

Abstract
Traumatic brain injury (TBI) increases the risk of neuropsychiatric disorders, particularly anxiety disorders. Yet, there are presently no therapeutic interventions to prevent the development of post-traumatic anxiety or effective treatments once it has developed. This is because, in large part, of a lack of understanding of the underlying pathophysiology. Recent research suggests that chronic neuroinflammatory responses to injury may play a role in the development of post-traumatic anxiety in rodent models. Acute peri-injury administration of immunosuppressive compounds, such as Ibudilast (MN166), have been shown to prevent reactive gliosis associated with immune responses to injury and also prevent lateral fluid percussion injury (LFPI)-induced anxiety-like behavior in rats. There is evidence in both human and rodent studies that post-traumatic anxiety, once developed, is a chronic, persistent, and drug-refractory condition. In the present study, we sought to determine whether neuroinflammation is associated with the long-term maintenance of post-traumatic anxiety. We examined the efficacy of an anti-inflammatory treatment in decreasing anxiety-like behavior and reactive gliosis when introduced at 1 month after injury. Delayed treatment substantially reduced established LFPI-induced freezing behavior and reactive gliosis in brain regions associated with anxiety and continued neuroprotective effects were evidenced 6 months post-treatment. These results support the conclusion that neuroinflammation may be involved in the development and maintenance of anxiety-like behaviors after TBI.
Key words: : glial cell response to injury, inflammation, traumatic brain injury
Introduction
Over 5.3 million people in the United States are living with traumatic brain injury (TBI)-related disabilities,1 including anxiety disorders, which are among the most prevalent.2–4
The rates of a variety of anxiety disorders reported by patients with TBI are consistently elevated relative to general population rates,3–5 and the risk for developing post-traumatic stress disorder (PTSD) remains elevated for years postinjury.6–8 Because the temporal pattern of onset is variable and the etiology unclear, there are currently few interventions for the treatment of PTSD.
The ongoing inflammatory response after TBI is an emerging target for the treatment of post-traumatic anxiety. Extensive research has shown that chronic neuroinflammation continues for months to years after injury,9–12 and evidence for chronic inflammation has been observed in a number of studies examining patients with PTSD, panic disorder, obsessive-compulsive disorder (OCD), and generalized anxiety disorder.13–18 Glial activation may be involved in the development and maintenance of PTSD,9–12 and mounting evidence supports the role of inflammatory processes in both TBI and anxiety disorders.
After injury, immune cells rapidly produce endogenous danger signals or “alarmins,” which function as potent effectors of innate defense and promote immune system activation by recruiting antigen-presenting cells (APCs) that relay and amplify the inflammatory response.19 The resident APCs of the cenral nervous system (CNS) are microglia, which undergo marked recruitment and activation in response to danger signals,20,21 triggering the onset of prolonged astrocytic activation through the production of proinflammatory cytokines, chemokines, and other proinflammatory mediators.22,23
Activated microglia are thought to contribute to the initiation and maintenance of astrogliosis, which is involved in neural cell damage and inhibition of regenerative responses through secretion of excessive neurotoxic substances, destabilization of neurotransmitter balance, disruption of synaptic connectivity, and excitotoxic neuronal death24–28 and therefore may contribute to functional alterations of brain areas involved in post-traumatic anxiety.
Several studies have reported increased anxiety-like behavior in rodent TBI models,29–32 including increased conditioned33 and unconditioned34 fear responses to both learned and novel stimuli. TBI in rodents also increases levels of activated glial cells and proinflammatory cytokines,34–38 and administration of these cytokines increases anxiety-like behaviors.29–32
The aim of the present study was therefore to determine whether neuroinflammation is associated with the long-term maintenance of post-traumatic anxiety in an animal model. We examined the efficacy of delayed, immunosuppressive treatment (with a glial cell activation inhibitor, Ibudilast) in reducing anxiety-like behaviors and TBI-induced immunological damage.
Methods
Twenty-four adult viral-free male Sprague-Dawley rats (275–325 g; Harlan Laboratories, Madison, WI) were housed in pairs in temperature- (23±3°C) and light- controlled (12-h light/dark) rooms with ad libitum access to food and water. All procedures were performed in accord with University of Colorado (Boulder, CO) Institutional Animal Care and Use Committee guidelines for the humane use of laboratory rats in biological research. Rats were randomly assigned to the following groups (n=6/group): sham+vehicle; sham+MN166; lateral fluid percussion injury (LFPI)+vehicle; and LFPI+MN166.
Lateral fluid percussion injury
LFPI is the most commonly used animal model of TBI and has been shown to reliably replicate many of the pathological changes observed after human TBI, validating its use as a clinically relevant model of human TBI.39 The LFPI used in this study has been described previously.40 Briefly, LFPI rats were anesthetized with halothane (3% induction, 2.0–2.5% maintenance) and mounted in a stereotaxic frame. A 3.0-mm-diameter craniotomy was centered at 3 mm caudal to the bregma and 4.0 mm lateral of the sagittal suture, with the exposed dura remaining intact. A female Luer-Loc hub (inside diameter of 3.5 mm) was secured over the craniotomy with cyanoacrylate adhesive. After hub implantation, rats were removed from the stereotaxic frame and connected to the LFPI apparatus. The LFPI apparatus delivered an impact force (2.0 atmospheres; 20 ms), resulting in a moderate TBI. The injury cap was then removed, the scalp sutured, and rats were returned to their home cages for recovery. Sham-operated rats underwent identical surgical preparation, but did not receive the brain injury.
