

Abstract
In retinitis pigmentosa (RP), the death of cones normally follows some time after the degeneration of rods. Recently, surviving cones in RP have been studied and reported in detail. These cones undergo extensive remodeling in their morphology. Here we report an extension of the remodeling study to consider possible modifications of spatial-distribution patterns. For this purpose we used S334ter-line-3 transgenic rats, a transgenic model developed to express a rhodopsin mutation causing RP. In this study, retinas were collected at postnatal (P) days P5–30, 90, 180, and P600. We then immunostained the retinas to examine the morphology and distribution of cones and to quantify the total cone numbers. Our results indicate that cones undergo extensive changes in their spatial distribution to give rise to a mosaic comprising an orderly array of rings. These rings first begin to appear at P15 at random regions of the retina and become ubiquitous throughout the entire tissue by P90. Such distribution pattern loses its clarity by P180 and mostly disappears at P600, at which time the cones are almost all dead. In contrast, the numbers of cones in RP and normal conditions do not show significant differences at stages as late as P180. Therefore, rings do not form by cell death at their centers, but by cone migration. We discuss its possible mechanisms and suggest a role for hot spots of rod death and the remodeling of Müller cell process into zones of low density of photoreceptors.
Indexing Terms: retinitis pigmentosa, cone mosaics, reorganization, retina
A wide variety of mutations that affect rods in retinitis pigmentosa (RP) first lead to their degeneration (Blanks et al., 1974; Farber and Lolley, 1974; Bowes et al., 1990; Rosenfeld et al., 1992; Marc et al., 2003). Then rod degeneration frequently results in the death of cones, although the extent of cone degeneration can vary between patients (Ripps, 2002; Delyfer et al., 2004; Hartong et al., 2006) and animal models (Carter-Dawson et al., 1978; LaVail et al., 1997). Eventually, these cones undergo almost complete degeneration in human and retinal degenerative animal models (Blanks et al., 1974; Berson, 1993; Chang et al., 1993; Farber et al., 1994; Li et al., 1994; Milam et al., 1996, 1998). Thus, understanding the cellular level of cones in degenerative animal models may influence therapeutic efforts of cone repopulation, transplantation, and retinal prosthesis.
Recently, some studies have shown in detail remodeling of cones in retinal degenerate animal models (Barhoum et al., 2008; Lin et al., 2009, Hombrebueno et al., 2010). In these studies, shortening or loss of cone outer segments (COS), loss of their normal vertical alignment, disorganization of axon terminals, and outgrowth of new processes from the cell body and the axon process were reported. These cones maintained much of their abnormal phenotype until postnatal (P)180 (Hombrebueno et al., 2010) of retinal degeneration. Furthermore, past studies reported on the discrepancies in the quantity or the densities of surviving M- and S-cones in different regions of the retinas of different retinal degenerate animal models: ventral versus dorsal (LaVail and Battelle, 1975; Carter-Dawson et al., 1978; Garcia-Fernandez et al., 1995; Jimenez et al., 1996; LaVail et al., 1997) and central versus peripheral regions (LaVail et al., 1982; Lin et al., 2009; Hombrebueno et al., 2010). However, the pattern of mosaic in which S- and M-cones are distributed in retinal degenerative models is not well studied. Hence, the present work focuses on examining the distribution patterns of cones in developing retinas of the S334ter-line-3 model. (This model shall be referred to as the RP model in the rest of the article.) Because the maintenance of prolonged survival of cones is important for the treatment of retinal degeneration, a thorough understanding of mosaic formation by cones in the progression of the disease will influence therapeutic efforts of cone repopulation, transplantation, and retinal prosthesis.
Materials and Methods
Animals
The third line of albino Sprague-Dawley rats homozygous for the truncated murine opsin gene (stop codon at residue 334; S334ter-line-3) was obtained from M.M. LaVail (University of California, San Francisco, CA). Homozygous S334ter-line-3 breeding pairs were mated with normal Copenhagen rats to produce offspring heterozygous for the S334ter transgene that was subsequently used in this study. Heterozygous animals were used instead of homozygous in order to avoid any changes in the retina due to albinism (O'Steen and Anderson 1972; O'Steen et al., 1974; Baker et al., 2005). A line of homozygous RP rats was also kept in breed for comparison study. For control, age-matched Sprague-Dawley rats (Harlan, Indianapolis, IN) were used. All rats were housed under cyclic 12/12-hour light/dark conditions with free access to food and water. Both sexes of normal (control) and RP rats were used. All procedures were in conformance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD). The University of Southern California Institutional Animal Care and Use Committee reviewed and approved all procedures.
Tissue preparation
The animals at P5-30, 90, 180, and P600 were used (n = 15 for each stage). All animals were dark-adapted for at least 1 hour prior to sacrifice in the dark. Animals were deeply anesthetized by intraperitoneal injection of pentobarbital (40 mg/kg body weight) and the eyes were enucleated. Animals were then killed with an overdose of pentobarbital. The anterior segment and crystalline lens were removed and the eyecups were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (PB), pH 7.4, for 30 minutes to 1 hour at 4°C. Following fixation, the retinas were carefully isolated from the eyecups and were transferred to 30% sucrose in PB for 24 hours at 4°C. For storage, all retinas (for cryostat sections and whole mounts) were then frozen in liquid nitrogen and stored at −70°C, thawed, and rinsed in 0.01 M phosphate-buffered saline (PBS; pH 7.4). For cryostat sections, eyecups were embedded in OCT embedding medium (Tissue-Tek, Elkhart, IN), then quickly frozen in liquid nitrogen and subsequently sectioned along the vertical meridian on a cryostat at a thickness of 20 μm.
Immunohistochemistry
For fluorescence immunohistochemistry, 20-μm-thick cryostat sections were incubated in 10% normal goat serum (NGS; Jackson ImmunoResearch Laboratories, West Grove, PA; dilution 1:1,000) or normal donkey serum (NDS, Jackson ImmunoResearch Laboratories; dilution 1:1,000) for 1 hour at room temperature. Sections were then incubated overnight with either marker for middle-wavelength-sensitive opsin (M-opsin) or short-wavelength-sensitive opsin marker (S-opsin) or rhodopsin marker (Rho 1D4) or proliferating cell nuclear antigen (PCNA). Each antiserum was diluted in PBS containing 0.5% Triton X-100 at 4°C. Retinas were washed in PBS for 45 minutes (3 × 15 minutes) and afterwards incubated for 2 hours at room temperature in either carboxymethylindocyanine-3 (Cy3)-conjugated affinity-purified donkey antirabbit IgG (Jackson ImmunoResearch Laboratories; dilution 1:500) or carboxymethylindocyanine-5 (Cy5)-conjugated affinity-purified donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories; dilution 1:300) or Alexa 488 antigoat IgG (Molecular Probes, Eugene, OR; dilution 1:300). The sections were washed for 30 minutes with 0.1 M PB and coverslipped with Vectashield mounting medium (Vector Labs, Burlingame, CA). For whole mount immunostaining the same immunocytochemical procedures described above were used. However, we used longer incubation times with primary antibodies (3 nights with anti-S-opsin, 2 nights with anti-M-opsin, rho 1D4, and PCNA) and secondary antibodies (4 hours either with Alexa 488 donkey antigoat IgG or with Cy3-conjugated donkey antirabbit IgG or Cy5-conjugated donkey antimouse IgG).
For double-label studies, whole mounts were incubated for 3 nights in a mixture of S-opsin and anti-M-opsin markers. Incubation with these antibodies used 0.5% Triton X-100 in 0.1 M PBS at 4°C. After this incubation, whole mounts were rinsed for 30 minutes with 0.1 M PBS. Afterwards, we incubated them with Alexa 488 donkey antigoat and Cy3-conjugated donkey antirabbit IgG for 2 nights at 4°C. For triple-label studies, whole mounts were first incubated for 2 nights in a mixture of anti-M-opsin and rho 1D4 antibody. Again, incubation with these antibodies used 0.5% Triton X-100 in 0.1 M PBS at 4°C. After this, the whole mounts were rinsed for 30 minutes with 0.1 M PBS before incubating them with Cy3-conjugated donkey antirabbit IgG and Cy5-conjugated goat antimouse IgG for 2 nights at 4°C. Finally, the whole mounts were stained with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL).
For nuclear layer staining, we used TOPRO-3 (Invitrogen, Carlsbad, CA; T3605, dilution 1:1,000). TOPRO-3 was incubated for 10 minutes then washed for 30 minutes with 0.1 M PB and cover-slipped with Vectashield mounting medium. In controls, the primary antibody was omitted from the incubation solution. Whole mounts were then washed again for 30 minutes with 0.1 M PB and coverslipped with Vectashield mounting medium. Sections and whole mounts were then analyzed using a Zeiss LSM 510 (Thornwood, NY) confocal microscope. Immunofluorescence images were processed in Zeiss LSM-PC software. The brightness and contrast of the images were adjusted using Adobe Photoshop 7.0 (Adobe Systems, Mountain View, CA). For presentation, all Photoshop adjustments (brightness and contrast only) were carried out equally across sections.
Antibody characterization
Please see Table 1 for a list of all antibodies used.
