

Abstract
Extracellular vesicles (EVs) are nano-sized vesicles released by normal and diseased cells as a novel form of intercellular communication, and can serve as an effective therapeutic vehicle for genes and drugs. Yet, much remains unknown about the in vivo properties of EVs such as tissue distribution, and blood levels and urine clearance - important parameters that will define their therapeutic effectiveness and potential toxicity. Here we combined Gaussia luciferase and metabolic biotinylation to create a sensitive EV reporter (EV-GlucB) for multimodal imaging in vivo, as well as monitoring of EV levels in the organs and biofluids ex vivo after administration of EVs. Bioluminescence and fluorescence-mediated tomography imaging on mice displayed a predominant localization of intravenously administered EVs in the spleen followed by the liver. Monitoring EV signal in the organs, blood and urine further revealed that the EVs first undergo a rapid distribution phase followed by a longer elimination phase via hepatic and renal routes within six hours, which are both faster than previously reported using dye-labeled EVs. Moreover, we demonstrate systemically injected EVs can be delivered to tumor sites within an hour following injection. Altogether, we show the EVs are dynamically processed in vivo with accurate spatiotemporal resolution, and target a number of normal organs as well as tumors with implications for disease pathology and therapeutic design.
Keywords: Exosomes, microvesicles, bioluminescence, fluorescence, biotin, biodistribution, delivery
Exosomes and microvesicles, collectively termed extracellular vesicles (EVs), are nanometer sized (40 – 1,000 nm diameter) particles endogenously released by cells, capable of delivering lipids, proteins, mRNAs, microRNAs (miRNAs) and other non-coding RNAs.1,2 While specialized physical conduits, such as membrane nanotubes and gap junctions are spatially limited and require direct cell-to-cell contacts,3,4 EVs can mediate communication between both neighboring and distant cells, thereby emerging as a novel form of intercellular communication and delivery vehicle. Under pathological conditions such as cancer, tumors produce an abundance of EVs that contain a select subset of cellular proteins, mRNAs and miRNAs that manipulate normal cells in their microenvironment to enhance angiogenesis, invasiveness, immune suppression and tumor growth.5-7 Further, EVs released from melanoma cells are capable of preconditioning sentinel lymph nodes and bone marrow progenitor cells to facilitate metastases of these tumors.8,9 In addition, recent studies have shown promising results in using EVs as novel therapeutic vehicles in cancer immunotherapy and suicide therapy, as well as delivery of RNA-interference and drugs.1,2,10-12 Unlike its artificial synthetic counterpart - liposomes, EVs are poorly immunogenic, while still capable of shielding “therapeutic cargoes” from rapid degradation in vivo, as well as overcoming biological barriers, such as the blood-brain barrier.3,4,13-17 However, much remains unexplored about the in vivo properties of EVs, including tissue distribution, blood levels and clearance dynamics – important parameters that will define their therapeutic effectiveness, and potential toxicity in clinical applications.
Here we designed a highly sensitive and versatile EV reporter system that enables multimodal in vivo imaging, as well as tracking of EV biodistribution and clearance of exogenous EVs over time. We engineered EVs to display a membrane reporter, termed EV-GlucB, consisting of Gluc fused to a biotin acceptor domain, which is metabolically biotinylated when expressed in mammalian cells in the presence of biotin ligase. These EVs exhibit a strong bioluminescent signal when incubated with the Gluc substrate, coelentrazine (CTZ). In addition, biotin on the surface allows EVs to be conjugated to any labeled streptavidin, which can then be imaged non-invasively in vivo using different techniques including fluorescence-mediated tomography (FMT). Furthermore, ex vivo analysis of tissues/blood/urine with the Gluc assay allows evaluation of biodistribution and clearance of EVs. Combing this new EV reporter system with non-invasive in vivo imaging and ex vivo analyses, we examined the fate of systemically injected EVs in mice.
RESULTS AND DISCUSSION
Generation of EV-GlucB
To display both Gluc and biotin on the surface of EVs, we took advantage of our previously published reporter consisting of a fusion between a membrane-bound variant of the Gluc reporter and a biotin acceptor peptide (BAP; GlucB), which exposes both on the surface of cells and vesicles.5-7,18 Human embryonic kidney (HEK) 293T cells were stably transduced with lentivirus vectors encoding two expression cassettes: 1) GlucB or Gluc (control, with Gluc being a secreted protein), and green fluorescent protein (GFP) separated by an internal ribosome entry site (IRES), and 2) sshBirA-IRES-mCherry, a biotin ligase codon optimized for mammalian gene expression and present within the secretory pathway (Figure 1, 2a).8,9,19 Western blot analysis of the cells confirmed GlucB and Gluc expression, and showed sshBirA greatly enhances biotinylation of the GlucB reporter (Figure 2b). EVs were next evaluated by transmission electron microscopy which demonstrated successful surface display of Gluc and biotin on EV-GlucB vesicles, and not on EV-Gluc vesicles (Figure 2c, d). To further confirm that GlucB labels and yields functional biotinylation of EVs, isolated EVs were dot blotted on nitrocellulose membranes followed by probing with streptavidin-horseradish peroxidase (HRP) which demonstrated a quantity-dependent biotinylation of EV-GlucB, and no biotinylation of EV-Gluc (Figure 2e). Nanoparticle tracking analysis (NTA) showed a similar size distribution pattern between EV-Gluc and EV-GlucB vesicles, indicating labeling of EVs with GlucB does not alter physical properties of EVs when compared to unlabeled EV-Gluc (control) (Figure 2f).
Figure 1. Schematic diagram for in vivo multimodal imaging of EVs.

(a) Membrane-bound Gluc (GlucB) or Gluc (control), and the secreted form of humanized bacterial biotin ligase (sshBirA) were delivered via lentivectors to HEK 293T cells for stable expression. (b) Upon expression, and EV production by the cells, the sshBirA tags the BAP sequence of GlucB with a single biotin moiety at a specific lysine residue which is then displayed on the cell surface,20 as well as on the EV surface. EVs were isolated from conditioned medium of cells and injected intravenously (IV) via tail or retro-orbital veins into nude mice for bioluminescence and fluorescent mediated tomography (FMT) imaging. For bioluminescence imaging, coelentrazine, a Gluc substrate, was IV-administered immediately prior to imaging. For FMT imaging, isolated EVs were conjugated with streptavidin-Alexa680 prior to administration into nude mice. (c) EVs derived from cells synthesizing naturally secreted Gluc were used as controls as the Gluc is not present in the EVs. Abbreviations: BAP, biotin acceptor peptide (BAP); CMV, cytomegalovirus; GFP, green fluorescent protein; hBirA, humanized biotin ligase; hGluc, humanized Gaussia luciferase; IRES, internal ribosome entry site; ss, signal peptide; TM, transmembrane domain of platelets-derived growth factor receptor.