Ibudilast (MN166) administration
MN166 (MediciNova, San Diego, CA) is a relatively nonselective phosphodiesterase inhibitor with anti-inflammatory actions by glial cell attenuation.41,42 Treated rats received a 5-day dosing regimen of once-daily MN166 injections (10 mg/kg, subcutaneously), beginning at 30 days after LFPI. Weight was recorded before each dosing and treatment administered at the same time each day to maintain constant levels across a 24-h period. Dose selection was based on previous animal pharmacology results43 showing MN166 to be safe and well tolerated, yielding plasma concentration-time profiles commensurate with high-dose regimens in clinical development. MN166 administered by this regimen yields plasma and CNS concentrations that are linked to molecular target actions, including, most potently, macrophage migration inhibitory factor (MIF) inhibition 44 and, secondarily, phosphodiesterases −4 and −10 inhibition.45 The relevance of MIF inhibition in disorders of neuroimmune function, such as neuropathic pain, has recently been well demonstrated.46
Neuromotor tests
Baseline testing of motor, vestibular, and locomotive performance in all groups was conducted immediately before surgery and again at 1 month after injury (Fig. 1). These tests included ipsi- and contralateral assessment of fore- and hindlimb use to assess motor function, locomotion, limb use, and limb preference,47,48 toe spread to assess gross motor response,49 placing to assess visual and vestibular function,50,51 catalepsy rod test to assess postural support and mobility,52 bracing to assess postural stability and catalepsy,53,54 and air righting to assess dynamic vestibular function.55,56 Scoring ranged from 0 (severely impaired) to 5 (normal strength and function). Individual test scores were summed, and a composite neuromotor score (0–45) was then generated for each animal. In addition to the composite neuromotor score, limb-use asymmetry was assessed during spontaneous exploration in the cylinder task, a common measure of motor forelimb function after CNS injury in rats,50,57 and postinjury locomotor activity was assessed through distance traveled on a running wheel; both tasks were scored for 5 min under red light (∼90 lux).
FIG. 1.

Experimental timeline. Neuromotor testing included measures of motor, vestibular, and locomotive performance and was conducted immediately before LFPI and again at 1 month postinjury. A single shock was delivered after neuromotor testing was completed at the 1-month time point, and freezing behavior was assessed at 1 through 6 months postinjury. Tissue was then collected for immunohistochemistry. LFPI, lateral fluid percussion injury.
Behavioral measures
The core features of anxiety in humans include feelings of apprehension or dread both with or without autonomic signs and symptoms, and in the case of post-traumatic anxiety (PTSD), also includes reexperiencing trauma, avoidant behavior, and hypervigilance.4 The immediate shock paradigm was chosen to elicit PTSD-related traits of abnormally elevated fear responses and hypervigilance. In traditional contextual fear conditioning, rats are placed in a context and shock is delayed for a period of time, allowing for association between the contextual cues and the shock, resulting in increased freezing during later testing. However, rats that are immediately shocked upon placement in a shock chamber show no increase in freezing behavior during later testing. This phenomenon is known as the immediate shock deficit and results in failure to display contextual fear conditioning because the rats do not have time to construct a representation of the context,58–60 indicating a lack of association between the context and shock. However, we previously found that LFPI rats show increases in freezing responses even in the absence of fear conditioning.34 These unconditioned freezing responses may reflect pathological anxiety, which involves exaggerated fear, hypervigilance, and readiness to respond to danger or negative events,61 because freezing behavior is part of an anticipatory response to stress or danger. Shock was chosen as the stressor because it resulted in anxiety-like freezing behavior, which is a simple reproducible response elicited as a defense reaction in both conditioned and unconditioned fearful situations.62
A novel environment was used for testing to ensure that there was no association between the context and shock, because rats were never tested in the context where the shock occurred. The novel context was a standard rat cage with one vertically and one horizontally striped wall. No aversive stimuli were introduced in this context and no conditioning occurred. Rats were tested (5 min) and the percent of freezing behavior was assessed. Freezing was defined as the absence of movement, except for heart beat/respiration, and was recorded in 10-sec intervals. Freezing behavior in the novel environment was measured after administration of a foot shock in a separate apparatus. The shock apparatus consisted of two chambers placed inside sound-attenuating chests. The floor of each chamber consisted of 18 stainless steel rods (4 mm in diameter), spaced 1.5 cm center to center and wired to a shock generator and scrambler (Colbourn Instruments, Allentown, PA). An automated program delivered a 2-sec/1.5-mA electric shock. Rats were transported in black buckets and shocked immediately upon entry to chambers. After shock, rats were returned to their home cages.
Timeline for behavioral testing
Testing was performed at 1 through 6 months postinjury. A single shock was delivered after neuromotor testing was completed at the 1-month time point.
Immunohistochemistry
Rats were intracardially perfused with 0.9% saline and tissue was collected, then fixed with 4% paraformaldehyde (PFA) overnight at 4°C. Tissue was transferred to a 30% sucrose phosphate-buffered saline solution for 1–2 days, then stored at −80°C. Brains were sectioned at 20 μm and mounted onto SuperFrost Plus slides (Fisher Scientific, Pittsburgh, PA) using a cryostat at −22°C. Brain sections were postfixed with 4% PFA for 15 min at room temperature, then treated with 0.3% H2O2 for 30 min at room temperature. Immunoreactivity in brain regions associated with anxiety (insula and amygdala) was assessed for markers of microglia (CD11b/c; OX42 labeling) and astrocytes (glial fibrillary acidic protein; GFAP), using an avidin-biotin-horseradish peroxidase (ABC) reaction.63 Sections were incubated at 4°C overnight in either mouse anti-rat OX-42 (1:100; BD Biosciences Pharmingen, San Jose, CA) or mouse anti-pig GFAP (1:100; MP Biomedicals, Aurora, OH). The next day, sections were incubated at room temperature for 2 h with biotinylated goat anti-mouse immunoglobulin G antibody (1:200; Jackson ImmunoResearch, West Grove, PA). Sections were washed and incubated for 2 h at room temperature in ABC (1:400; Vector Laboratories, Burlingame, CA) and reacted with 3’,3-diaminobenzidine (Sigma-Aldrich, St. Louis, MO). Sections were air-dried overnight and then dehydrated with graded alcohols, cleared in Histoclear and cover-slipped with Permount (Fisher Scientific, Fairlawn, NJ).