Table 1. Primary Antibodies.
| Antigen (what is being stained for) | Immunogen (what the antibody was raised against; full sequence and species) | Manufacturer, species antibody was raised in, mono- vs. polyclonal, catalog or lot number | Dilution used |
|---|---|---|---|
| M-opsin | 14 amino acids of mouse M-opsin peptide (residues 3-16, QRLTGEQTLDHYED; Zhu et al., 2003) | Gift of Dr. C. Craft, Doheny Eye Institute, University of Southern California, Los Angeles, rabbit polyclonal | 1:2,000 |
| S-opsin | 20 amino acids synthetic peptide (EFYLFKNISSVGPWDGPQYH) within the first 50 amino acids at the N-terminus of the human S-opsin (Schiviz et al., 2008) | Santa Cruz Biotechnology, Santa Cruz, CA, goat polyclonal, # SC-14363 | 1:1,500 |
| Rho 1D4 | 9 amino acids of the carboxy-terminal segment of bovine rhodopsin (TETSQVAPA; Molday and MacKenzie, 1983) | Gift of Dr. B. Thomas, Doheny Eye Institute, University of Southern California, Los Angeles, mouse monoclonal | 1:100 |
| PCNA | 10 amino acids synthetic peptide (LVFEAPNQEK) of the rat PCNA (Waseem and Lane, 1990; Zabouri et al., 2011) | Dako, Carpinteria, CA, Clone PC10, mouse monoclonal, #M0879 | 1:100 |
The M-opsin antiserum recognized a major band of 39 kDa molecular weight on western blots of mouse retinal extract and stained the M-cones in mouse and rat retina (Zhu et al., 2003; Xu et al., 2011; Lee et al., 2011). However, no western blot bands or immunostaining was observed in a peptide-blocking assay, which applied an excess of the specific peptide used to generate the M-opsin antibody (Zhu et al., 2003).
The S-opsin antiserum recognized a major band of ≈40 kDa and minor band of ≈42 kDa molecular weight on western blots of normal mouse retinal extract (Sato et al., 2010) and stained S-cones in mouse, rat, artiodactyl, and primate retina (Haverkamp et al., 2005; Wässle et al., 2006; Schiviz et al., 2008; Hombrebueno et al., 2010; Ray et al., 2010; Lee et al., 2011; Puller and Haverkamp, 2011). The staining pattern seen in our RP rat retina was the same as in previous reports (refer to Hombrebueno et al., 2010; Lee et al., 2011).
The development of a cell line for the Rho 1D4 and the isolation of the antibody were achieved by Dr. Robert Molday and coworkers (University of British Columbia, Vancouver). Rho 1D4 antiserum recognized a band of ≈40 kDa molecular weight on western blots of rat retinal extract (Li et al., 2003; Tanito et al., 2007), and stained a pattern of cellular morphology in the RP rat retina that is identical with previous reports (Cideciyan et al., 1998; Lee et al., 2011).
PCNA is a polymerase δ accessory protein (Tan et al., 1986; Bravo et al., 1987) and its marker targets cellular DNA synthesis (Jaskulski et al., 1988). The PCNA antiserum recognized a single band of 36 kDa molecular weight on western blot of rat retinal extract (Gordon et al., 2002). Waseem and Lane (1990) produced the antiserum and found that it stained the nuclei in an immortalized line of monkey epithelial cells. The antibody was later used to mark proliferating cells in mouse (Rakoczy et al., 2003; Zeiss and Johnson, 2004; Sigulinsky et al., 2008) and rat retina (Yamamoto et al., 1996). The pattern of cellular morphology observed in the RP rat retina is identical with the aforementioned previous reports.
Hematoxylin staining
The anterior segments of the eyeballs were removed and the eyecups fixed in 4% paraformaldehyde in 0.1 M PB for 2 hours at 4°C. Following fixation, eyecups were then washed by several changes of PB and transferred to 30% sucrose in PB for 5 hours at 4°C. We then embedded the eyecups in OCT embedding medium (Tissue-Tek, Elkhart, IN). They were next fast-frozen in liquid nitrogen and sectioned along the vertical meridian on a cryostat at a thickness of 10 μm. We then collected sections on gelatin-coated slides for hematoxylin staining and dipped them in hematoxylin for 5 minutes. They were then washed in tap water, dehydrated in alcohol, cleared in xylene, and mounted in xylene-based medium (Richard-Allan Scientific, Kalamazoo, MI).
TUNEL staining
Cell death was visualized by a modified TUNEL technique, according to the manufacturer's instructions (In Situ Cell Detection kit, Boehringer Mannheim, Mannheim, Germany). The P15 RP whole mount retinas harvested were incubated with proteinase K (10 μg/ml in 10 mM Tris/HCl, pH 7.4-8.0) for 10 minutes at 37°C. After rinsing in PBS the sections were incubated with TUNEL reaction mixture (terminal deoxynucleotidyl transferase plus nucleotide mixture in reaction buffer) for 60 minutes at 37°C. Sections were then washed again for 30 minutes with 0.1 M PB and coverslipped with Vectashield mounting medium.
Quantification and statistics
The size (n = 2 for all stages) and the total number (n = 3 for all stages) of rings formed by M-opsin- and S-opsin-immunoreactive cones in RP retinas were measured at P30 and P90. For P30 RP retinas (n = 3) the numbers of rings were also measured separately for the dorsal and the ventral hemispheres (divided by an imaginary line running through the optic disc horizontally). The size of the ring was defined as the mean distance of two cell bodies separated in the opposite side of the arrangement, completely across the ring. For rings that were not always completely circular in shape, the Zeiss LSM Image Browser Software was used to estimate the largest and the smallest diameter for each ring and the values were averaged to get the mean size of the ring. The total of 123 rings from P30 RP retinas and 148 rings from P90 RP retinas were arbitrarily selected for the measurement of their diameters. The retinal-area for these retinas were also measured, P30 (n = 3), 180 (n = 2), and P600 (n = 3) by ImageJ (National Institutes of Health, Bethesda, MD). In addition, the total number of M-opsin- and S-opsin-immunoreactive cones in both normal and RP retinas were manually counted at different stages: P30 (n = 2), 180 (n = 3) and P600 (n = 3). For comparison study, the homozygous RP rat retinas (n = 2) were also examined for the total number of M-opsin-immunoreactive cones at P180. Finally, both heterozygous and homozygous RP retinas (n = 4 each) were examined to compare their densities of M-opsin-immunoreactive cone cell body at P180 in whole mounts. An area of 0.16 mm2 in the middle region of dorsal retina was selected from each retina for measurement. We made sure that no more than three of what seemed to have been rings were within the tested areas. All the measurements were expressed as mean ± standard errors. Student's t-tests were used to examine the difference between two different means. The tests were performed with MatLab v. 7.4.0 (MathWorks, Natick, MA) and all graphs were generated by Microsoft Excel spreadsheet, 2010 (Redmond, WA). A difference between the means of separate experimental conditions was considered statistically significant at P < 0.05.
Results
Remodeling of M-opsin-immunoreactive cones in developing RP retinas
Recently, S-opsin-immunoreactive cones were reported to undergo extensive morphological modifications in RP retinas (Hombrebueno et al., 2010). We examined M-opsin-immunoreactive cones in vertical sections of normal (N) and RP retinas at P15, 30, and P90. In normal retinas at P15 (data not shown), P30, and P90 (data not shown), we observed M-opsin immunoreactivity in the segments, cell bodies, axon processes, and pedicles of cones (Fig. 1A). All M-opsin-immunoreactive cones were upright and vertically aligned. These results are consistent with previous data (Rohrer et al., 2005; Fujieda et al., 2009; Hombrebueno et al., 2010). The entire M-opsin-immunoreactive cones are labeled in RP retinas (Fig. 1B–D). In P15 RP retina, M-opsin-immunoreactive cones were upright (Fig. 1B), similar to that seen in normal retinas. This image was taken from the central part of the retinal section. The cone outer segments (COS) are shortened and distorted in orientation (arrow) compared to that in normal condition. By P30, M-opsin-immunoreactive cones have shortened remarkably in length, from the COS to the pedicle (Fig. 1C). The COS were shortened and distorted. The overall orientation of some cones were not vertical but were slightly aquiline/curved. By P90, all M-opsin-immunoreactive cones have lost their upright orientation completely (Fig. 1D). All cones were positioned “flat” against the outer part of the inner nuclear layer (INL). We also observed separate regions full of clusters of cell bodies followed by regions devoid of cell bodies but rich in processes. These two regions of different cellular structures alternated along the length of the vertical retinal sections.
Figure 1.
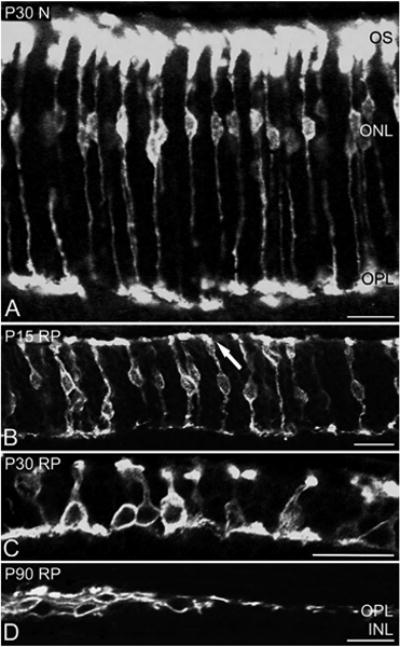
Confocal micrographs taken from vertical sections of retinas processed for M-opsin immunoreactivity. The micrographs are for P30 N (A), P15RP (B), P30RP (C), and P90 RP (D). In P30 N retinas, entire M-opsin-immunoreactive cones are labeled. In P15 RP retinas, the OS are distorted in orientation (arrow). In P30 RP retinas, M-opsin immunoreactive cones are shortened in length and show disorganized axon terminals (C). In P90 RP retinas, M-opsin-immunoreactive cones are positioned “flat” against the outer part of the INL. ONL, outer nuclear layer; OPL, outer plexiform layer; INL, Inner nuclear layer; OS, outer segment; N, normal; RP, retinitis pigmentosa. Scale bars = 20 μm.