Figure 2. GlucB and sshBirA label and biotinylate EVs on the surface.

(a, b) Stable HEK293T cells expressing sshBirA with GlucB or Gluc. (a) Live-cell imaging of HEK293T cells stably transduced with GlucB-IRES-GFP or Gluc-IRES-GFP vectors, both with sshBirA-IRES-mCherry. Bar, 100 μm. (b) Western blot analysis showing enhanced biotinylation of cells stably expressing GlucB with sshBirA, as compared to GlucB alone. No biotinylation was detected in cells expressing Gluc alone or Gluc with sshBirA. Immunoblotting with anti-Gluc antibodies showed Gluc and GlucB at expected sizes (Gluc: 20 kDa; GlucB: 42 kDa). A low level of biotinylated BAP domain (22 kDa) was also detected in GlucB and GlucB + sshBirA samples. Mock transduced HEK293T were used as a negative control. GAPDH was immunoprobed as a loading control. (c, d) Transmission electron micrograph (TEM) demonstrating biotinylation and Gluc labeling of EV-GlucB on the membrane. (c) Sectioned EVs were immunolabeled with either anti-CD63, an exosome marker,21 or anti-Gluc antibody followed by gold-conjugated secondary antibody to visualize GlucB labeling of EVs on the membrane, with EV-Gluc showing no Gluc signal but having the CD63 signal. Bar, 100 nm. (d) EVs in suspension were immunolabeled with an anti-biotin antibody followed by 10 nm gold-conjugated secondary antibody and biotinylation of EV-GlucB, but not EV-Gluc surface was detected. (e) Dot blot detection of biotinylated EVs. EVs isolated from HEK293T cells stably expressing sshBirA with either GlucB (top) or Gluc (bottom) were dot blotted on nitrocellulose membranes in a dose range followed by probing with streptavidin-HRP and chemiluminescence detection. EV-GlucB showed quantity-dependent biotinylated EVs, whereas EV-Gluc control exhibited no biotinylation background signal. (f) Nanoparticle tracking analysis (NTA) of EVs. Similar size distribution between EV-Gluc (peaks: 67, 100 and 177 nm) and EV-GlucB (81, 104 and 175 nm) vesicles was detected.
EV-GlucB exhibited EV-specific Gluc activity and stability in biofluids ex vivo
To examine whether EV-GlucB contained active luciferase, isolated EVs were subjected to sucrose gradient fractionation followed by Gluc activity assay. EV-GlucB exhibited a greater than 106-fold increase in Gluc activity when compared to EV-Gluc (control) in vesicle-containing fractions (Figure 3a; #3-6). Moreover, the majority of Gluc activity in EV-Gluc was detected in the top layer above the sucrose gradient, indicating that the naturally secreted Gluc is not incorporated into EVs and remain mostly as free protein (Figure 3b). Proteins collected from the pelleted fractions were further analyzed by Western blotting and showed specific EV labeling of both Gluc and biotin in fractions 3-6 of the EV-GlucB samples, coinciding with fractions containing the exosomal marker, Alix (Figure 3c, d).21 To test whether EV-GlucB is stable or may degrade rapidly in biofluids, blood and urine samples obtained from athymic nude mice spiked with EV-GlucB and incubated at 37 °C over 24 h showed no significant loss in Gluc activity. (Figure 3e, f). Therefore, biofluid samples collected at different time points from the following in vivo experiments should reveal the level of EV-GlucB biofluids at the time of collection, but not necessarily the stability of EV-GlucB in circulation in vivo. These findings confirm EV-specificity of the EV-GlucB reporter, as well as stability of the luciferase activity in biofluids ex vivo.
Figure 3. EV-GlucB exhibited EV-specific Gluc activity, and was stable in blood and urine ex vivo over time.

(a, b) Gluc activity assay of EV-GlucB (a) and EV-Gluc (b) vesicles following sucrose gradient fractionation. Note distinct increase in Gluc activity of EV-GlucB in EV-containing fractions (#3, 4, 5) when compared to EV-Gluc. (c, d) Western blot analysis of proteins extracted from pelleted fractions demonstrated significant Gluc expression and biotinylation of EV-GlucB (c), but not on EV-Gluc (d). Exosomal marker, Alix (95 kDa), was immunoprobed to identify exosome-containing fractions. Cell lysates of HEK293T cells stably expressing sshBirA with GlucB and Gluc were used as positive controls. (e, f) Gluc activity of EV-GlucB was stable in biofluids ex vivo over 24 h. Blood (e) and urine (f) collected from untreated animals were spiked with EV-GlucB vesicles, incubated at 37°C, and samples collected at different time points over 24 h. No significant loss of EV-GlucB signal was detected in either type of biofluid. P > 0.05 by one-way analysis of variance (ANOVA) at all time points.
Multimodal in vivo imaging of IV-administered EV-GlucB
To visualize and track the distribution of intravenously administered EVs in vivo, EV-GlucB or phosphate buffered saline (PBS; control) were injected into athymic nude mice via the retro-orbital vein. Thirty min post-EV treatment, CTZ injection revealed a significant amount of Gluc signal in the spleen and liver in EV-GlucB-injected mice, but not the controls (Figure 4a, b). This observation was confirmed by quantitation of average bioluminescent radiance from ventral side images, which showed a significantly higher signal in the liver and spleen of EV-GlucB-treated mice when compared to controls (Figure 4c).
Figure 4. In vivo imaging of IV-administered EVs.