Image analysis
Slides were viewed with an Olympus BX-61 microscope, using Olympus Microsuite software (Olympus America, Melville, NY), with bright-field illumination at 10× magnification. Densitometric analysis was performed using Scion Image software. Images were analyzed, under blinded conditions, in National Institutes of Health ImageJ (NIH; Bethesda, MD) using grayscale. Signal pixels of a region of interest (ROI) were defined as having gray values 3.5 standard deviations above the mean gray value of a cell-poor area close to the ROI. The number of pixels and the average gray values above the set background were then computed for each ROI and multiplied, giving an integrated densitometric measurement. Six measurements were made for each ROI; measurements were then averaged to obtain a single integrated density value per rat, per region.
Statistical analyses
Results are expressed as mean±standard error of the mean. Analyses for behavioral measures used analysis of variance (ANOVA) with repeated measures (time after injury) and treatment as the independent variable. The integrated density was measured at one time point (6 months postinjury) and utilized one-way ANOVAs to compare regions between groups. Tukey's honestly significant difference was used to conduct planned pair-wise comparisons to follow up significant overall ANOVAs. Data were analyzed using SPSS® Statistics software (SPSS, Inc., Chicago, IL), and, in all cases, statistical significance was set at p<0.05.
Results
LFPI-induced increases in freezing behavior were observed when rats were placed in a novel context after shock in a separate environment (Fig. 2; F(3, 20)=9.029; p=0.001). Exposed only to this minor additional stressor and before treatment with either MN166 or vehicle, LPFI rats (Fig. 2, white and black bars) froze approximately twice as long as sham-operated rats (Fig. 2, light and dark gray bars) at the 1-month time point: LFPI+vehicle (p<0.03 vs. sham+vehicle and sham+MN166) and LFPI+MN166 (p<0.03 vs. sham+vehicle and sham+MN166), whereas both LFPI groups (before treatment or vehicle) did not differ (p=0.94) statistically.
FIG. 2.
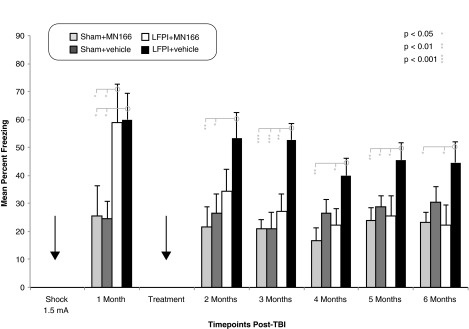
Freezing behavior in novel context. Sham-operated rats froze approximately 25% before treatment with MN166 or vehicle, whereas LFPI rats froze at significantly higher rates (∼60%), indicating greater anxiety-like behavior. After treatment, anxiety-like freezing behavior in LFPI+MN166 rats was reduced, (∼25%), compared to LFPI+vehicle rats (∼50%). This effect was significant at 3 months and remained through 6 months postinjury. Freezing in sham+MN166 and sham+vehicle rats could not be distinguished from LFPI+MN166-treated rats at all time points after treatment, whereas LFPI+vehicle-injected rats froze significantly more than both sham groups at all post-treatment time points, with the exception of the sham vehicle group at the 4- and 6-month time points. Data represent mean±standard error of the mean. LFPI, lateral fluid percussion injury; TBI traumatic brain injury.
At 2 months postinjury, after treatment with MN166 or vehicle, freezing in both sham-operated groups remained constant at approximately 25%. There was a drastic reduction (25%) in freezing behavior in MN166-treated rats, with untreated LFPI+vehicle rats freezing approximately 20% more than LFPI+MN166 rats. Freezing differences between sham+vehicle and sham+MN166 control groups and LFPI+MN166-treated rats no longer reached significance (p=0.49 and p=0.26, respectively). Freezing behavior in vehicle-injected LFPI rats remained consistently higher than sham controls (p<0.03 vs. sham+vehicle and sham+MN166), with untreated rats freezing approximately 30% more than sham-operated controls at 2 months postinjury.
At 3 through 6 months postinjury, freezing averages for sham+MN166 and sham+vehicle control groups again remained constant (20 and 25%, respectively). Freezing behavior in vehicle-injected LFPI rats remained consistently higher than drug-treated controls at all post-treatment time points (3 through 6 months; p<0.03 vs. sham+MN166). Freezing behavior in untreated LFPI rats remained higher than sham+vehicle controls at the 3- and 5-month time points postinjury (3 months, p=0.00; 4 months, p=0.10; 5 months, p=0.04; 6 months, p=0.13).
The behavior of MN166-treated LFPI rats remained indistinguishable from controls. Remarkably, freezing differences between sham+MN166- and sham+vehicle-injected control groups and LFPI+MN166-treated rats did not reach significance at any of the post-treatment time points (3 through 6 months; p>0.30 vs. sham+vehicle and sham+MN166). In contrast, untreated LFPI+vehicle-injected rats froze significantly more than treated rats, approximately twice that of the treated rats across all post-treatment time points (3 through 6 months; p<0.03 vs. LFPI+MN166).