Reorganization of M-opsin- and S-opsin-immunoreactive cones in orderly array of rings
To investigate the distribution pattern of cones in RP retina, we used M-opsin and S-opsin antibodies to identify cones in whole mount retinas. Figure 2 shows example of whole mounts processed for M-opsin (Fig. 2A,D,G) and S-opsin (Fig. 2B,E,H) immunoreactivities. The images were taken from the mid-peripheral part of the inferior (3 mm away from the optic disk) of P90 normal (Fig. 2A–C) and P90 RP (Fig. 2D–I) whole mount retinas. The results showed that there were more M-opsin-immunoreactive cones compared to S-opsin-immunoreactive cones in all retinas. In P90 normal retinas, M-opsin- (Fig. 2A) and S-opsin- (Fig. 2B) immunoreactive cones were distributed homogeneously throughout the retinas. Double-labeling experiments showed no colocalization of M-opsin and S-opsin immunoreactivity (Fig. 2C). In P90 RP retinas we observed strikingly different mosaic of opsin-immunoreactive cones (Fig. 2D–F). M-opsin- (Fig. 2D) and S-opsin- (Fig. 2E) immunoreactive cones were distributed in arrangements that resembled rings. When both M-opsin and S-opsin immunoreactivity are shown together (Fig. 2F), one can see that M-opsin- and S-opsin-immunoreactive cones formed rings at the same locations of the retina. A high-magnification view of part of a ring marked by the inset rectangle revealed that M-opsin- (Fig. 2G) and S-opsin- (Fig. 2H) immunoreactive cones share a specific orientation. Almost all the COS and the cell bodies were near the rims of the rings, whereas the processes were extended toward the center of the rings. Some M-opsin-immunoreactive cones and few S-opsin-immunoreactive cones showed abnormal processes sprouting from either their cell bodies or their axon processes. Double exposure showed how cell bodies were aligned very close to each other (Fig. 2I). Also, there was no colocalization of M-opsin and S-opsin-immunoreactive cones.
Figure 2.
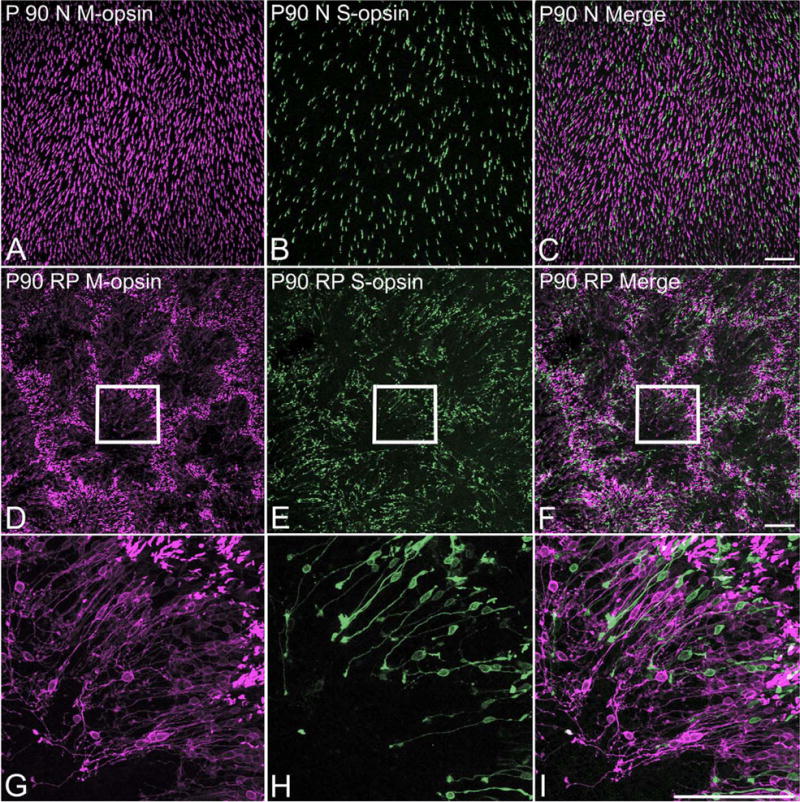
Confocal micrographs taken from whole mounts processed for M-opsin and S-opsin immunoreactivities. Low-power micrographs illustrate the homogeneous distributions of M-opsin (A) and S-opsin (B) cones in P90 normal retina. Double exposure (C) demonstrates no colocalization of M-opsin and S-opsin immunoreactivity. Low-power micrographs show that M-opsin (D) and S-opsin (E) cones in P90 RP retinas exhibit spatial organizations in matrices of rings. Double exposures (F) demonstrates that both types of cones form rings at the same locations in the RP retinas. High-power micrographs of part of a ring marked with inset rectangles in D–F, are shown in G–I, respectively. The orientation of M-opsin (G) and S-opsin (H) immunoreactive cones in rings are shown. Double exposures (I) demonstrates the same orientation of M-opsin and S-opsin cones. Scale bars = 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Cone rings are not associated with retinal foldings
We aimed to investigate whether cones rearranging themselves in rings have any relationship with rosettes first described by Flexner (1891) and Wintersteiner (1897). Mostly observed in retinoblastoma, rosettes are spherical folding of the ONL, mostly composed of photoreceptors (Tansley, 1933; Ts'o et al., 1970; Gallie et al., 1999). In order to study whether the arrangements of cones in rings in RP retinas reflect rosettes or not, we examined for any presence of physical folding at the level of the ONL. Rings arranged of M-opsin-immunoreactive cones were seen in P30 whole mount RP retina (Fig. 3A). Light micrographs taken under differential interference contrast (DIC) mode at the same focal plane as M-opsin-immunoreactive cones showed no obvious retinal folds (Fig. 3B). The merged image of the two micrographs confirmed that rings in P30 RP retina were not associated with physical foldings of the ONL of the retina (Fig. 3C). Similarly, rings of M-opsin-immunoreactive cones in P90 RP retinas (Fig. 3D) were also not associated with physical retinal foldings (Fig. 3E). The merged image of the two modes confirmed that rings in P90 RP retina were not rosettes (Fig. 3F). In order to examine then in vertical sections, we processed P15 and P30 retinal sections with hematoxylin stain. The result showed all the retinal layers (Fig. 3G–J). Hematoxylin staining of P15 RP retinal sections showed multiple rows of nuclei in the ONL (Fig. 3G). A high-magnification view of the ONL indicated within the inset rectangle showed that its thickness along the length of the section was uniform (Fig. 3H). However, multiple spaces empty of cell bodies were spotted within the ONL (arrow). P30 RP retinal sections, on the other hand, indicated that the thickness of the ONL was not uniform (Fig. 3I). Grooves were often observed in the ONL, as marked within the inset rectangle. At the trough of the groove (arrow), cell bodies were lacking (Fig. 3J). To study the arrangement of cell bodies in more detail, we processed P30 RP whole mount retinas with antibodies against Mopsin plus TOPRO-3 nuclear stain. When the image was taken at the level of the M-opsin-immunoreactive cell bodies at the ONL, nuclei at the center of the ring were out of focus (Fig. 3K). A high-magnification view of the center of the ring confirmed this (Fig. 3L). These nuclei were in the INL just below the level of M-opsin-immunoreactive cones. Such results suggest that the center of the ring is the trough of the grooves seen in the vertical sections. Taken together, our rings are not the same as rosettes in their physical architecture.
Figure 3.
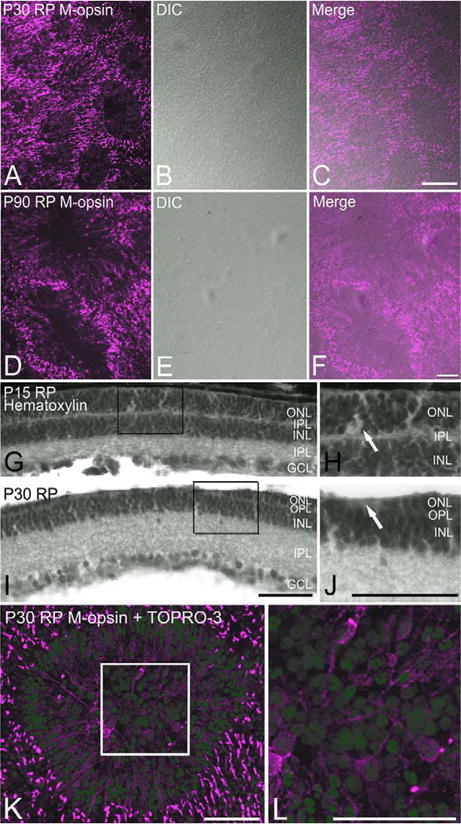
Confocal micrograph taken from P30 (A–C) and P90 (D–F) whole mount RP retinas processed for M-opsin immunoreactivities showing rings in their distribution (A,D). Light micrograph taken at the same retinal location under DIC mode shows no retinal folds (B,E). Double exposure (C,F) confirms no retinal folds are associated with rings. Light micrographs taken from RP vertical retinas processed with hematoxylin staining (G–J). At P15, the thickness of the ONL is uniform (G). H: Higher-power micrograph of G is shown. At P30, the ONL show “grooves.” J: Higher-power micrograph of groove is shown. No nuclei are visible at the trough of the groove (arrow). Confocal micrograph taken from P30 whole mount RP retina processed with M-opsin antibody (red) and TOPRO-3 (blue). Nuclei at the center of the ring are not in the same focal plane as M-opsin-immunoreactive cones (K). L: Higher-power micrograph of K is shown. DIC, differential interference contrast; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars = 100 μm in A–J; 50 μm in K,L. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Clusters of cell death are observed inside the rings
We processed P15 whole mount RP retinas for M-opsin (Fig. 4A) with rhodopsin (Fig. 4B) and TUNEL (Fig. 4C) labeling to observe the spatial correlation of cones, rods, and dying rods. We observed colocalization of local zones with low densities of cones and rods (Fig. 4A,B,D). Triple labeling of M-opsin, rhodopsin, and TUNEL showed clusters of dying cells inside zones with no cones and rods (Fig. 4D). Such clusters were consistent with our previous study showing massive rod cell death around P15 (Ray et al., 2010; Lee et al., 2011; Li et al., 2011). Hence, the holes emerged at least in part because of clusters of rod death.