(a, b) Bioluminescence imaging of EV-GlucB in athymic nude mice. Animals were administered a bolus of either PBS (control) or EV-GlucB via the retro-orbital vein. CTZ was injected by the same route at 30 min post-initial administration to image EV-GlucB. (a) Representative image showing dorsal side of nude mouse with a prominent signal at regions corresponding to the spleen (arrow) in EV-GlucB-administered animals. (b) Imaging of ventral side showing a significant signal at regions corresponding to the spleen (arrow) and liver (arrowhead) in EV-GlucB-treated mice. No appreciable signal was detected on either side of PBS-injected mice. (c) Quantitation of EV-GlucB signal from bioluminescence imaging at ventral regions corresponding to the liver and spleen at 60 min post-EV administration. Sr, steradian. *, P < 0.05 by Student’s t-test. (d, e) FMT imaging of EV-GlucB in athymic nude mice. Alexa680-conjugated EV-GlucB or PBS (control) were administered via the tail vein and imaged with FMT at 30 min post-injection. (d) Single Z plane of FMT imaging showing elevated fluorescence signal predominantly at the spleen (arrow) and the liver (arrowhead) in Alexa680-EV-GlucB, but not in control-treated mice. (e) 3D representation of FMT imaging illustrating Alexa680-EV-GlucB localizing mainly to the spleen (arrow) and the liver (arrowhead). A low level of background signal was also detected in the control.
To test the multimodality imaging capability of this EV reporter, EV-GlucB or PBS were labeled with streptavidin-Alexa680 conjugate followed by centrifugal filtration to remove unbound residues. The labeled EVs were then injected via the tail vein into athymic nude mice and imaged with FMT (30 min later) which revealed EV localization to the spleen and the liver similar to the distribution seen by Gluc bioluminescence imaging (Figure 4d, e).
Biodistribution of IV-administered EV-GlucB
To confirm findings from in vivo EV imaging and further investigate tissue distribution of IV-injected EVs, organs were collected at different time points post-EV treatment to assess for Gluc activity. In agreement with results from in vivo imaging experiments, highest EV signal was detected in the spleen followed by the liver, then the lungs and kidneys (Figure 5a). By comparison the brain, heart and muscle showed lower amounts of signal across all time points (Figure 5b). All the above tissues were evaluated in their entirety, while a portion of skeletal muscle (hind leg) was sampled as an internal control to assess EV signal in non-organ tissues. Notably, whereas EV signal decreased by more than half from 30 to 60 min in the liver and the kidneys (liver: 27,792 ± 4,171 to 12,620 ± 2,589 RLU; kidneys: 5,448 ± 1102 to 2162 ± 192 RLU), the spleen and the lungs levels stayed relatively constant with no significant reduction in EV signal during this period (spleen: 51,227 ± 18,146 to 50,899 ± 8,285 RLU; lungs: 3,438 ± 335 to 4,090 ± 821 RLU). At 360 min, the spleen and the heart had the most EV signal remaining as compared to the 30 min time point (spleen: 46.9 ± 22.4 %; heart: 31.6 ± 9 % of initial levels), whereas the lungs, the liver and the kidneys showed the least (lung: 13.3 ±2.4 %; liver: 3.9 ± 1.3 %; kidneys: 3.6 ± 0.6 %). By contrast, the brain reached baseline signal at 120 min time point (Supporting Information Figure 1), indicating minimal EV distribution to this organ. Interestingly, the muscle retained a significant fraction of activity (muscle: 45.1 ± 9 %) out to 120 min.
Figure 5. Biodistribution and retention of IV-administered EV-GlucB.

(a, b) Biodistribution of IV-injected EV-GlucB via tail vein over time. Gluc activity was measured from organs collected from mice at different time points following EV-GlucB injection without transcardial perfusion with PBS. (c, d) Tissue retention of IV-injected EV-GlucB via tail vein over time in perfused tissues. Organs were collected from EV-injected animals at the same time points as above following transcardial perfusion with PBS. (e) Subcellular visualization of EV-GlucB in perfused kidneys by confocal microscopy. Kidney samples were collected from mice 30 min after IV-administration of EV-GlucB or EV-Gluc and transcardial perfusion with PBS, then cryosectioned and immunostained with anti-Gluc (rabbit) and Alexa Fluor® 647 goat anti-rabbit antibodies. EV-GlucB (arrow) was detected in a punctuate pattern in the perinuclear region of renal cells. EV-Gluc was used as a negative control for nonspecific binding or packaging of secreted Gluc protein in EVs and unspecific binding of the anti-Gluc antibodies to EVs. Nuclei were visualized by 4,6-diamidino-2-phenylindole (DAPI). DIC, differential interference contrast. Bar, 10 μm.
Previous studies have used lipophilic dyes to label EVs to study their in vivo properties as they, at least initially, provide a robust signal for EV detection.8,13,14,17,22 However, these dyes, including PKH26 (red fluorescent dye), PKH2 and PKH67 (green fluorescent dyes) are reported to have an in vivo half-life ranging from 5 to >100 days. Thus while the dyes may assist in “marking a trail” of where the administered EVs have been trafficked to in vivo, the persistence of the dye may outlast the labeled EVs in vivo. That is dye-labeled EVs may be degraded and/or recycled in vivo while the dyes themselves remained intact and visible in the tissues over time, yielding inaccurate spatiotemporal information regarding the fate of the EVs in vivo. The reporter described here employs Gaussia luciferase (Gluc) which emits flash bioluminescence (480 nm peak) that is over 1,000-fold more sensitive than commonly used Renilla and firefly luciferases.23 Moreover, metabolic biotinylation of surface receptors using BAP and bacterial biotin ligase allows multimodal imaging in vivo, including magnetic resonance imaging (MRI), single-photon emission computed tomography/positron emission tomography (SPECT/PET) and FMT (this study).18 Combining both Gluc and biotin, we created an EV-specific reporter with high sensitivity and multimodal imaging capacity to study in vivo dynamics of systemically administered EVs.
Biodistribution analysis of GlucB labeled EVs also revealed a short half-life of less than 30 minutes in vivo in most tissues. Whereas most EV-GlucBs were cleared from the animals by 6 hours post-injection, previous reports detected a significant amount of dye-labeled EVs in tissues out to 24 h.8,22 Similar to studies with systemically delivered dye-labeled EVs at early time points,8,22 we observed the highest EV-GlucB levels in the spleen followed by the liver, kidneys, and lungs. On the other hand, melanoma-derived EVs (approximately 5 μg; < 50 nm in diameter) were found to localize mostly to the liver followed by the lungs, kidneys and spleen.24 The difference may be attributed to the cell type producing the EVs used, as well as a variation in EV isolation methods, both of which dictate the shape, size, surface protein, lipid composition and population of purified EVs.25,26 In addition, un-processed dye following EV clearance and/or degradation may remain in tissues, resulting in inaccurate spatiotemporal resolution of EVs in vivo. Based on a previous study carried out with different sized liposomes,27 it is inferred that ≤220 nm-sized EVs have an overall higher cargo encapsulation ratio than ≤ 50 nm-sized EVs, making ≤ 220 nm a model size range for EVs used here. While it is beyond the scope of the current study, it remains to be investigated whether EV donor cell type and preparation protocol affect subsequent biodistribution and clearance in vivo.