The behavioral effects of surgery alone, independent of LFPI, were observed in the sham+vehicle group (Fig. 2, dark gray bars). This group froze more than the sham+MN166-treated group across most time points, in spite of having no significant differences in freezing behavior at baseline. Additionally, whereas the LFPI+vehicle group did freeze more than the sham+vehicle group across the entire study, they did not statistically differ at the 4- and 6-month time points, which may reflect behavioral, immunological, and morphological damage noted by other researchers in response to craniotomy alone.64 Disruption of blood vessels and nerves along the scalp suture, mechanical pressure of the drill, and atmospheric exposure when the bone flap is removed can alter vascular physiology and lead to structural and functional impairments after surgery alone.64,65 Though traditional sham control groups are the standard in TBI research, these results suggest that injury resulting from surgery may increase freezing behavior, although sham+MN166-treated rats showed reductions in freezing behavior from baseline, which does indicate efficacy of MN166 treatment in reducing behavioral effects resulting from craniotomy alone. However, based on these results, future studies will need to include naïve or anesthesia-only controls to determine whether MN166 treatment can reduce anxiety-like freezing behavior to levels of uninjured controls.
LFPI-induced freezing responses were not influenced by motor, vestibular, or locomotive impairments, because neuromotor composite scores of the brain-injured groups (LFPI+MN166 and LFPI+vehicle) did not significantly differ from controls (F(3, 20)=0.383; p=0.766). Rats in all groups consistently received normal scores on fore- and hindlimb use, toe spread, placing, catalepsy rod, bracing, and air righting tests, indicating no impairments in motor, vestibular, or locomotive functioning as a result of TBI. There were also no significant between-group differences in limb-use asymmetry observed for contra- (F(3, 20)=0.058; p=0.981) and ipsilateral (F(3, 20)=0.285; p=0.836) forelimb use during vertical exploratory behavior in the cylinder task, indicating no limb-use bias resulting from injury (Fig. 3A). No significant between-group differences were found in locomotor performance evidenced by distance traveled during the running wheel activity (F(3, 20)=0.152; p=0.464), revealing no postinjury impairments in locomotion (Fig. 3B).
FIG. 3.

Cylinder task and running wheel activity at 1 month postinjury. (A) LFPI rats mean number of spontaneous forelimb placements (ipsi- and contralateral) during exploratory activity in the cylinder test did not differ from controls at 1 month postinjury, indicating no deficits in limb use resulting from injury. (B) LFPI rats mean change in distance traveled in the running wheel activity did not significantly differ from controls at 1 month postinjury, indicating no impairments in locomotion resulting from injury. Data represent mean±standard error of the mean. LFPI, lateral fluid percussion injury.
OX-42 and GFAP immunoreactivity (reflecting microglia and astrocytic activation, respectively) was assessed in the insula and amygdala in MN166- and vehicle-injected LFPI rats for comparison to sham-operated controls. Representative images (20×), showing GFAP immunoreactivity in several of these regions, are shown in Figure 4, revealing normal astrocyte morphology in both MN166- and vehicle-injected sham controls. LFPI+vehicle rats showed clear signs of reactive astrocytes (Fig. 4; bottom row), whereas LFPI rats treated with MN166 (Fig. 4, third row) were difficult to differentiate from sham-operated control groups.
FIG. 4.

Representative photomicrographs (20×) depicting GFAP immunoreactivity assessed in the insula and amygdala at 6 months postinjury. Vehicle-injected LFPI rats showed clear signs of reactive astrocytes (bottom row), whereas astrocytes from sham-operated rat tissue appeared to have normal morphology (top rows). LFPI rats treated with MN166 (third row) were difficult to differentiate from sham-operated groups. Rhinal fissure (rf ) and commissural stria terminalis (cst). GFAP, glial fibrillary acidic protein; LFPI, lateral fluid percussion injury.
Immunohistochemistry (IHC) was conducted to assess TBI-induced increases in gliosis and efficacy of MN166 in reducing reactive gliosis in brain regions associated with anxiety. Results revealed increased GFAP labeling in both brain regions examined, confirming that astroglial activation was significantly greater in LFPI+vehicle, compared to other, groups in insula (Fig. 5A, left graph; F(3, 140)=3.761; p=0.012) and amygdala (Fig. 5B, left graph; F(3, 140)=6.025; p=0.001). In contrast, no differences in GFAP labeling were observed between sham-operated and LFPI+MN166 groups in either region (insula, p>0.60 vs. sham+vehicle and sham+MN166; amygdala, p>0.40 vs. sham+vehicle and sham+MN166). Whereas MN166-treated LFPI rats were not distinguishable from sham-operated controls, post-hoc analyses revealed that LFPI+vehicle rats had significantly greater astrocytic activation in both brain regions, as compared to controls (Fig. 5A,B, left graphs: insula, p<0.02 vs. LFPI+MN166, sham+vehicle, and sham+MN166; amygdala, p<0.01 vs. LFPI+MN166, sham+vehicle, and sham+MN166).
FIG. 5.

Astro- and microglial activation in insula and amygdala at 6 months postinjury. LFPI+vehicle rats had increased reactive gliosis postinjury, as evidenced by increased glial activation, compared to controls, and treatment with MN166 reduced gliosis levels in LFPI rats to those of sham-operated controls. (A and B) LFPI+vehicle rats had significantly increased in GFAP labeling in both regions, indicating higher astroglial activation, compared to sham-operated and LFPI+MN166-treated rats. (C) In the CE, microglial activation was greater in LFPI+vehicle-injected rats, compared to both sham-operated groups and LFPI+MN166-treated rats, and was approaching significance in BLA. Data represent mean±standard error of the mean. LFPI, lateral fluid percussion injury; GFAP, glial fibrillary acidic protein; CE, central amagdala; BLA, basolateral amygdala.