Figure 4.
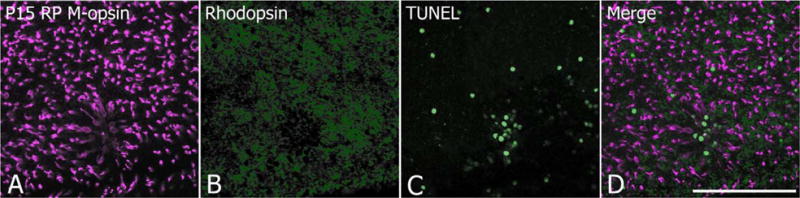
Confocal micrograph taken from P15 whole mount RP retinas processed for M-opsin (A), rhodopsin (B) immunoreactivities and for apoptotic cells (C). Triple exposure (A–C) indicates a cluster of apoptotic cells inside the ring. Where rings are not observed, apoptotic cells are scattered randomly. Scale bar = 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Rings first develop at around P15 and start to lose their form from around P180
In order to examine when the rings first develop and how they change over time, we immunostained whole mount RP retinas at P5-30 (n ≥ 5), 180, and P600 (n = 3). The distribution of M-opsin- and S-opsin-immunoreactive cones in P5-14 RP retinas (data not shown) resembled that of in normal retinas (Fig. 2A-C). At P15, a small region of abnormal distribution of cones was observed for the first time (Fig. 5A). The relative position of the initial ring-formation in the retinas was random (data not shown). The immature ring in P15 was much smaller in size in comparison to ones found in later postnatal stages. A high-magnification view showed that the orientation of all cones was the same as previously observed; COS forming the rims of the rings and the other parts of the cones being near the center of the rings (Fig. 5B). By P30, rings have grown larger in both their number and their size (Fig. 5C). M-opsin- and S-opsin-immunoreactive cones formed rings at the same regions in the RP retina. All cones had the same orientation and the center of the ring was filled with processes (Fig. 5D). Compared with cones in P15 RP retinas, it was much easier to view the entire cones (from the COS to the pedicle) in one focal plane, suggesting that cones in P30 RP retinas were closer to losing their vertical alignment within the ONL. By P180, rings have somewhat lost their shape (Fig. 5E). A higher-magnification view of a part of what seemingly used to be a ring revealed that cones were comparatively more disorderly (Fig. 5F). The orientation shared by all cones until P90 (Fig. 2I) was not as evident anymore. Most processes were no longer extended straight toward the center of the ring, thereby leaving a large area in the central zone of the ring devoid of cell processes. Most cones were clustered together and were far from arranged in orderly array. In addition, new processes emerged from either the cell bodies or from the remaining axon processes. Also, a lot of cones had lost their COS. In P600 whole mount RP retinas, there were larger areas of space devoid of M-opsin- and S-opsin-immunoreactive cones compared to P180 RP retinas (Fig. 5G). A higher-magnification view on some cones indicated that they have generated extensive branches in their processes that were not typical in the morphology of normal cones (Fig. 5H). Their morphology seemed to resemble bipolar or amacrine cells. Also, most have lost their COS.
Figure 5.
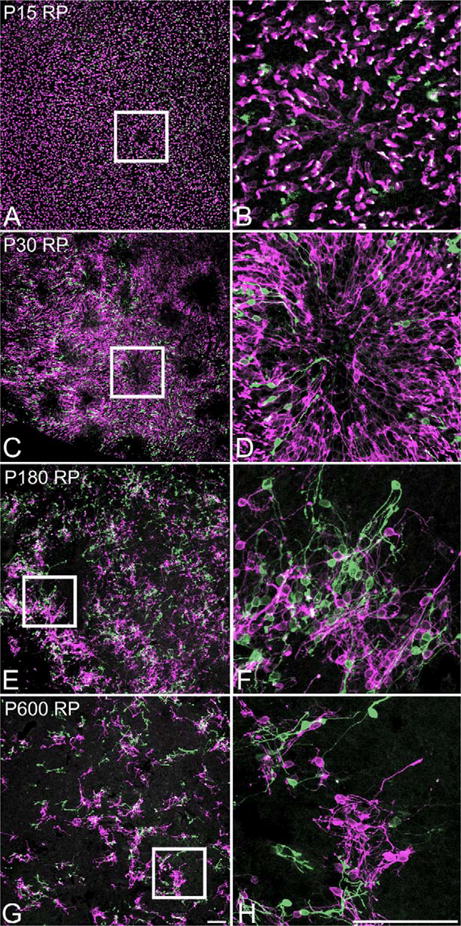
Confocal micrograph taken from whole mount RP retina processed for M-opsin (red) and S-opsin (green) immunoreactivities at P15 (A,B), 30 (C,D), 180 (E,F), and P600 (G,H). Double exposure shows a ring of M-opsin and S-opsin immunoreactive cone at P15 (A). A higher-power micrograph of a ring is shown (B). It illustrates the change of orientation of cones; starting to lie flat with their processes pointing toward the center of the ring. Many rings are visible by P30 (C). D: Higher-power micrograph of a ring is shown (C). All the COS and the cell bodies are near the rims of the rings, whereas the processes are pointing toward the center of the ring. At P180, rings start to lose their form (E). Higher-power micrograph of a part of what probably used to be a ring reveal M-opsin- and S-opsin-immunoreactive cones are no longer organized in the previously observed orientation (F). A lot of cones show growth of abnormal processes and loss of their OS. At P600, rings are no longer clear (G). Higher-power micrograph illustrates the cones' extensive growth of processes and their loss of OS (H). Scale bars = 100 μm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Rings in RP retinas grow significantly with age in both sizes and numbers
The sizes and quantities of rings at P30 and P90 were examined. We did not use P180 retinas for these measurements since the distribution pattern has mostly lost its form of rings and we did not want to introduce personal sampling errors to data. Composite images of P30 (Fig. 6A) and P90 (Fig. 6B) whole mount RP retinas showing M-opsin immunoreactivity were constructed. In all P30 RP retinas, more rings were observed in the peripheral region of the retina compared to the areas closer to the optic disk. Also, when the mean total counts of rings in the dorsal region (138 ± 5; mean ± standard error) were compared with those in the ventral region (54 ± 4; Fig. 6C), the dorsal region had a significantly greater number of rings (P < 0.002, one-tailed Student's t-test). The dorsal and the ventral regions are shown as areas divided by the dotted lines in Figure 6A. The mean total number of rings in P90 RP retinas (350 ± 10) retinas was significantly larger compared to that in P30 RP retinas (191 ± 6, P < 0.003, one-tailed Student's t-test; Fig. 6D). By P90, rings were seen throughout the retina. The mean diameter of rings formed by M-opsin- and S-opsin-immunoreactive cones were significantly larger in P90 (275 ± 3 μm) compared to P30 (168 ± 1 μm, P < 7e-023, one-tailed Student's t-test; Fig. 6E). These results showed that rings increase significantly in both their mean size and their quantity between P30 and P90 with progression of the disease.
Figure 6.
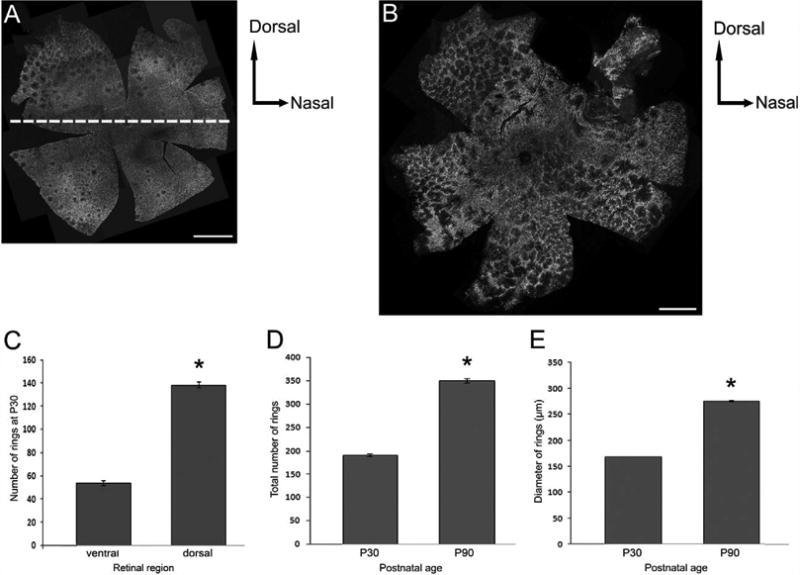
Composite image of confocal micrographs taken from the whole mount RP retinas processed for M-opsin immunoreactivities at P30 (A) and at P90 (B). At P30 there are comparatively more rings in the dorso-peripheral region of the retina. At P90 rings are seen throughout the entire retina. A graph of the mean total number of rings versus retinal regions of the P30 RP retinas (n = 3) suggest significantly greater number of rings in the dorsal region of the retinas compared to the ventral region (C). A graph of mean total number of rings versus postnatal age (n = 3; D) and a graph of mean diameter of rings (μm) versus postnatal age (n = 2; E) indicate rings grow both in their number and size from P30 to P90. Data presented as mean ± standard error. *P < 0.005 or better. Scale bars = 1 mm.