Retention of IV-administered EV-GlucB in organs
To elucidate organ uptake and retention of IV-administered EVs into tissues per se rather than into the combination of tissues and blood within them, EV injected animals were transcardially perfused with PBS before collecting the organs/muscle at different time points. Surprisingly, perfused kidneys, and not the spleen, showed the highest EV signal followed by the liver, lung, heart, brain and muscle (Figure 5c, d). Perfused spleen showed minimal EV-GlucB signal across all time points, suggesting EVs are not efficiently taken up into these cells despite the high amount of EVs present in blood passing through the highly vascularized spleen (Figure 5a, c). Perfused liver and lungs showed a similar trend in EV signal reduction from 30 to 60 min (liver: 60,783 ± 7,565 to 20,118 ± 1,666; lung: 4,532 ± 1,114 to 4,148 ± 861 RLU) as compared to their non-perfused counterparts, indicating the EVs are actively taken up by these organs, likely for processing and degradation (Figure 5c). The brain, heart and muscle also showed a comparable trend in EV signal reduction during this period (brain: 910 ± 152 to 529 ± 39; heart: 2,849 ± 1,814 to 1,814 ± 376 RLU; muscle: 410 ± 40 to 50 ± 3 RLU), showing EV-GlucB are delivered and taken up by these organs, albeit in low amounts (Figure 5d).
To verify elevated EV-GlucB localization in the perfused kidneys, cryosections of the kidneys were immunolabeled with anti-Gluc antibodies, and a robust signal was detected only in EV-GlucB-injected, and not EV-Gluc control samples (Figure 5e). Interestingly, most EV-GlucB staining was detected as punctate in the perinuclear region of the renal cells, similar to previous in vitro finding when recipient cells were treated with dye labeled EVs.5,8,28 However, we speculate the heightened EV retention in the perfused kidneys is an artifact of perfusion where EVs in the blood are forced into and trapped by the kidneys pending urination, resulting in greatly elevated EV retention in the perfused kidneys.
Very limited EV signals were detected in the brain and heart, indicating that only a small amount of EVs are delivered (possibly due to the blood-brain barrier for the brain) and/or taken up there.13 Notably, since hind legs muscle samples (with an average weight of 87 mg per sample; approximately 1% of total skeletal muscle) consistently showed an appreciable level of EV-GlucB signal, and considering that skeletal muscles in a female mouse makes up to 64% of body weight,29 a significant portion of EV-GlucB may be delivered to the skeletal muscles via IV administration.
The predominant EV localization in the non-perfused spleen may be attributed to: i) the applied EV dosage (100 μg) which may be an excess amount resulting in saturation of liver macrophages, which bind and endocytose particles, resulting in higher “free” EV level in the blood and consequently “spillover” into the spleen vasculature27, and/or ii) lymphocytes or macrophages, which bind or absorb the EVs in the blood and traffic to the spleen. In fact, transcardial perfusion revealed that the spleen tissue itself retained little EV signals, suggesting that it serves as a transitory reservoir under conditions of excess EVs. It remains to be determined if lower dosages of EVs would abolish this phenomenon with increasing levels in the liver relative to the spleen for clinical considerations. On the other hand, and if this interpretation is valid, it may be possible to utilize the high dosage as a means to prolong EV exposure to the spleen for therapeutic strategies and use it as a “storage depot” for subsequent delivery to other tissues.
Blood level and urine clearance of IV-administered EV-GlucB
To investigate the dynamics of exogenously administered EVs in vivo, blood and urine were collected over time from EV-GlucB-injected animals, and luciferase activity was measured (Figure 6a, b). Highest EV-GlucB signal was detected in the blood at the earliest time point measured, 30 min, followed by a rapid decrease at 60 min and then a gradual decrease from 90 min to 360 min (Figure 6a). A two-phase exponential decay equation model revealed that EVs in the blood first undergo a distribution phase with a short half-life of 19.9 ± 7.7 min followed by an elimination phase with a longer half-life of 184.5 ± 7.4 min. By contrast, EV-GlucB signal in the urine peaked at 60 min followed by a rapid reduction from 60 to 120 min (2.07 ± 0.58 × 10−3 to 0.63 ± 0.08 ×10−3 % ID) and then a gradual decrease from 120 to 360 min (0.09 ± 0.04 ×10−3 % ID) (Figure 6b). While levels of EV-GlucB activity in the blood and organs were highest at 30 min followed by a steady reduction out to 360 min, highest levels of EV-GlucB in urine occurred at 60 and 120 min, suggesting that a small amount of EV-derived GlucB is cleared via the renal route following the distribution phase and hepatic clearance. At 360 min, minimal signal was detected in the blood while some organs (notably the spleen) and the urine still retained a small portion of signal, indicating a small proportion of EV-GlucB remains active in the organs and then is cleared via the kidneys.
Figure 6. EV-GlucB distribution in biofluids.
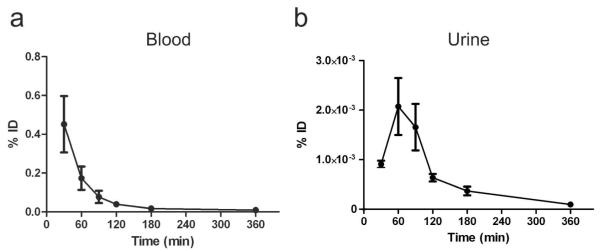
Blood (a) and urine (b) were collected at various time points after IV-injection of EV-GlucB, and Gluc activity was measured to evaluate blood levels and urine clearance of EV-GlucB. % ID, percent of initial dose.
These findings also support the heightened EV retention in the perfused kidneys probably as an artifact of perfusion, where EVs in the blood are forced into and trapped by the kidneys. This is shown by the significant reduction in EV-GlucB levels in the perfused kidneys from 30 to 60 min post treatment, which coincides with the increasing levels of Gluc activity detected in the urine during the same time period, indicating a small but appreciable fraction of vesicular protein is removed via renal clearance.