Analysis of GFAP immunoreactivity in subregions of the insula (Fig. 5A, right graph) also revealed that LFPI+vehicle rats had increased GFAP labeling in agranular (F(3, 140)=2.493; p=0.063), dysgranular (F(3, 140)=7.388; p=0.000), and granular (F(3, 140)=2.998; p=0.033) insular regions. No significant differences between sham-operated and LFPI+MN166 groups were found in subregions of the insula (agranular, p>0.70 vs. sham+vehicle and sham+MN166; dysgranular, p>0.20 vs. sham+vehicle and sham+MN166; granular, p>0.20 vs. sham+vehicle and sham+MN166). Untreated, LFPI+vehicle rats had greater astrocytic activation in all three subregions, as compared to controls (Fig. 5A, right graph: agranular, p<0.03 vs. LFPI+MN166 and sham+vehicle and p=0.079 vs. sham+MN166; dysgranular, p<0.01 vs. LFPI+MN166, sham+vehicle, and sham+MN166; granular, p=0.124 vs. LFPI+MN166, p=0.003 vs. sham+vehicle, and p=0.087 vs. sham+MN166).
In the subregions of the amygdala (Fig. 5B, right graph), GFAP labeling in LFPI+vehicle rats was significantly increased in basolateral amagdala (BLA; F(3, 140)=39.154; p=0.000) and central amygdala (CE; F(3, 140)=12.073; p=0.000) nuclei, compared to controls. Post-hoc analyses revealed that LFPI+vehicle rats had significantly greater astrocytic activation in both subregions (CE, p<0.001 vs. LFPI+MN166, sham+vehicle, and sham+MN166; BLA, p=0.001 vs. LFPI+MN166, sham+vehicle, and sham+MN166). MN166-treated LFPI rats had significantly less GFAP expression than sham+vehicle controls in CE (p=0.03), but did not differ from sham+MN166-treated rats (p=0.58). LFPI+MN166-treated rats also did not differ from sham controls in the BLA (p=0.58 vs. sham+vehicle and p=0.06 vs. sham+MN166).
LFPI+vehicle rats also showed significantly increased microglial activation, as measured by OX-42 labeling, compared to control groups (Fig. 5C), but this was restricted to subregions of the amygdala, such as the CE (F(3, 140)=9.290; p=0.000), and also approached significance in BLA (F(3, 140)=2.399; p=0.071) nuclei. Post-hoc analysis revealed significant increases in microglial activation for LFPI+vehicle rats in CE (p<0.001 vs. LFPI+MN166, sham+vehicle, and sham+MN166) and BLA (p=0.009 vs. sham+MN166). No differences in OX-42 labeling were observed between sham-operated and LFPI+MN166 groups in amygdala, nor were any significant between-group differences found in OX-42 expression for the insula.
Discussion
LFPI-induced anxiety-like behaviors are found at long-term, postinjury time points in untreated brain-injured rats, as compared to those treated with MN166. Pharmacological suppression of immune responses at 1 month postinjury, when anxiety-like behavior has fully developed, markedly reduces long-term behavioral and immunological impairments after TBI (out to 6 months) and restores MN166-treated rats to levels indistinguishable from sham-operated controls. These findings not only implicate chronic neuroinflammation in the development of anxiety-like behaviors after TBI, but also show that delayed treatment is capable of reversing established post-traumatic anxiety behaviors. To our knowledge, this is the first study to examine delayed immunosuppression at long-term injury time points, because other immunosuppressive treatments targeting anxiety-like behaviors have been administered before or within hours of injury.34,37,38,66–69 These results indicate that the persistence of post-traumatic anxiety may reflect chronic neuroinflammatory neuropathy, a possibility supported by the observation of activated microglia and astrocytes, key cells mediating inflammatory processes, many years after injury in long-term survivors of TBI.9–12
Chronic post-traumatic neuroinflammation suggests the presence of a self-perpetuating positive feedback loop, possibly involving reactivation and further promotion of inflammatory mediators after injury in an inflammation-damage-inflammation cycle.70 Stressed, damaged, and injured cells release endogenous danger signals, which trigger local inflammatory responses needed for tissue repair and regeneration.21,71–73 Damage-associated molecular patterns (DAMPs) play an important role in the propagation of the proinflammatory cascade of innate immunity, promoting the release of cytokines and other inflammatory mediators.70,74 DAMPs initiate the innate immune response through the activation of APCs, which up-regulate costimulatory and major histocompatibility complex molecules.21,71,75 APCs respond to endogenous signals through Toll-like receptors (TLRs), which recognize a variety of DAMPs and act as pattern recognition receptors for endogenous molecules.