M-opsin and S-opsin-immunoreactive cones in RP retinas do not die until P180
We aimed to examine when cones degenerate in RP retinas. M-opsin- and S-opsin-immunoreactive cones in P30, P180, and P600 normal and RP whole mount retinas were counted (Fig. 7A). One-tailed Student's was used to test the significance between the different means. In P30 normal retinas the mean total numbers of M-opsin- and S-opsin-immunoreactive cones were 88,700 ± 700 and 35,000 ± 2,000 (mean ± standard error), respectively. In P30 RP retinas, the mean total numbers of M-opsin and S-opsin-immunoreactive cones were 87,000 ± 4,000 and 36,000 ± 4,000, respectively. These mean counts for the normal and the RP retinas were not statistically significantly different. In P180 normal retinas, the mean total numbers of M-opsin- and S-opsin-immunoreactive cones were 88,600 ± 400 and 32,600 ± 100, respectively. In P180 RP retinas, the mean total numbers of M-opsin- and S-opsin-immunoreactive cones were 87,500 ± 300 and 31,200 ± 700, respectively. Again, the test showed no significant differences between the counts in the normal and RP retinas. In P600 normal retinas, the mean total numbers of M-opsin- and S-opsin-immunoreactive cones were 70,000 ± 10,000 and 26,000 ± 7,000, respectively. The decrease in the counts seen in P600 normal retinas compared to P30 and P180 normal retinas were not statistically significant. In P600 RP retinas the mean total numbers of M-opsin- and S-opsin-immunoreactive cones were 12,000 ± 2,000 and 9,100 ± 900, respectively. The mean count for both M-opsin- and S-opsin-immunoreactive cones in the RP retinas were significantly lower compared to the normal retinas (P < 0.004 and 0.04, one-tailed Student's t-test). The counts for P180 RP retinas showed no significant difference from the counts for P30 RP retinas. Taken together, these results suggest that cones of the S334ter-line-3 transgenic rat do not degenerate until after P180. However, a significant degree of degeneration is detected by P600. Furthermore, we examined proliferating cells using PCNA labeling in RP whole mount retinas (P15, 30, and 180). We have not observed proliferating cells in RP retinas, certainly not within the arrangement of rings (data not shown). Hence, the steady total cell counts mean that there has been no cone death in RP retinas until P180.
Figure 7.
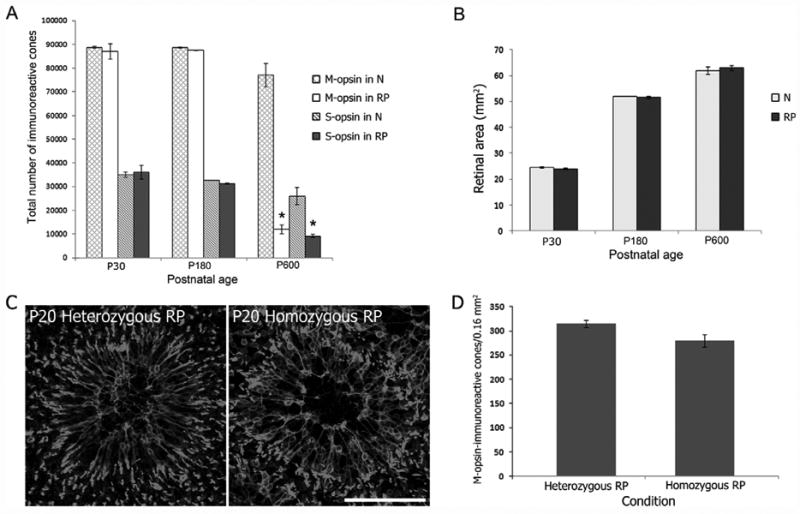
A graph of mean total number of immunoreactive cells versus postnatal age (A) shows no significant differences between the normal and the RP retinas at P30 (n = 2) and P180 (n = 3). Significant reduction is seen in P600 RP retinas (n = 3) for both M-opsin- and S-opsin-immunoreactive cone counts. A graph of RP retinal area (mm2) versus postnatal age indicates growth of retina in size with age (B: P30, n = 3; P180, n = 2; P600, n = 3). There are no significant differences in the retinal area between the normal and the RP retinas. Confocal micrographs taken from whole mount heterozygous (left) and homozygous (right) RP retinas processed for M-opsin immunoreactivities (C). Both show the same morphology, arrangement, and orientation of M-opsin-immunoreactive cones—the COS forming the rim of the ring and their processes in the inside of the ring. The densities of M-opsin-immunoreactive cones cell bodies in the dorsal wing of P180 heterozygous and homozygous RP retinas (n = 4 each) indicated no significant difference (D). Data presented as mean ± standard error. *P < 0.005 or better. Scale bar = 100 μm.
For control, we also measured the areas of the retinas used for counting cones to ensure that no sampling errors occurred between the normal and the RP retinas (Fig. 7B). The mean retinal areas for P30 normal and RP retinas were 24.5 ± 0.5 mm2 and 23.9 ± 0.6 mm2, respectively. For P180 normal and RP retinas, they were 52 ± 1 mm2 and 51.7 ± 0.9 mm2, respectively. And for P600 normal and RP retinas, they were 62 ± 3 mm2 and 63 ± 2 mm2, respectively. Therefore, the retinas were shown to grow with age. However, the Student's t-test (two-tailed) indicated no significant differences in the areas between the normal and the RP retinas at all the postnatal stages measured. Hence, the drops in the P600 RP cone-counts were neither due to selection of unusually small RP retinas nor due to shrinkage in their areas due to disease.
To compare the morphology, distribution, and orientation of M-opsin-immunoreactive cones in homozygous RP rat retinas with those in heterozygous RP rats, we processed their retinas using M-opsin antibody. The whole mount retinas from both heterozygous and homozygous P20 RP rats (Fig. 7C, left panel) showed what we have previously observed (Figs. 2D–I, 5). The COS of the M-opsin-immunoreactive cones were in the rim of the ring and the processes were pointed toward the center of the ring (Fig. 7C). However, there were more rings in homozygous retinas than in heterozygous ones (data not shown).
To compare the survival of M-opsin-immunoreactive cones in heterozygous and homozygous RP retinas, we further counted the total number of the cells in the P180 homozygous RP whole mounts. The mean total number of M-opsin-immunoreactive cones was 85,000 ± 2,000 (mean ± standard error). The two-tailed Student's t-test revealed that this number was not statistically significantly different from that from P180 heterozygous retinas. Furthermore, we examined the regional densities of the M-opsin-immunoreactive cone cell bodies in heterozygous and homozygous RP rat retinas at P180. The mean density of M-opsin-immunoreactive cone cell bodies in the dorsal wing in heterozygous RP retinas was 310 ± 20. The mean density in the homozygous RP retinas was 290 ± 30. The two-tailed Student's t-test indicated that the two density measures were not statistically significantly different from each other.
Discussion
Remodeling of cone morphology in RP retinas
In the present study we observed entire parts of cones stained by S-opsin and M-opsin antibodies in normal (Fig. 1A) and RP retinas (Fig. 1B–D). This expression pattern of cone opsin proteins has been found in normal mice and rats (Rohrer et al., 2005; Fujieda et al., 2009; Hombrebueno et al., 2010), retinal pigment epithelium-specific 65 kDa protein (RPE65)-deficient mice (Rohrer et al., 2005; Karan et al., 2008; Zhang et al., 2008), cyclic nucleotide gated channel alpha 3 (CNGA3)-deficient mice (Michalakis et al., 2005), and guanylate cyclase (GC)-knockout mice (Karan et al., 2008). Such expression patterns aided in examining how cones remodel in RP retinas as the disease progressed. We found that the remodeling of M-opsin-immunoreactive cones was similar to that of S-opsin-immunoreactive cones previously described in the same RP animal model (Hombrebueno et al., 2010). Many of the main morphological changes we observed were also shared with different animal models of RP. For example, the shortening and distortion of the COS observed from P15 in our RP retinas (Fig. 1B), were reported also in rd1 mice from as early as P8 (Fei, 2002; Lin et al., 2009), and in rd10 mice from around P30 (Barhoum et al., 2008). The loss of the COS we observed at P90, 180, and P600 (Figs. 2I, 5F,H) was reported in rd1 mice at P12 (Fei, 2002; Lin et al., 2009). The abnormal sprouting of processes from either the cell bodies or the axon processes of M-opsin-immunoreactive cells (Figs. 2I, 5F,H) was also observed from S-opsin-immunoreactive cones in the same animal model (Hombrebueno et al., 2010). Such abnormal sprouting from cones was also reported in rd1 mice (Fei, 2002; Lin et al., 2009). Furthermore, the loss of vertical alignment and positioning of cones at the outer part of the INL (Fig. 1D) was described in P90 RP rats (S-opsin-immunoreactive cones; Hombrebueno et al., 2010), and also in rd1 mice at P75 (Lin et al., 2009). Thus, we can conclude that the way that cones remodel in RP rats is similar to that seen in some other animal models of RP. It is likely that similar signal from the affected cascade from mutation in RP initiate such morphological changes of cones.
Remodeling of the spatial distribution of cones into orderly array of rings in RP retinas
Contrary to the pattern of morphological remodeling that is shared across some animal models of RP, the distribution pattern of cones in rings is not ubiquitous. We found orderly array of rings formed by both M-opsin- and S-opsin-immunoreactive cones in S334ter-line-3 RP retinas (Figs. 2, 5). Such distinct distribution patterns of cones was also observed in human patients with eye disease due to retinal dystrophy, inherited retinal degeneration, central ring scotomas, and genetic perturbations in the photopigment in M-cones (Carroll et al., 2004; Choi et al., 2006; Duncan et al., 2007; Joeres et al., 2008; Rossi et al., 2011). In these patients, circular areas of dark spaces or patchy regions on the ONL of their retinas were observed. In addition, cyclin D1-deficient mice (Ma et al., 1998) showed “holes of photoreceptors” that resemble our rings (Ma et al., 1998). We do not yet know why such ring-like cone distribution is present in these various cases. Previously, Lee et al. (2011) have hypothesized that remodeled Müller cell processes, which contribute to the formation of the rings in S334ter-line3 RP retinas, are significant for the survival of the cones. Thus, studying the cellular basis of ring formation in our RP model may help understand the neural-glial interactions, survival, and reorganization of photoreceptors in some human patients.