Altogether, most EV-GlucB was cleared from the organs and biofluids by 360 min post-IV administration, indicating active cellular uptake and degradation of the EVs by different cell types. Since the liver and lung showed the highest retention, we hypothesis the EVs, similar to liposomes, are actively taken up by phagocytic cells such as Kupffer cells and alveolar macrophages in the liver and lung, respectively, with eventual lysosomal degradation.30 Albeit in a lower amount, a significant level of the reporter signal was detected in the urine. Combined with the observation that the EV signal was greatly elevated in the kidneys following transcardial perfusion, showing the EVs do circulate to that organ, we speculate that a small portion of the IV-administered EVs may be internalized by the kidney cells, such as podocytes and subjected to degradation for release into the urine. The detailed cellular mechanism for internalization and processing of circulating EVs in vivo in different organs remains to be examined and warrants future investigations.
Systemic administered EV-GlucB was quickly trafficked to tumors
Since solid tumors are often densely vascularized due to aberrant angiogenesis,31 we speculated that solid tumors, like highly vascularized spleen, may accumulate EVs following systemic administration. To examine this possibility, athymic nude mice with subcutaneous Gli36-mCherry human glioma xenograft tumors in the left and right chest regions were injected with EV-GlucB via the tail vein, and showed a prevalent level of EV signals at the tumor sites at 60 min post-administration as revealed by bioluminescence imaging (Figure 7a). Upon quantitation of EV-GlucB signals in isolated liver, spleen and tumors, the tumors showed a significant amount of Gluc signal (Figure 7b), suggesting IV-injected EV-GlucB can be actively delivered to these tumors via systemic administration.
Figure 7. EV-GlucB localization to subcutaneous xenograft tumors.
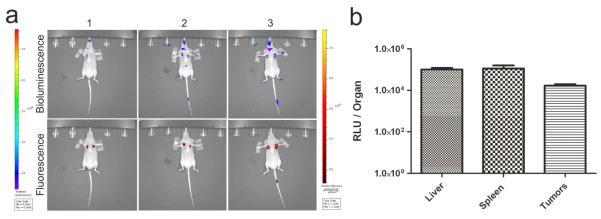
(a) Bioluminescence imaging for EV-GlucB administered IV (top row) into nude mice bearing subcutaneous xenograft tumors visualized by fluorescence imaging (bottom row; images shown for three mice). Stable Gli36 human glioma cells expressing mCherry (Gli36-mCherry) were implanted subcutaneously into left and right chest regions of athymic nude mice. Upon tumor development, animals were administered with a bolus of 100 μg EV-GlucB via the tail vein, and CTZ was introduced via the retro-orbital vein at 60 min post-EV injection to track EV-GlucB by bioluminescence (left bar; photon/sec/cm2/sr). Tumor locations were verified by mCherry signal in radiant efficiency (right bar; [photon/sec/cm2/sr]/[μm/cm2]) via fluorescence imaging. (b) Quantitation of EV-GlucB distribution in five tumor-bearing mice. Gluc activity was determined from the livers, spleen and tumors collected at 60 min post-EV administration. P < 0.09 by one-way ANOVA with Tukey’s post-hoc test.
CONCLUSIONS
In this study, we addressed the in vivo properties of IV-administered EVs with accurate spatial and temporal resolution. For this, one needs a reporter that enables the following functions: i) specific labeling of EVs; ii) a multimodal platform for non-invasive EV imaging in vivo to accommodate different imaging techniques; and iii) a robust and stable reporter that can signal nanometer-sized EVs in the organs, blood and urine over time. The biotinylated EV-GlucB system described here fulfilled these demands and enabled tracking and monitoring of EVs, thereby revealing dynamic processing of EVs through different organs and clearance from blood and urine over time.
As EVs are negatively charged with phospholipids on their outer surface,32 our findings are in line with pharmacokinetic studies using negatively charged, radiolabeled liposomes that reported an association of IV-administered liposomes with the mononuclear phagocyte system – mainly in the liver and spleen.27,33 Moreover, only liposomes with 160 and 460 nm diameters showed an elevated disposition to the spleen and a decline in the liver with increasing concentration following IV-administration when compared to that of 58 nm-sized liposomes,27 inferring a comparable size and dose-dependent biodistribution of EV-GlucB. Different amounts of EVs ranging from 4 to 150 μg protein content have been administered in mouse models,8,13,22,24 and 100 μg protein of EVs (≤ 220 nm in diameter) per mouse was used here as an experimental dosage, making these studies comparable. Future studies on the potential correlation between EV size, dosage and pharmacokinetic properties will provide additional insights into EV-mediated therapies.
Notably, since cellular uptake of EVs depends on specific surface lipids and ligand-receptor pairing,34-36 human HEK293T-derived EVs used in this study may exhibit a distinct biodistribution profile from that of mouse cell-derived EVs in the current mouse model and this warrants future investigation. Moreover, as vesicular proteins vary from one cell type to the other,37 it would be of interest to employ the high sensitivity of the current method to compare in vivo properties of endogenously released EV from different cell types, including normal and disease-associated cells. Given that cancer cells are known to produce an abundant amount of EVs and can display onco-proteins such as EGFRvIII on their surface,7,8 cancer-derived EVs will likely exhibit different circulation, biodistribution and clearance properties from their normal counterparts, with additional changes associated with tumor progression and response to treatment.
We also found IV administered EV-GlucB to be quickly trafficked to subcutaneously xenograft tumors at one hour post-EV injection in vivo. Given that most solid tumors, and even premalignant neoplastic cells, are known to induce angiogenesis and exhibit leaky vasculature,31 these findings suggest EVs may serve as an effective vehicle to deliver therapeutic genes/agents to sites of tumor neovascularization.
In conclusion, our studies revealed a dynamic distribution and clearance process of systemically administered EVs with accurate spatiotemporal resolution previously unachievable using dye-based methods. The labeling method developed will provide insights into the role of EVs in different fields, including cancer, and their use in clinical therapy.
METHODS
Cell culture
HEK293T (American Type Culture Collection, Manassas, VA) and Gli36 human glioma cells (Dr. Anthony Capanogni, UCLA, Los Angeles, CA) were cultured in high glucose Dulbecco’s modified Eagle’s medium (Corning Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO) and 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen) in a humidified cell culture-grade incubator with 5% CO2 at 37 °C. EV-depleted FBS was prepared by centrifuging FBS at 100,000 × g at 4 °C for 16 hr followed by supernatant collection and filtration through a 0.22 μm filter (Millipore, Billerica, MA).