Microglia are the resident immunological cells and primary APCs of the CNS, remaining quiescent until activated through TLR engagement with DAMPs to perform effector inflammatory and APC functions.76 Microglial cells contribute to neuroinflammation in response to DAMPS by secreting proinflammatory cytokines, such as interleukin (IL)-1 and tumor necrosis factor alpha (TNF-α), which amplify the inflammatory response by initiating the production of other cytokines and promoting microglial proliferation and activation of astrocytes.70 The early phase of TBI-induced reactive gliosis has been reported to begin in with predominant microglia activation that peaks within 1 week,28,77–82 but continues for several weeks and overlaps later with persistent astrocytic activation.28,82,83
Our IHC results support this time course, indicating injury-induced astrogliosis in the insula and amygdala at 6 months postinjury, but less activation of microglia (only significant in the amygdala), suggesting that microglial activation may precede astrocytic activation and modulate the onset and maintenance of astrogliosis.27,84–88 Lower levels of microglia expression could be the result of assessment at 6 months postinjury, when microglia may have returned to a quiescent or surveying state,28,89 whereas astrocytic activation persists in a long-lasting, self-perpetuating inflammatory response in the brain that exceeds early neuroprotection and results in neurodegenerative changes capable of continuing the inflammatory cycle.9,27,90
Chronic inflammation has been observed in a number of studies examining patients with trauma-related anxiety disorders, reporting increases in downstream mediators, such as peripheral elevations of TNF-α, interferon-gamma (IFN-γ), IL-1β, and IL-6, in patients with PTSD,13–16 elevations of TNF-α and IL-6 in patients with OCD,17 and elevations in proinflammatory cytokines and chemokines (monocyte chemoattractant protein 1, macrophage inflammatory protein 1 alpha, IL-1α, IL-1β, IL-6, IL-8, Eotaxin, granulocyte macrophage colony-stimulating factor, and IFN-γ) in individuals with panic disorder and PTSD.18 Despite compelling evidence implicating excessive inflammatory actions and a generalized inflammatory state in the development of anxiety disorders after TBI, central measures of proinflammatory cytokine elevations specifically related to human PTSD and other anxiety disorders have not yet been performed. However, the current results provide evidence for chronic neuroinflammation in the development and maintenance of post-traumatic anxiety in an animal model, as indicated by elevated immunoreactivity in the amygdala and insula at 6 months postinjury.
The amygdala and insula have long been associated with human anxiety disorders. Studies in patients with PTSD implicate exaggerated responses of the amygdala and insula,91–95 impaired inhibition of medial prefrontal cortex and anterior cingulate,94,96–98 and decreased hippocampal volume.97,99,100 Other neuroimaging reports of patients with non-trauma-related OCD and phobia, as well as those with PTSD, have revealed that aversive anticipation (a hallmark of anxiety) involves increased activation of both the amygdala and insula.92 Evidence for the specific involvement of these brain areas in human post-traumatic anxiety is complemented by animal models, including findings of increased PTSD-related traits and increased Stathmin 1 (a protein known to increase fear responses) expression in the amygdala after blast injury,101 increased fear conditioning and up-regulation of excitatory N-methyl-D-aspartate receptors (crucial receptors for normal fear learning and memory) in the amygdala after concussive injury,33 and our current results showing increased anxiety-like behavior and reactive gliosis in insula and amygdala at long-term time points after LFPI. However, though these results support findings from our previous study34 indicating increased gliosis in these areas, the insula and amygdala are not the only regions involved in anxiety and do not exclude the possibility that other regions may be contributing to the results reported here. Future studies will be aimed at characterizing the model and inclusion of other brain regions involved in human anxiety, including cingulate cortex, hippocampus, medial prefrontal cortex, locus coeruleus, and the bed nucleus of the stria terminalis.94,97,99,102,103
Although the exact role of the immune system in the pathogenesis of anxiety disorders after TBI remains unknown, neuroinflammation is emerging as a potential target. The present findings of treatment-related reductions in anxiety-like behaviors and reactive gliosis in brain regions associated with anxiety support the use of immunosuppression to improve functional outcome after TBI. Peri-injury and immediate postinjury immunosuppression have been found to be neuroprotective after TBI in rodents, resulting in increased structural preservation and improved functional outcomes.104 Early administration of the immunosuppressant drugs, minocycline, statins, cyclosporin A, and FK506, have been shown to exert anti-inflammatory effects through suppression of micro- and astroglial production of IL-1β, TNF-α, and IL-6, resulting in reduced functional deficits, cerebral edema, and brain lesion volumes,35–38,66 improving mitochondrial preservation, reducing dendritic spine loss, and improving cognitive performance and functional motor recovery.105,106 Our previous investigation found that peri-injury Ibudilast treatment attenuated glial cell activation at the time of injury, resulting in reduced anxiety-like behaviors and immunological impairments after LFPI. 34
These immunosuppressant drugs all have direct inhibitory effects on microglia and astrocytes, leading to better functional recovery after TBI; however, these treatments require rapid administration and reduce the therapeutic window to the day of injury. The current work shows reversal of established anxiety-like behaviors and reactive gliosis at 1 month postinjury, delayed treatment time points that have not been tested with any other immunosuppressive interventions, in spite of substantial evidence that many molecular, biochemical, and immunological changes occur for many months after injury and that clinical intervention may not be possible at early stages of TBI. Ongoing inflammation represents a window of opportunity for therapeutic intervention to prevent progressive tissue damage, loss of function, and possibly interrupt the progression of neuropathological conditions after injury.
Our finding that delayed immunosuppression is capable of reversing established post-traumatic anxiety behaviors and immunological impairments through 6 months after TBI contributes to growing evidence that the critical window for treatment after TBI can be expanded to include those suffering from long-term TBI-related disabilities. Studies have shown that delayed treatment (24 h) with erythropoietin, a novel neuroprotective cytokine found to improve neuronal survival through attenuation of cytokine production and inflammation, improved sensorimotor functional recovery, reduced hippocampal cell loss, enhanced neurogenesis, and improved neurological outcomes after controlled cortical impact (CCI) and weight-drop rodent TBI models.107,108 Similarly, a recent study reported reduced chronic inflammation and neurodegeneration after activation of metabotropic glutamate receptor 5 with the specific agonist, (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), which has been shown to decrease microglial activation and release associated proinflammatory mediators. The study delayed treatment until 1 month after CCI in mice, delivering a single intracerebroventricular injection of CHPG. The results revealed reductions in reactive gliosis, hippocampal cell loss, reduced lesion progression, and improved motor and cognitive recovery, compared to untreated controls.109
Immunosuppression of chronic neuroinflammation may hold promise as a therapeutic target in treatment of established anxiety disorders after TBI. It has been shown here that inflammation produced by neuroimmune responses after injury plays a role in TBI-induced anxiety, and delayed immunosuppression leads to better functional outcomes at long-term postinjury treatment points after TBI. Delayed, postinjury suppression of glial cell activation could therefore expand the clinical window for treatment of TBI-induced anxiety disorders in humans.
