, esters, or oximes, see: Zaitsev VG, Shabashov D, Daugulis O. J. Am. Chem. Soc. 2005;127:13154. doi: 10.1021/ja054549f. Wasa M, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2009;131:9886. doi: 10.1021/ja903573p. Stowers KJ, Kubota A, Sanford MS. Chem. Sci. 2012;3:3192. doi: 10.1039/C2SC20800H. Renaudat A, Jean-Gérard L, Jazzar R, Kefalidis CE, Clot E, Baudoin O. Angew. Chem. Int. Ed. 2010;49:7261. doi: 10.1002/anie.201003544.
- 5.(a) Yamamoto H. Lewis Acids in Organic Synthesis. New York: Wiley-VCH; 2000. [Google Scholar]; (b) Crabtree RH. The Organometallic Chemistry of the Transition Metals. Hoboken: Wiley-Interscience; 2005. [Google Scholar]; (c) Noyori R. Asymmetric Catalysis in Organic Synthesis. New York: Wiley-Interscience; 1994. [Google Scholar]; (d) Ojima I. Catalytic Asymmetric Synthesis. New York: Wiley-VCH; 2000. [Google Scholar]; (e) Lelais G, MacMillan DWC. Aldrichim. Acta. 2006;39:79. [Google Scholar]; (f) Taylor MS, Jacobsen EN. Angew. Chem. Int. Ed. 2006;45:1520. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- 6.(a) Mori Y, Sakaguchi Y, Hayashi H. J. Phys. Chem. A. 2000;104:4896. [Google Scholar]; (b) Mangion D, Arnold DR. Acc. Chem. Res. 2002;35:297. doi: 10.1021/ar010108z. [DOI] [PubMed] [Google Scholar]
- 7.For reviews on aldol reactions see: Evans DA, Nelson JV, Taber TR. Topics in Stereochemistry. New York: Wiley; 1982. Nelson SG. Tetrahedron: Asymmetry. 1998;9:357. Denmark SE, Stavenger RA. Acc. Chem. Res. 2000;33:432. doi: 10.1021/ar960027g. Bisai V, Bisai A, Singh VK. Tetrahedron. 2012;68:4541. Schetter B, Mahrwald R. Angew. Chem. Int. Ed. 2006;45:7506. doi: 10.1002/anie.200602780. Trost BM, Brindle CS. Chem. Soc. Rev. 2010;39:1600. doi: 10.1039/b923537j. Mahrwald R. Modern Aldol Reactions. Berlin: Wiley-VCH; 2004. Heathcock CH. Asymmetric Synthesis 3. New York: Academic Press; 1984. Heathcock CH. Comprehensive Organic Synthesis 1. Oxford: Pergamon Press; 1991.
- 8.Wurtz A. Bull. Soc. Chim. Fr. 1872;17:436. [Google Scholar]
- 9.(a) Burstein C, Glorius F. Angew. Chem. Int. Ed. 2004;43:6205. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]; (c) Nair V, Vellalath S, Poonoth M, Mohan R, Suresh E. Org. Lett. 2006;8:507. doi: 10.1021/ol052926n. [DOI] [PubMed] [Google Scholar]; (d) Zeitler K. Angew. Chem. Int. Ed. 2005;44:7506. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]; (e) Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lettan RB, II, Galliford CV, Woodward CC, Scheidt KA. J. Am. Chem. Soc. 2009;131:8805. doi: 10.1021/ja808811u. [DOI] [PubMed] [Google Scholar]
- 10.(a) Fu Z, Xu J, Zhu T, Leong WWY, Chi YR. Nat. Chem. 2013;5:835. doi: 10.1038/nchem.1710. [DOI] [PubMed] [Google Scholar]; (b) Mo J, Shen L, Chi YR. Angew. Chem. Int. Ed. 2013;52:8588. doi: 10.1002/anie.201302152. [DOI] [PubMed] [Google Scholar]
- 11.Büechi G, Wuest H. J. Org. Chem. 1969;34:1122. [Google Scholar]
- 12.(a) Hoppe D, Hense T. Angew. Chem. Int. Ed. Engl. 1997;36:2282. [Google Scholar]; (b) Hoppe D. Angew. Chem. Int. Ed. Engl. 1984;23:932. [Google Scholar]; (c) Weisenburger GA, Beak P. J. Am. Chem. Soc. 1996;118:12218. [Google Scholar]