EV production and isolation
Stable HEK293T cells expressing Gluc or GlucB with sshBirA were generated by transducing cells with previously described lentivirus vectors, CSCW-Gluc-internal ribosome entry site (IRES)-GFP or CSCW-GlucB-IRES-GFP, followed by infection with CSCW-sshBirA-IRES-mCherry lentiviruses18,19. Stable expression in cells was confirmed by Western blot analysis and microscopic examination using an inverted epifluorescence microscope (TE 200-U, Nikon, Melville, NY) coupled to a digital camera.
To isolate EVs, briefly, conditioned medium was collected from cells incubated with culture medium supplemented with 10% EV-depleted FBS for 48 h followed by sequential centrifugation of the supernatant at 300 × g for 10 min, 2,000 × g for 10 min at 4 °C, and filtration through a 0.22 μm filter (Millipore). The filtrate was then subjected to ultracentrifugation at 100,000 × g for 90 min at 4 °C5, and the EV pellets were resuspended with double-0.22 μm-membrane-filtered PBS. EV protein concentration was determined by Bradford protein assay (Bio-Rad, Hercules, CA).
Protein isolation and Western blot analysis
Stable HEK293T cells expressing Gluc or GlucB with sshBirA were lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (complete, Mini, Roche Diagnostics, Indianapolis, IN), and DNA was sheared by sonication. Protein concentration was determined by Bradford protein assay (Bio-Rad). Twenty μg of total protein was boiled for 2 min in SDS sample buffer, resolved by 10% SDS-PAGE gel with molecular weight standards (Precision Plus Protein™ All Blue Standards, Bio-Rad), and transferred onto nitrocellulose membranes.19,38 The membranes were blocked with 5% bovine serum albumin (BSA) fraction V (Invitrogen, Grand Island, NY) and immunoblotted with streptavidin-HRP conjugate (Pierce, Rockford, IL), anti-Gluc (rabbit; Nanolight, Pinetop, AZ) and anti-GAPDH antibodies (mouse; Calbiochem, San Diego, CA). This was followed by binding of secondary antibodies conjugated to HRP (Molecular Probes, Eugene, OR) and signal detection with an enhanced chemiluminescent substrate (Pierce).
Transmission electron microscopy
Isolated EVs were pelleted at 20,000 × g for 30 min at 4 °C followed by fixation with 4% formaldehyde in PBS for 2 hr. Fixed EVs were cryosectioned, immunolabeled with anti-Gluc (mouse; Nanolight) and anti-CD63 (mouse; BD Biosciences, San Hose, CA) followed by rabbit anti-mouse conjugated with 5 nm protein A-gold secondary antibodies (University Medical Center, Utrecht, Netherlands). To detect EV surface biotinylation, fixed EVs were resuspended and immunolabeled with anti-biotin antibodies (rabbit, Rockland Immunochemicals, Gilbertsville, PA) followed by 5 nm gold-labeled secondary antibodies (Sigma). Images were captured using Technai G2 Spirit Bio TWIN transmission electron microscope.
Dot blot detection of biotinylated EVs
Gluc or GlucB labeled EVs in PBS were spotted onto nitrocellulose membranes at 16, 31, 63, 125, 250, 500 and 1,000 ng protein followed by overnight incubation in 5% BSA fraction V (Invitrogen), immunoblotting with streptavidin-HRP conjugate (Pierce) and chemiluminescence detection of biotinylated EVs with SuperSignal West Pico Chemiluminescent Substrate kit (Pierce) on autoradiography films (Denville Scientific, Metuchen, NJ).
Nanoparticle tracking analysis (NTA)
Isolated EV-Gluc and EV-GlucB were diluted 1,000 fold with double-0.22 μm-membrane-filtered PBS and subjected to LM10 nanoparticle analyzer (NanoSight, Duxbury, MA) to determine size distribution using NTA software version 2.2.
Sucrose gradient ultracentrifugation
Isolated EVs were layered onto a sucrose density gradient consisted of 8, 30, 45 and 60% layers in PBS, and centrifuged at 232,500 × g for 38 min at 4 °C.39 The top layer and the subsequent 10 fractions were collected. Twenty μl of the top layer and each fraction were sampled for Gluc activity assay. To collect EV-containing pellets, the fractions were diluted 1:10 in PBS and centrifuged at 100,000 × g for 1 h at 4 °C. Pellets were then resuspended in 25 μl PBS or RIPA with protease inhibitor (mini, Complete, Roche Diagnostics) for Gluc activity assay or Western blot analysis, respectively.
Gluc activity assays
Fractions collected following sucrose gradient ultracentrifugation of EVs were diluted 1:50 in PBS, and 20 μl of the mixture was plated in triplicates into a white 96-well luminometer plate. Gluc activity was measured by MLX Microtiter Plate Luminometer (Dynex Technologies, Chantilly, VA) with automated injection of 50 μl CTZ (8 ng/ml; Nanolight) followed by photon counts for 10 s for each sample. EV-GlucB activity in organs was similarly measured with 20 μl organ lysates in triplicates. To monitor EV-GlucB activity in blood in vivo, blood was sampled over time by creating a small nick at the mouse tail, mixed with 25 mM EDTA, and plated in triplicates of 5 μl. Urine sampled over time was similarly plated in triplicates of 10 μl. Fifty μl CTZ (50 ng/ml; Nanolight) was used to detect EV-GlucB activity in blood and urine samples. To examine stability of EV-GlucB in biofluids, 15 μl of blood and urine were sampled from untreated athymic nude mice, spiked with 0.5 μg of EV-GlucB and incubated for 1-3, 6, 12 and 24 hr at 37°C before measuring EV-GlucB activity as described above.
In vivo bioluminescence and FMT imaging of EVs
All animals (6 weeks old) were handled under practices and operating procedures complying with the policies of the MGH Review Board. For in vivo EV bioluminescence, three athymic nude mice were injected with a bolus of 100 μg EV-GlucB or PBS via retro-orbital vein. Thirty min post-EV injection, 75 μl CTZ mixture from “Inject-A-Lume” kit (Nanolight) was injected via a different retro-orbital vein for bioluminescence imaging (at 1 min post-CTZ injection) using an IVIS® Spectrum connected to XGI-8 Anesthesia System (PerkinElmer, Waltham, MA).