Acknowledgments
This work was supported by the U.S. Army Medical Research and Material Command (grant PR100040), the Craig Hospital Gift Fund, a University of Colorado Innovative Seed Grant, the Autism Speaks Pilot Study (grant 7153), and the National Institutes of Health (grant nos. NS36981 [to D.S.B.] and DA024044 and DA01767 [to L.R.W.].
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Faul M., Xu L., Wald M.M., and Coronado V.G. (2010). Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Available at: http://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf Accessed November5, 2013
- 2.Rao V., and Lyketsos C. (2000). Neuropsychiatric sequelae of traumatic brain injury. Psychosomatics 41, 95–103 [DOI] [PubMed] [Google Scholar]
- 3.Hiott D.W., and Labbate L. (2002). Anxiety disorders associated with traumatic brain injuries. NeuroRehabilitation 17, 345–355 [PubMed] [Google Scholar]
- 4.Vaishnavi S., Rao V., and Fann J.R. (2009). Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics 50, 198–205 [DOI] [PubMed] [Google Scholar]
- 5.van Reekum R., Cohen T., and Wong J. (2000). Can traumatic brain injury cause psychiatric disorders? J. Neuropsychiatry Clin. Neurosci. 12, 316–327 [DOI] [PubMed] [Google Scholar]
- 6.Morton M.V., and Wehman P. (1995). Psychosocial and emotional sequelae of individuals with traumatic brain injury: a literature review and recommendations. Brain Inj. 9, 81–92 [DOI] [PubMed] [Google Scholar]
- 7.Deb S., Lyons I., Koutzoukis C., Ali I., and McCarthy G. (1999). Rate of psychiatric illness 1 year after traumatic brain injury. Am. J. Psychiatry 156, 374–378 [DOI] [PubMed] [Google Scholar]
- 8.Koponen S., Taiminen T., Portin R., Himanen L., Isoniemi H., Heinonen H., Hinkka S., and Tenovuo O. (2002). Axis I and II psychiatric disorders after traumatic brain injury: a 30-year follow-up study. Am. J. Psychiatry 159, 1315–1321 [DOI] [PubMed] [Google Scholar]
- 9.Gentleman S.M., Leclercq P.D., Moyes L., Graham D.I., Smith C., Griffin W.S., and Nicoll J.A. (2004). Long-term intracerebral inflammatory response after traumatic brain injury. Forensic. Sci. Int. 146, 97–104 [DOI] [PubMed] [Google Scholar]
- 10.Streit W.J., Mrak R.E., and Griffin W.S. (2004). Microglia and neuroinflammation: a pathological perspective. J. Neuroinflammation 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagamoto-Combs K., McNeal D.W., Morecraft R.J., and Combs C.K. (2007). Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 24, 1719–1742 [DOI] [PubMed] [Google Scholar]
- 12.Ramlackhansingh A.F., Brooks D.J., Greenwood R.J., Bose S.K., Turkheimer F.E., Kinnunen K.M., Gentleman S., Heckemann R.A., Gunanayagam K., Gelosa G., and Sharp D.J. (2011). Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 70, 374–383 [DOI] [PubMed] [Google Scholar]
- 13.Spivak B., Shohat B., Mester R., Avraham S., Gil-Ad I., Bleich A., Valevski A., and Weizman A. (1997). Elevated levels of serum interleukin-1 beta in combat-related posttraumatic stress disorder. Biol. Psychiatry 42, 345–348 [DOI] [PubMed] [Google Scholar]
- 14.Tucker P., Ruwe W.D., Masters B., Parker D.E., Hossain A., Trautman R.P., and Wyatt D.B. (2004). Neuroimmune and cortisol changes in selective serotonin reuptake inhibitor and placebo treatment of chronic posttraumatic stress disorder. Biol. Psychiatry 56, 121–128 [DOI] [PubMed] [Google Scholar]
- 15.Rohleder N., Joksimovic L., Wolf J.M., and Kirschbaum C. (2004). Hypocortisolism and increased glucocorticoid sensitivity of pro-Inflammatory cytokine production in Bosnian war refugees with posttraumatic stress disorder. Biol. Psychiatry 55, 745–751 [DOI] [PubMed] [Google Scholar]
- 16.von Kanel R., Hepp U., Kraemer B., Traber R., Keel M., Mica L., and Schnyder U. (2007). Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J. Psychiatr. Res. 41, 744–752 [DOI] [PubMed] [Google Scholar]
- 17.Konuk N., Tekin I.O., Ozturk U., Atik L., Atasoy N., Bektas S., and Erdogan A. (2007). Plasma levels of tumor necrosis factor-alpha and interleukin-6 in obsessive compulsive disorder. Mediators Inflamm. 2007, 65704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoge E.A., Brandstetter K., Moshier S., Pollack M.H., Wong K.K., and Simon N.M. (2009). Broad spectrum of cytokine abnormalities in panic disorder and posttraumatic stress disorder. Depress. Anxiety 26, 447–455 [DOI] [PubMed] [Google Scholar]
- 19.Pugin J. (2012). How tissue injury alarms the immune system and causes a systemic inflammatory response syndrome. Ann. Intensive Care 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matzinger P. (1998). An innate sense of danger. Semin. Immunol. 10, 399–415 [DOI] [PubMed] [Google Scholar]