Possible reasons why rings were not observed in some other animal models of RP could be due either to the way that different mutations affect photoreceptors or to the experimental protocol used to reach the conclusion. If the degeneration rate is fast, and most rods and cones die a short time after birth, such as in rd1 mice (Carter-Dawson et al., 1978; Jiménez et al., 1996), cones may not have enough time to form rings. It could also be possible that the period during which cones show rings before they quickly degenerate is short. Thus, if an experiment sampled retinas at relatively coarse time bins, then it might not catch the moment when rings are present. We have shown that our moderately slow-degenerative RP rats represent an apt model to study the progression of cone spatial rearrangement into a distribution pattern that is also reported in some important cases, including human patients.
Ring formation is triggered by rod deaths, not by the mechanical disruption of the ONL cones in RP retinas begin forming rings at P15 (Fig. 5A). Because almost all pups used for this experiment had opened their eyes at P16, patterned visual input does not appear to be necessary for ring formation. One must thus find nonvisual, epigenetic mechanisms for the emergence of rings. We observed numerous spaces empty of photoreceptor nuclei in the ONL of P15 RP vertical retinal sections (Fig. 3H; arrow). And there were apoptotic cells in the ONL of P15 RP whole mount retinas (Fig. 4C). These apoptotic cells are dying rod cells as rod deaths but no cone deaths have been reported at around P15 in RP retinas (Li et al., 2010; Ray et al., 2010; Lee et al., 2011). Previously, Ray et al. (2010) reported the largest number of apoptotic cells in the ONL of RP retinas at P15. Our results indicated a close temporal and spatial correlation of dying rods with cone rings formation. Where rings were not present, rod death was scattered randomly in the retina (Fig. 4D). However, where a ring was present, the focal death of rods was observed in a cluster (Fig. 4D). Taken together, our results suggest that the clusters of rod deaths may be causing ring formation. Clusters of photoreceptor deaths were also reported to lead to the formation of “holes of photoreceptors” in cyclin D1-deficient mice (Ma et al., 1998). Rod degeneration in hot spots could be due to local reduction of survival factors intrinsically secreted by rods after their death (LaVail et al., 1998). Previous studies have also reported how degenerating rods can often lead to apoptosis in their neighbors (Huang et al., 1993; Kedzierski et al., 1998).
Furthermore, our results also indicated that our rings were not formed by mechanical collapse nor foldings of the ONL (Fig. 3A–J). Our rings were very regular and consistent in terms of their spatial arrangement and orientation of cones (Fig. 2D–I). We have also observed alternating regions full of cell bodies and regions full of processes along the length of the RP vertical sections (Fig. 1D). Such preciseness suggests that rings are not formed by mere mechanical disruption following rod loss. Continual expansion of rings in their quantity and size until P90 (Fig. 6C–E), long after all rods are degenerated in RP retinas (Li et al., 2010; Ray et al., 2010), also support this. In all, we can conclude that ring formation is triggered by rod deaths not by the mechanical disruption of the ONL.
Spatial and temporal distribution of rings in RP retina
Our results indicated that despite the initial formation of a ring being random in its relative position in the RP retina at P15, there was a greater occurrence of rings in the peripheral and dorsal regions at P30 (Fig. 6A,C) before spreading throughout the retina by P90 (Fig. 6B). This may be due to the intrinsic disparity in these regions as previously described in rats with retinal dystrophy (LaVail and Battelle, 1975). Previous studies have shown that the degeneration of cones in the central retina precedes the peripheral region in rd1 mice (Carter-Dawson et al., 1978; Lin et al., 2009). Also, hemispheric asymmetry in the number of surviving cones was previously reported in rd1 mice (Garcia-Fernandez et al., 1995; Jimenez et al., 1996; LaVail et al., 1997). These studies reported slower rate of cone degeneration in the dorsal retina compared to the ventral retina. Therefore, it is possible that ring formation is also influenced by the same mechanisms that induce such regional differences in survival of cones. Further studies are required to examine whether cones survive better in these regions in late stage of RP.
No cone degeneration until rings start to lose their form
Interestingly, our total cone cell body counts indicated no cell death until P180 (Fig. 7A)—that is, until when rings start to lose their clear form and cones lose their specific orientation (Fig. 5E–H). The growth in the retinal area we observed from P30 to P600 in both normal and RP rats (Fig. 7B) is consistent with previous studies (McCall et al., 1987; Harman et al., 2003). Thus, no sampling errors were introduced to our cone counts. We also did not observe proliferating cells in the ONL of RP retinas at P15, 30, and 180 (data not shown). Proliferation of photoreceptors from P15 was also not reported in any RP models. Although Müller cells are known to exhibit some neuronal stem cell properties (Fischer and Reh, 2001, 2003; Ooto et al., 2004), Müller cells transdifferentiating into photoreceptors was not reported to date in mammalian retinas. Hence, these findings collectively indicate that there was no cone degeneration until P180. This was reported otherwise in Li et al. (2010). Li et al. (2010) immunostained cones in homozygous S334ter-line-3 rat retinas using peanut agglutinin (PNA, marks COS) and cone arrestin (CAR, marks entire cones). Li et al. (2010) supported cone death using their evidence of falling COS densities and weakening of CAR immunoreactivity with the progression of the disease. However, for examining the degeneration of cones it is more accurate to measure by cone cell bodies rather than by the COS, as many cones were found to lose their COS in later stages (Figs. 2I, 5F,H). Also, one needs to take extra caution when measuring the density instead of the total cell counts in the RP retinas because the distribution of cones is not homogenous. Depending on which regions you select for measurement, the density may vary.
We have neither observed significant differences in the total number of M-opsin-immunoreactive cones nor observed differences in their cell body densities in heterozygous versus homozygous P180 RP retinas (Fig. 7D). Hence, some discrepancies between Li et al. (2010) and our study could have been due to different staining protocols. If the staining was relatively weaker, the cellular structures inside the rings could have been difficult to discern.
Taken together, our data suggest that the formation of rings is not a product of secondary degeneration of cones.
Possible mechanisms underlying reorganization of cones in rings in RP retinas
The absence of cone degeneration and proliferation until P180 (Fig. 7A) and their expansion in rings (Fig. 6D,E) suggest that cones are migrating. As to how the cones may migrate, one mechanism was proposed by Lee et al. (2011). In their study, they showed that remodeled processes of Müller cells filled the inside of the rings. Also, the cone processes in RP retinas were intertwined with the processes of Müller cells (Lee et al., 2011). Moreover, when glial-cell toxin DL-alpha-aminoadipic acid (AAA) was injected intravitreally, the rings disappeared (Lee et al., 2011). These results suggest that Müller cells interact with cones, compelling them to migrate into the array of rings in RP retinas. Another significant finding regarding Müller cells was reported by Xia et al. (2011). They applied intravitreal injection of Oncostatin M (OSM), a member of the IL-6 family of cytokines (Rose and Bruce, 1991), to P20 and 35 homozygous RP rats and observed the abolishment of rings. Xia et al. (2011) interpreted the resulting homogenous immunoreactivity of cones as OSM protecting and regeneration the COS of cones inside the rings. They also reported that the effects of OSM on cones were mediated by Müller cells. Because we do not normally observe cone cell death in RP retinas until P180, our data suggest that OSM is not inducing further survival of photoreceptors in the Xia et al. study (2011). An alternate interpretation of their and our data is that OSM causes the redistribution of cones, perhaps by regulating tight junctions between Müller cells and cones (Lee et al., 2011). The disappearance of rings upon application of OSM could have been due to loosening of tight junctions between Müller cells and cones. Several past studies have reported on the role of cytokines on tight junction alteration in retina (Zech et al., 1998; Abe et al., 2003; Villarroel et al., 2009; Aveleira et al., 2010).
At P600, we observed significant cone death in RP retinas (Fig. 7A). The decline of the total cone number in the normal retinas at P600 compared to those at their earlier stages (Fig. 7A) was statistically not significant and was probably due to the effect of aging (McCall et al., 1987; Dorey et al., 1989; Harman et al., 2003). Cones in rings maintained their abnormal phenotypes for prolonged periods until their degeneration (Fig. 5F,H). Such extended cone survival in RP retinas long after massive rod death was previously reported. In rd1 mice, cones were reported to survive up to at least 18 months (Carter-Dawson, 1978). There must be some mechanisms that help cones survive for long periods in harsh conditions. An intriguing possibility is that close-gathering of cones may aid in their survival. Within rings, cones are aligned very close to one another (Fig. 2I). This may allow them better share of self-secreted trophic factors. Future studies on the effects of different distribution pattern of cones on their survival in RP should enlighten what possible effects rings may have on cones and what mediates their rearrangement. Understanding whether and how rings in RP retinas improve the survival of cones would provide the scientific and clinical communities with better knowledge of how to prolong cone survival in RP.
Acknowledgments
We thank Professor Cheryl Craft for providing the M-opsin antibody and for helpful discussions during the performance of this work. We also thank Professor Biju Thomas for providing the Rhodopsin antibody. In addition, we thank Robert-Marlo Bautista for technical support, and Nadav Ivzan, Arvind Iyer Junkwan Lee, and Wan-Qing Yu for discussions of the materials during several lab meetings. We thank Denise Steiner for administrative support.
Grant sponsor: Fight for Sight (grant to E.-J.L.); Grant sponsor: National Science Foundation; Grant number: 0310723 (to N.M.G.); Grant sponsor: National Eye Institute; Grant number: EY016093 and EY11170 (to N.M.G.).