To study EV-GlucB distribution in tumor-bearing mice, five athymic nude mice were subcutaneously implanted with Gli36 cells (4 × 106) stably expressing mCherry on the left and right chest regions under anesthesia by intraperitoneal (i.p.) injection of a mixture of ketamine (100 mg/kg) and xylazine (3 mg/kg). When tumors reached a size of 3-5 nm in diameter (as monitored by mCherry fluorescence imaging), a bolus of 100 μg EV-GlucB was administered via the tail vein, followed by an injection of 75 μl CTZ mixture via the retro-orbital vein at 60 min post-EV injection. Bioluminescence images were captured as described above, and fluorescence images of stable Gli36-mCherry tumors were acquired using the same system under 605 nm excitation and 660 nm emission filters.
For FMT imaging of EVs, EV-GlucB (800 μg) was first conjugated with Alexa Fluor® 680-streptavidin (80 μg) in 5% BSA-containing PBS for 30 min at 4 °C, and excess dye was removed using Amicon Ultra-15 100K centrifugal filter device (Millipore). One hundred μg of Alexa Fluor® 680-conjugated EV-GlucB or PBS was injected via tail vein, and FMT imaging was carried out at 30 min post-treatment with FMT2500 system connected to RC2+ integrated anesthesia system (PerkinElmer).
Biodistribution analysis and immunocytochemistry
Athymic nude mice were injected with 100 μg EV-GlucB via the tail vein, and euthanized with i.p. injection of ketamine (300 mg/kg) and xylazine (30 mg/kg) at 30, 60, 120, and 360 min post-treatment. Transcardial perfusion with PBS was performed as previously described.40 Brain, heart, lungs, liver, kidneys, spleen and hind leg muscle (average of 87 mg of latter) were harvested from 3 non-perfused and 4 PBS-perfused mice per time point, and snap frozen in liquid nitrogen. Organs were then weighed, finely diced, and 100 mg of each organ was homogenized with M-PER lysis buffer supplemented with protease inhibitors (Thermo Scientific, Rockford, IL) for Gluc activity analysis. Baseline Gluc signal of each organ was determined as described above with non-perfused and PBS-perfused mice at 30 min following IV-injection with PBS. Total RLU per organ was calculated as follows and adjusted to baseline signal: (RLU/20 μl) × (500 μl M-PER lysis buffer/100 mg of organ) × (average organ weight in mg).
To detect and visualize EV-GlucB in kidney tissues, organs were harvested from EV-injected animals at 30 min and snap frozen with optimal cutting temperature (OCT) compound in liquid nitrogen. Embedded kidneys were cryosectioned (12 μm thick) and immunostained with anti-Gluc (rabbit; Nanolight) and Alexa Fluor® 647 goat anti-rabbit antibodies. Samples were imaged with a LSM510 confocal microscope and 63X Zeiss Plan-APOCHROMAT oil, 1.4NA objective (Zeiss, Thornwood, NY).
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants from NIH/NCI CA141150, CA069246, U19CA179563, and Voices Against Brain Cancer (VABC) (X.O.B.). C.P.L was supported by the Canadian Institute of Health Research (CIHR). We would like to thank: R. Amante for his suggestions and discussion, G. Lewandroski and D. Morse for the production of lentiviruses (MGH Vector Core, Boston, MA-supported by NIH/NINDS P30NS045776 [B.A.T., X.O.B.]), Ran C. for IVIS® Spectrum imaging assistance (Martinos Center for Biomedical Imaging, Boston, MA), T. Diefenbach for microscope assistance (Ragon Institute Microscopy Core, Boston, MA).
Funding Sources This work was supported by grants from NIH/NCI CA141150, CA069246, U19CA179563, and Voices Against Brain Cancer (VABC) (X.O.B.), NIH/NINDS P30NS045776 (X.O.B. and B.A.T.). C.P.L was supported by the Canadian Institute of Health Research (CIHR).
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Supporting Information Available: Baseline Gluc signal of non-perfused and PBS-perfused organs. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Lai CP, Breakefield XO. Role of Exosomes/Microvesicles in the Nervous System and Use in Emerging Therapies. Front. Physiol. 2012;3:228. doi: 10.3389/fphys.2012.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raposo G, Stoorvogel W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis DM, Sowinski S. Membrane Nanotubes: Dynamic Long-Distance Connections Between Animal Cells. Nat. Rev. Mol. Cell Biol. 2008;9:431–436. doi: 10.1038/nrm2399. [DOI] [PubMed] [Google Scholar]
- 4.Goodenough DA, Paul DL. Gap Junctions. Cold Spring Harbor Perspect. Biol. 2009;1:a002576. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Curry WT, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma Microvesicles Transport RNA and Proteins That Promote Tumour Growth and Provide Diagnostic Biomarkers. Nat. Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ. Embryonic Stem Cell-Derived Microvesicles Reprogram Hematopoietic Progenitors: Evidence for Horizontal Transfer of mRNA and Protein Delivery. Leukemia. 2006;20:847–856. doi: 10.1038/sj.leu.2404132. [DOI] [PubMed] [Google Scholar]
- 7.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular Transfer of the Oncogenic Receptor EGFRvIII by Microvesicles Derived From Tumour Cells. Nat. Cell Biol. 2008;10:619–624. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 8.Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar CM, et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells Toward a Pro-Metastatic Phenotype Through MET. Nat. Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hood JL, San RS, Wickline SA. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Research. 2011;71:3792–3801. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- 10.EL Andaloussi S, Mäger I, Breakefield XO, Wood MJA. Extracellular Vesicles: Biology and Emerging Therapeutic Opportunities. Nat. Rev. Drug Discovery. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 11.Tan A, La Peña, De H, Seifalian AM. The Application of Exosomes as a Nanoscale Cancer Vaccine. Int. J. Nanomed. 2010;5:889–900. doi: 10.2147/IJN.S13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizrak A, Bolukbasi MF, Ozdener GB, Brenner GJ, Madlener S, Erkan EP, Ströbel T, Breakefield XO, Saydam O. Genetically Engineered Microvesicles Carrying Suicide mRNA/Protein Inhibit Schwannoma Tumor Growth. Mol. Ther. 2012;21:101–108. doi: 10.1038/mt.