- 13.Nakamura E, Aoki S, Sekiya K, Oshino H, Kuwajima I. J. Am. Chem. Soc. 1987;109:8056. [Google Scholar]
- 14.Kang JY, Connell BT. J. Am. Chem. Soc. 2010;132:7826. doi: 10.1021/ja910057g. [DOI] [PubMed] [Google Scholar]
- 15.Hosomi A, Hashimoto H, Sakurai H. J. Org. Chem. 1978;43:2551. [Google Scholar]
- 16.(a) Nakamura E, Kuwajima I. J. Am. Chem. Soc. 1977;99:7360. [Google Scholar]; (b) Burke ED, Lim NK, Gleason JL. Synlett. 2003:390. [Google Scholar]
- 17.The formation of this β-coupling product was previously observed with the preformed enamine 4 and benzophenone using a high-energy light-promoted protocol: Kawanisi M, Kamogawa K, Okada T, Nozaki H. Tetrahedron. 1968;24:6557.
- 18.(a) Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nagib DA, MacMillan DWC. Nature. 2011;480:224. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dedeian K, Djurovich PI, Garces FO, Carlson G, Watts RJ. Inorg. Chem. 1991;30:1685. [Google Scholar]
- 21.(a) Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top. Curr. Chem. 2007;281:143. [Google Scholar]; (b) Dixon IM, Collin J-P, Sauvage J-P, Flamigni L, Encinas S, Barigelletti F. Chem. Soc. Rev. 2000;29:385. [Google Scholar]
- 22.Wagner PJ, Truman RJ, Puchalski AE, Wake R. J. Am. Chem. Soc. 1986;108:7727. doi: 10.1021/ja00284a041. [DOI] [PubMed] [Google Scholar]
- 23.(a) Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tarantino KT, Liu P, Knowles RR. J. Am. Chem. Soc. 2013;135:10022. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- 24.Schoeller WW, Niemann J. J. Chem. Soc. Perkin Trans. 2. 1988:369. [Google Scholar]
- 25.Bard AJ. Encyclopedia of Electrochemistry of the Elements. New York: Marcel Dekker; 1978. [Google Scholar]
- 26.The use of DMPU is related to the requirement of high-dielectric media for efficient electron transfer processes (e.g. DMF, DMSO, etc. are also viable although slightly less effective).
- 27.It is well known that azepane and pyrrolidine amines are nucleophilic, due to the capacity to donate electrons from nitrogen to alleviate ring strain (not found to the same level with piperidine). We believe the use of an azepane catalyst leads to a more nucleophilic 5πe− system as a result of this phenomenon. Work is ongoing to further define this electronic effect.
- 28.We have tried several additives such as LiCl, LiClO4, LiBF4, KAsF6, etc; however, slightly improved yields were obtained with LiAsF6
- 29.(a) Weinaug UJ, Ammermann S, Gargouri H, Hoping M, Erk P, Kahle K, Lennartz C, Molt O, Münster I, Tamm M, Kowalsky W, Johannes H-H. Proc. of SPIE. 2008;7051:705108. [Google Scholar]; (b) Grushin VV, Herron N, LeCloux DD, Marshall WJ, Petrov VA, Wang Y. Chem. Commun. 2001:1494. [Google Scholar]
- 30.This experiment reveals that proton coupled electron transfer (PCET) is not operative in the reduction of acetophenone via the excited state of Ir(p-MeO-ppy)3. This does not rule out the possibility of PCET in the reduction of acetophenone from the Ir(II) species arising from oxidation of enamine 4.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.