Literature Cited
- Abe T, Sugano E, Saigo Y, Tamai M. Interleukin-1beta and barrier function of retinal pigment epithelial cells (ARPE-19): aberrant expression of junctional complex molecules. Invest Ophthalmol Vis Sci. 2003;44:4097–4104. doi: 10.1167/iovs.02-0867. [DOI] [PubMed] [Google Scholar]
- Aveleira CA, Lin CM, Abcouwer SF, Ambrósio AF, Antonetti DA. TNF-á signals through PKCæ/NF-êB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GE, Dovey M, Davda P, Guibal C, Jeffery G. Protein kinase C immunoreactivity in the pigmented and albino rat retina. Eur J Neurosci. 2005;22:2481–2488. doi: 10.1111/j.1460-9568.2005.04453.x. [DOI] [PubMed] [Google Scholar]
- Barhoum R, Martínez-Navarrete G, Corrochano S, Germain F, Fernandez-Sanchez L, de la Rosa EJ, de la Villa P, Cuenca N. Functional and structural modifications during retinal degeneration in the rd10 mouse. Neuroscience. 2008;155:698–713. doi: 10.1016/j.neuroscience.2008.06.042. [DOI] [PubMed] [Google Scholar]
- Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- Blanks JC, Adinolfi AM, Lolley RN. Photoreceptor degeneration and synaptogenesis in retinal-degenerative (rd) mice. J Comp Neurol. 1974;156:95–106. doi: 10.1002/cne.901560108. [DOI] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Bravo R, Frank R, Blundell PA, Macdonald-Bravo H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta. Nature. 1987;326:515–517. doi: 10.1038/326515a0. [DOI] [PubMed] [Google Scholar]
- Carroll J, Neitz M, Hofer H, Neitz J, Williams DR. Functional photoreceptor loss revealed with adaptive optics: an alternate cause for color blindness. Proc Natl Acad Sci U S A. 2004;101:8461–8466. doi: 10.1073/pnas.0401440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–498. [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Hawes NL, Roderick TH. New mouse primary retinal degeneration (rd-3) Genomics. 1993;16:45–49. doi: 10.1006/geno.1993.1138. [DOI] [PubMed] [Google Scholar]
- Chen J, Nathans J. Genetic ablation of cone photoreceptors eliminates retinal folds in the retinal degeneration 7 (rd7) mouse. Invest Ophthalmol Vis Sci. 2007;48:2799–2805. doi: 10.1167/iovs.06-0922. [DOI] [PubMed] [Google Scholar]
- Choi SS, Doble N, Hardy JL, Jones SM, Keltner JL, Olivier SS, Werner JS. In vivo imaging of the photoreceptor mosaic in retinal dystrophies and correlations with visual function. Invest Ophthalmol Vis Sci. 2006;47:2080–2092. doi: 10.1167/iovs.05-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, Jacobson SG. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delyfer MN, Léveillard T, Mohand-Saïd S, Hicks D, Picaud S, Sahel JA. Inherited retinal degenerations: therapeutic prospects. Biol Cell. 2004;96:261–269. doi: 10.1016/j.biolcel.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci. 1989;30:1691–1699. [PubMed] [Google Scholar]
- Duncan JL, Zhang Y, Gandhi J, Nakanishi C, Othman M, Branham KE, Swaroop A, Roorda A. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3283–3291. doi: 10.1167/iovs.06-1422. [DOI] [PubMed] [Google Scholar]
- Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451. doi: 10.1126/science.186.4162.449. [DOI] [PubMed] [Google Scholar]
- Farber DB, Flannery JG, Bowes-Rickmann C. The rd mouse story: seventy years of research on an animal model of inherited retinal degeneration. Prog Ret Eye Res. 1994;13:31–64. [Google Scholar]
- Fei Y. Cone neurite sprouting: an early onset abnormality of the cone photoreceptors in the retinal degeneration mouse. Mol Vis. 2002;27:306–314. [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Müller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat Neurosci. 2001;4:247–252. doi: 10.1038/85090. [DOI] [PubMed] [Google Scholar]
- Fischer AJ, Reh TA. Potential of Müller glia to become neurogenic retinal progenitor cells. Glia. 2003;43:70–76. doi: 10.1002/glia.10218. [DOI] [PubMed] [Google Scholar]
- Flexner S. A peculiar glioma (neuroepithelioma?) of the retina. Johns Hopkins Hospital Bull. 1891;15:115. [Google Scholar]
- Fujieda H, Bremner R, Mears AJ, Sasaki H. Retinoic acid receptor-related orphan receptor alpha regulates a subset of cone genes during mouse retinal development. J Neurochem. 2009;108:91–101. doi: 10.1111/j.1471-4159.2008.05739.x. [DOI] [PubMed] [Google Scholar]
- Gallie BL, Campbell C, Devlin H, Duckett A, Squire JA. Developmental basis of retinal-specific induction of cancer by RB mutation. Cancer Res. 1999;59(7 Suppl):1731s–1735s. [PubMed] [Google Scholar]
- García-Fernández JM, Jimenez AJ, Foster RG. The persistence of cone photoreceptors within the dorsal retina of aged retinally degenerate mice (rd/rd): implications for circadian organization. Neurosci Lett. 1995;187:33–36. doi: 10.1016/0304-3940(95)11330-y. [DOI] [PubMed] [Google Scholar]
- Gordon WC, Casey DM, Lukiw WJ, Bazan NG. DNA damage and repair in light-induced photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2002;43:3511–3521. [PubMed] [Google Scholar]
- Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471. doi: 10.1016/s0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- Harman AM, MacDonald A, Meyer P, Ahmat A. Numbers of neurons in the retinal ganglion cell layer of the rat do not change throughout life. Gerontology. 2003;49:350–355. doi: 10.1159/000073762. [DOI] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Wässle H, Duebel J, Kuner T, Augustine GJ, Feng Ge, Euler T. The primordial, blue-cone color system of the mouse retina. J Neurosci. 2005;25:5438–5445. doi: 10.1523/JNEUROSCI.1117-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombrebueno JR, Tsai MM, Kim HL, De Juan J, Grzywacz NM, Lee EJ. Morphological changes of short-wavelength cones in the developing S334ter-3 transgenic rat. Brain Res. 2010;1321:60–66. doi: 10.1016/j.brainres.2010.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PC, Gaitan AE, Hao Y, Petters RM, Wong F. Cellular interactions implicated in the mechanism of photoreceptor degeneration in transgenic mice expressing a mutant rhodopsin gene. Proc Natl Acad Sci U S A. 1993;90:8484–8488. doi: 10.1073/pnas.90.18.8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- Jaskulski D, deRiel JK, Mercer WE, Calabretta B, Baserga R. Inhibition of cellular proliferation by antisense oligodeoxynucleotides to PCNA cyclin. Science. 1988;240:1544–1546. doi: 10.1126/science.2897717. [DOI] [PubMed] [Google Scholar]
- Jiménez AJ, García-Fernández JM, González B, Foster RG. The spatio-temporal pattern of photoreceptor degeneration in the aged rd/rd mouse retina. Cell Tissue Res. 1996;284:193–202. doi: 10.1007/s004410050579. [DOI] [PubMed] [Google Scholar]
- Karan S, Zhang H, Li S, Frederick JM, Baehr W. A model for transport of membrane-associated phototransduction polypeptides in rod and cone photoreceptor inner segments. Vision Res. 2008;48:442–452. doi: 10.1016/j.visres.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedzierski W, Bok D, Travis GH. Non-cell-autonomous photoreceptor degeneration in rds mutant mice mosaic for expression of a rescue transgene. J Neurosci. 2010;18:4076–4082. doi: 10.1523/JNEUROSCI.18-11-04076.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Battelle BA. Influence of eye pigmentation and light deprivation on inherited retinal dystrophy in the rat. Exp Eye Res. 1975;21:167–192. doi: 10.1016/0014-4835(75)90080-9. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Blanks JC, Mullen RJ. Retinal degeneration in the pcd cerebellar mutant mouse. I. Light microscopic and autoradiographic analysis. J Comp Neurol. 1982;212:217–230. doi: 10.1002/cne.902120302. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Matthes MT, Yasumura D, Steinberg RH. Variability in rate of cone degeneration in the retinal degeneration (rd/rd) mouse. Exp Eye Res. 1997;65:45–50. doi: 10.1006/exer.1997.0308. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung CH, Steinberg RH. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- Lee EJ, Ji Y, Zhu CL, Grzywacz NM. Role of Müller cells in cone-mosaic rearrangement in a rat model of retinitis pigmentosa. Glia. 2011;59:1107–1117. doi: 10.1002/glia.21183. [DOI] [PubMed] [Google Scholar]
- Li ZY, Jacobson SG, Milam AH. Autosomal dominant retinitis pigmentosa caused by the threonine-17-methionine rhodopsin mutation: retinal histopathology and immunocytochemistry. Exp Eye Res. 1994;58:397–408. doi: 10.1006/exer.1994.1032. [DOI] [PubMed] [Google Scholar]
- Li F, Cao W, Anderson RE. Alleviation of constant-light-induced photoreceptor degeneration by adaptation of adult albino rat to bright cyclic light. Invest Ophthalmol Vis Sci. 2003;44:4968–4975. doi: 10.1167/iovs.03-0140. [DOI] [PubMed] [Google Scholar]