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 14.Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, Ju S, Mu J, Zhang L, Steinman L, et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-Inflammatory Drugs From the Nasal Region to the Brain. Mol. Ther. 2011;19:1769–1779. doi: 10.1038/mt.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malam Y, Loizidou M, Seifalian AM. Liposomes and Nanoparticles: Nanosized Vehicles for Drug Delivery in Cancer. Trends Pharmacol. Sci. 2009;30:592–599. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, Barnes S, Grizzle W, Miller D, Zhang H-G. A Novel Nanoparticle Drug Delivery System: the Anti-Inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010;18:1606–1614. doi: 10.1038/mt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva AKA, Kolosnjaj-Tabi J, Bonneau S, Marangon I, Boggetto N, Aubertin K, Clément O, Bureau MF, Luciani N, Gazeau F, et al. Magnetic and Photoresponsive Theranosomes: Translating Cell-Released Vesicles Into Smart Nanovectors for Cancer Therapy. ACS Nano. 2013;7:4954–4966. doi: 10.1021/nn400269x. [DOI] [PubMed] [Google Scholar]
- 18.Niers JM, Chen JW, Lewandrowski G, Kerami M, Garanger E, Wojtkiewicz G, Waterman P, Keliher E, Weissleder R, Tannous BA. Single Reporter for Targeted Multimodal In Vivo Imaging. J. Am. Chem. Soc. 2012;134:5149–5156. doi: 10.1021/ja209868g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niers JM, Chen JW, Weissleder R, Tannous BA. Enhanced In Vivo Imaging of Metabolically Biotinylated Cell Surface Reporters. Anal. Chem. 2011;83:994–999. doi: 10.1021/ac102758m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tannous BA, Grimm J, Perry KF, Chen JW, Weissleder R, Breakefield XO. Metabolic Biotinylation of Cell Surface Receptors for In Vivo Imaging. Nat. Meth. 2006;3:391–396. doi: 10.1038/nmeth875. [DOI] [PubMed] [Google Scholar]
- 21.Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as Mediators of Intercellular Communication in Cancer--the Emerging Science of Cellular ‘Debris’. Semin. Immunopathol. 2011;33:455–467. doi: 10.1007/s00281-011-0250-3. [DOI] [PubMed] [Google Scholar]
- 22.Ohno S-I, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor microRNA to Breast Cancer Cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badr CE, Tannous BA. Bioluminescence Imaging: Progress and Applications. Trends Biotechnol. 2011;29:624–633. doi: 10.1016/j.tibtech.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, Takakura Y. Visualization and In Vivo Tracking of the Exosomes of Murine Melanoma B16-BL6 Cells in Mice After Intravenous Injection. J. Biotechnol. 2013;165:77–84. doi: 10.1016/j.jbiotec.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 25.Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ. Comparison of Ultracentrifugation, Density Gradient Separation, and Immunoaffinity Capture Methods for Isolating Human Colon Cancer Cell Line LIM1863-Derived Exosomes. Methods. 2012;56:293–304. doi: 10.1016/j.ymeth.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Chaput N, Théry C. Exosomes: Immune Properties and Potential Clinical Implementations. Semin. Immunopathol. 2011;33:419–440. doi: 10.1007/s00281-010-0233-9. [DOI] [PubMed] [Google Scholar]
- 27.Abra RM, Hunt CA. Liposome Disposition In Vivo. Biochim. Biophys. Acta, Lipids Lipid Metab. 1981;666:493–503. doi: 10.1016/0005-2760(81)90311-8. [DOI] [PubMed] [Google Scholar]
- 28.Tian T, Wang Y, Wang H, Zhu Z, Xiao Z. Visualizing of the Cellular Uptake and Intracellular Trafficking of Exosomes by Live-Cell Microscopy. J. Cell. Biochem. 2010;111:488–496. doi: 10.1002/jcb.22733. [DOI] [PubMed] [Google Scholar]
- 29.Griffin GE, Goldspink G. The Increase in Skeletal Muscle Mass in Male and Female Mice. Anat. Rec. 1973;177:465–469. doi: 10.1002/ar.1091770311. [DOI] [PubMed] [Google Scholar]
- 30.Ahsan F. Targeting to Macrophages: Role of Physicochemical Properties of Particulate Carriers—Liposomes and Microspheres—on the Phagocytosis by Macrophages. J. Controlled Release. 2002;79:29–40. doi: 10.1016/s0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of Cancer: the Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Zwaal RFA, Bevers EM, Comfurius P, Rosing J, Tilly RHJ, Verhallen PFJ. Loss of Membrane Phospholipid Asymmetry During Activation of Blood Platelets and Sickled Red Cells; Mechanisms and Physiological Significance. Mol. Cell. Biochem. 1989;91:23–31. doi: 10.1007/BF00228075. [DOI] [PubMed] [Google Scholar]
- 33.Colley CM, Ryman BE. Liposomes as Carriers In Vivo for Methotrexate. Biochem. Soc. Trans. 1975;3:157–159. doi: 10.1042/bst0030157. [DOI] [PubMed] [Google Scholar]
- 34.Théry C, Ostrowski M, Segura E. Membrane Vesicles as Conveyors of Immune Responses. Nat. Rev. Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 35.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a Phosphatidylserine Receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 36.Escrevente C, Keller S, Altevogt P, Costa J. Interaction and Uptake of Exosomes by Ovarian Cancer Cells. BMC Cancer. 2011;11:108. doi: 10.1186/1471-2407-11-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson RJ, Kalra H, Mathivanan S. ExoCarta as a Resource for Exosomal Research. J. of Extracell. Vesicles. 2012;1:18374. doi: 10.3402/jev.v1i0.18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai CPK, Bechberger JF, Thompson RJ, MacVicar BA, Bruzzone R, Naus CC. Tumor-Suppressive Effects of Pannexin 1 in C6 Glioma Cells. Cancer Res. 2007;67:1545–1554. doi: 10.1158/0008-5472.CAN-06-1396. [DOI] [PubMed] [Google Scholar]
- 39.Maguire CA, Balaj L, Sivaraman S, Crommentuijn MH, Ericsson M, Mincheva-Nilsson L, Baranov V, Gianni D, Tannous BA, Sena-Esteves M, et al. Microvesicle-Associated AAV Vector as a Novel Gene Delivery System. Mol. Ther. 2012;20:960–971. doi: 10.1038/mt.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gage GJ, Kipke DR, Shain W. Whole Animal Perfusion Fixation for Rodents. J. Vis. Exp. 2012;65:e3564. doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.