- Li Y, Tao W, Luo L, Huang D, Kauper K, Stabila P, Lavail MM, Laties AM, Wen R. CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PLoS One. 2010;5:e9495. doi: 10.1371/journal.pone.0009495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Masland RH, Strettoi E. Remodeling of cone photoreceptor cells after rod degeneration in rd mice. Exp Eye Res. 2009;88:589–599. doi: 10.1016/j.exer.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Papermaster D, Cepko C. A unique pattern of photoreceptor degeneration in cyclin D1 mutant mice. Proc Natl Acad Sci U S A. 1998;95:9938–9943. doi: 10.1073/pnas.95.17.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE, Jones BW. Retinal remodeling in inherited photoreceptor degenerations. Mol Neurobiol. 2003;28:139–147. doi: 10.1385/MN:28:2:139. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Moriuchi T, Koji T, Nakane PK. Molecular cloning of cDNA coding for rat proliferating cell nuclear antigen (PCNA)/cyclin. EMBO J. 1987;6:637–642. doi: 10.1002/j.1460-2075.1987.tb04802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MJ, Robinson SR, Dreher B. Differential retinal growth appears to be the primary factor producing the ganglion cell density gradient in the rat. Neurosci Lett. 1987;79:78–84. doi: 10.1016/0304-3940(87)90675-6. [DOI] [PubMed] [Google Scholar]
- Michalakis S, Geiger H, Haverkamp S, Hofmann F, Gerstner A, Biel M. Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest Ophthalmol Vis Sci. 2005;46:1516–1524. doi: 10.1167/iovs.04-1503. [DOI] [PubMed] [Google Scholar]
- Milam AH, Li ZY, Cideciyan AV, Jacobson SG. Clinicopathologic effects of the Q64ter rhodopsin mutation in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1996;37:753–765. [PubMed] [Google Scholar]
- Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Ret Eye Res. 1998;17:175–205. doi: 10.1016/s1350-9462(97)00012-8. [DOI] [PubMed] [Google Scholar]
- Molday RS, MacKenzie D. Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry. 1983;22:653–660. doi: 10.1021/bi00272a020. [DOI] [PubMed] [Google Scholar]
- Ooto S, Akagi T, Kageyama R, Akita J, Mandai M, Honda Y, Takahashi M. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc Natl Acad Sci U S A. 2004;101:13654–13659. doi: 10.1073/pnas.0402129101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Steen WK, Shear CR, Anderson KV. Retinal damage after prolonged exposure to visible light. A light and electron microscopic study. Am J Anat. 1972;134:5–21. doi: 10.1002/aja.1001340103. [DOI] [PubMed] [Google Scholar]
- O'Steen WK, Anderson KV, Shear CR. Photoreceptor degeneration in albino rats: dependency on age. Invest Ophthalmol. 1974;13:334–339. [PubMed] [Google Scholar]
- Puller C, Haverkamp S. Cell-type-specific localization of protocadherin β16 at AMPA and AMPA/kainate receptor-containing synapses in the primate retina. J Comp Neurol. 2011;519:467–479. doi: 10.1002/cne.22528. [DOI] [PubMed] [Google Scholar]
- Rakoczy PE, Brankov M, Fonceca A, Zaknich T, Rae BC, Lai CM. Enhanced recombinant adeno-associated virus mediated vascular endothelial growth factor expression in the adult mouse retina: a potential model for diabetic retinopathy. Diabetes. 2003;52:857–863. doi: 10.2337/diabetes.52.3.857. [DOI] [PubMed] [Google Scholar]
- Ray A, Sun GJ, Chan L, Grzywacz NM, Weiland J, Lee EJ. Morphological alterations in retinal neurons in the S334ter-line3 transgenic rat. Cell Tissue Res. 2010;339:481–491. doi: 10.1007/s00441-009-0916-5. [DOI] [PubMed] [Google Scholar]
- Ripps H. Cell death in retinitis pigmentosa: gap junctions and the ‘bystander’ effect. Exp Eye Res. 2002;74:327–336. doi: 10.1006/exer.2002.1155. [DOI] [PubMed] [Google Scholar]
- Roberts MR, Hendrickson A, McGuire CR, Reh TA. Retinoid X receptor (gamma) is necessary to establish the S-opsin gradient in cone photoreceptors of the developing mouse retina. Invest Opthalmol Vis Sci. 2005;46:2897–2904. doi: 10.1167/iovs.05-0093. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW, Crouch RK. Cone opsin mislocalization in Rpe65-/-mice: a defect that can be corrected by 11-cis retinal. Invest Ophthalmol Vis Sci. 2005;46:3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- Rose TM, Bruce AG. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc Natl Acad Sci U S A. 1991;88:8641–8645. doi: 10.1073/pnas.88.19.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld PJ, Cowley GS, McGee TL, Sandberg MA, Berson EL, Dryja TP. A null mutation in the rhodopsin gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet. 1992;1:209–213. doi: 10.1038/ng0692-209. [DOI] [PubMed] [Google Scholar]
- Rossi EA, Chung M, Dubra A, Hunter JJ, Merigan WH, Williams DR. Imaging retinal mosaics in the living eye. Eye (Lond) 2011;25:301–308. doi: 10.1038/eye.2010.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Nakazawa M, Takeuchi K, Mizukoshi S, Ishiguro S. S-opsin protein is incompletely modified during N-gly-can processing in Rpe65(-/-) mice. Exp Eye Res. 2010;91:54–62. doi: 10.1016/j.exer.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Schiviz AN, Ruf T, Kuebber-Heiss A, Schubert C, Ahnelt PK. Retinal cone topography of artiodactyl mammals: influence of body height and habitat. J Comp Neurol. 2008;507:1336–1350. doi: 10.1002/cne.21626. [DOI] [PubMed] [Google Scholar]
- Sigulinsky CL, Green ES, Clark AM, Levine EM. Vsx2/Chx10 ensures the correct timing and magnitude of Hedgehog signaling in the mouse retina. Dev Biol. 2008;317:560–575. doi: 10.1016/j.ydbio.2008.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CK, Castillo C, So AG, Downey KM. An auxiliary protein for DNA polymerase-delta from fetal calf thymus. J Biol Chem. 1966;261:12310–12316. [PubMed] [Google Scholar]
- Tanito M, Kaidzu S, Anderson RE. Delayed loss of cone and remaining rod photoreceptor cells due to impairment of choroidal circulation after acute light exposure in rats. Invest Ophthalmol Vis Sci. 2007;48:1864–1872. doi: 10.1167/iovs.06-1065. [DOI] [PubMed] [Google Scholar]
- Tansley K. The formation of rosettes in the rat retina. Br J Opthalmol. 1933;17:321–336. doi: 10.1136/bjo.17.6.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ts'o MO, Zimmerman LE, Fine BS. The nature of retinoblastoma. I. Photoreceptor differentiation: a clinical and histopathologic study. Am J Ophthalmol. 1970;69:339–349. doi: 10.1016/0002-9394(70)92263-4. [DOI] [PubMed] [Google Scholar]
- van de Pavert SA, Kantardzhieva A, Malysheva A, Meuleman J, Versteeg I, Levelt C, Klooster J, Geiger S, Seeliger MW, Rashbass P, Le Bivic A, Wijnholds J. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci. 2004;117(Pt 18):4169–4177. doi: 10.1242/jcs.01301. [DOI] [PubMed] [Google Scholar]
- Villarroel M, García-Ramírez M, Corraliza L, Hernández C, Simó R. Effects of high glucose concentration on the barrier function and the expression of tight junction proteins in human retinal pigment epithelial cells. Exp Eye Res. 2009;89:913–920. doi: 10.1016/j.exer.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Waseem NH, Lane DP. Monoclonal antibody analysis of the proliferating cell nuclear antigen (PCNA). Structural conservation and the detection of a nucleolar form. J Cell Sci. 1990;96(Pt 1):121–129. doi: 10.1242/jcs.96.1.121. [DOI] [PubMed] [Google Scholar]
- Wässle H, Regus-Leidig H, Haverkamp S. Expression of the vesicular glutamate transporter vGluT2 in a subset of cones of the mouse retina. J Comp Neurol. 2006;496:544–555. doi: 10.1002/cne.20942. [DOI] [PubMed] [Google Scholar]
- Wintersteiner H. Das Neuroepithelioma retinae: Eine anatomische und klinische Studie. Leipzig: Franz Deuticke; 1897. [Google Scholar]
- Xu J, Morris L, Fliesler SJ, Sherry DM, Ding XQ. Early onset, slow progression of cone photoreceptor dysfunction and degeneration in CNG channel subunit CNGB3 deficiency. Invest Ophthalmol Vis Sci. 2011;52:3557–3566. doi: 10.1167/iovs.10-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto C, Ogata N, Yi X, Takahashi K, Miyashiro M, Yamada H, Uyama M, Matsuzaki K. Immunolocalization of basic fibroblast growth factor during wound repair in rat retina after laser photocoagulation. Graefes Arch Clin Exp Ophthalmol. 1996;234:695–702. doi: 10.1007/BF00292356. [DOI] [PubMed] [Google Scholar]
- Zabouri N, Bouchard JF, Casanova C. Cannabinoid receptor type 1 expression during postnatal development of the rat retina. J Comp Neurol. 2011;519:1258–1280. doi: 10.1002/cne.22534. [DOI] [PubMed] [Google Scholar]
- Zech JC, Pouvreau I, Cotinet A, Goureau O, Le Varlet B, de Kozak Y. Effect of cytokines and nitric oxide on tight junctions in cultured rat retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1998;39:1600–1608. [PubMed] [Google Scholar]
- Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Invest Ophthalmol Vis Sci. 2004;45:971–976. doi: 10.1167/iovs.03-0301. [DOI] [PubMed] [Google Scholar]
- Zhang H, Fan J, Li S, Karan S, Rohrer B, Palczewski K, Frederick JM, Crouch RK, Baehr W. Trafficking of membrane-associated proteins to cone photoreceptor outer segments requires the chromophore 11-cis-retinal. J Neurosci. 2008;28:4008–4014. doi: 10.1523/JNEUROSCI.0317-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Brown B, Li A, Mears A, Swaroop A, Craft CM. GRK1-dependent photophorylation of S and M opsins and their binding to cone arrestin during cone phototransduction in the mouse retina. J Comp Neurol. 2003;23:6152–6160. doi: 10.1523/JNEUROSCI.23-14-06152.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]