- 21.Hirsiger S., Simmen H.P., Werner C.M., Wanner G.A., and Rittirsch D. (2012). Danger signals activating the immune response after trauma. Mediators Inflamm. 2012, 315941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gehrmann J., Banati R.B., and Kreutzberg G.W. (1993). Microglia in the immune surveillance of the brain: human microglia constitutively express HLA-DR molecules. J. Neuroimmunol. 48, 189–198 [DOI] [PubMed] [Google Scholar]
- 23.Gehrmann J., Matsumoto Y., and Kreutzberg G.W. (1995). Microglia: intrinsic immuneffector cell of the brain. Brain Res. 20, 269–287 [DOI] [PubMed] [Google Scholar]
- 24.Sternberg E.M. (1997). Neural-immune interactions in health and disease. J. Clin. Invest. 100, 2641–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raison C.L., and Miller A.H. (2003). When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am. J. Psychiatry 160, 1554–1565 [DOI] [PubMed] [Google Scholar]
- 26.Szelenyi J., and Vizi E.S. (2007). The catecholamine cytokine balance: interaction between the brain and the immune system. Ann. N. Y. Acad. Sci. 1113, 311–324 [DOI] [PubMed] [Google Scholar]
- 27.Zhang D., Hu X., Qian L., O'Callaghan J.P., and Hong J.S. (2010). Astrogliosis in CNS pathologies: is there a role for microglia? Mol. Neurobiol. 41, 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colangelo A.M., Cirillo G., Lavitrano M.L., Alberghina L., and Papa M. (2012). Targeting reactive astrogliosis by novel biotechnological strategies. Biotechnol. Adv. 30, 261–271 [DOI] [PubMed] [Google Scholar]
- 29.Connor T.J., Song C., Leonard B.E., Merali Z., and Anisman H. (1998). An assessment of the effects of central interleukin-1beta, −2, −6, and tumor necrosis factor-alpha administration on some behavioural, neurochemical, endocrine and immune parameters in the rat. Neuroscience 84, 923–933 [DOI] [PubMed] [Google Scholar]
- 30.Cragnolini A.B., Schioth H.B., and Scimonelli T.N. (2006). Anxiety-like behavior induced by IL-1beta is modulated by alpha-MSH through central melanocortin-4 receptors. Peptides 27, 1451–1456 [DOI] [PubMed] [Google Scholar]
- 31.Sokolova E.S., Lyudyno V.I., Simbirtsev A.S., and Klimenko V.M. (2007). The psychomodulatory action of subpyrogenic doses of interleukin-1beta in conditions of chronic administration to rats. Neurosci. Behav. Physiol. 37, 499–504 [DOI] [PubMed] [Google Scholar]
- 32.Zubareva O.E., and Klimenko V.M. (2009). Long-term disorders of behavior in rats induced by administration of tumor necrosis factor during early postnatal ontogenesis. Neurosci. Behav. Physiol. 39, 21–24 [DOI] [PubMed] [Google Scholar]
- 33.Reger M.L., Poulos A.M., Buen F., Giza C.C., Hovda D.A., and Fanselow M.S. (2012). Concussive brain injury enhances fear learning and excitatory processes in the amygdala. Biol. Psychiatry 71, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodgers K.M., Bercum F.M., McCallum D.L., Rudy J.W., Frey L.C., Johnson K.W., Watkins L.R., and Barth D.S. (2012). Acute neuroimmune modulation attenuates the development of anxiety-like freezing behavior in an animal model of traumatic brain injury. J. Neurotrauma 29, 1886–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen S.F., Hung T.H., Chen C.C., Lin K.H., Huang Y.N., Tsai H.C., and Wang J.Y. (2007). Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci. 81, 288–298 [DOI] [PubMed] [Google Scholar]
- 36.Li B., Mahmood A., Lu D., Wu H., Xiong Y., Qu C., and Chopp M. (2009). Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery 65, 179–185; discussion, 185–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homsi S., Federico F., Croci N., Palmier B., Plotkine M., Marchand-Leroux C., and Jafarian-Tehrani M. (2009). Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. 1291, 122–132 [DOI] [PubMed] [Google Scholar]
- 38.Homsi S., Piaggio T., Croci N., Noble F., Plotkine M., Marchand-Leroux C., and Jafarian-Tehrani M. (2010). Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J. Neurotrauma 27, 911–921 [DOI] [PubMed] [Google Scholar]
- 39.Thompson H.J., Lifshitz J., Marklund N., Grady M.S., Graham D.I., Hovda D.A., and McIntosh T.K. (2005). Lateral fluid percussion brain injury: a 15-year review and evaluation. J. Neurotrauma 22, 42–75 [DOI] [PubMed] [Google Scholar]
- 40.Frey L.C., Hellier J., Unkart C., Lepkin A., Howard A., Hasebroock K., Serkova N., Liang L., Patel M., Soltesz I., and Staley K. (2009). A novel apparatus for lateral fluid percussion injury in the rat. J. Neurosci. Methods 177, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizuno T., Kurotani T., Komatsu Y., Kawanokuchi J., Kato H., Mitsuma N., and Suzumura A. (2004). Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology 46, 404–411 [DOI] [PubMed] [Google Scholar]
- 42.Rolan P., Hutchinson M., and Johnson K. (2009). Ibudilast: a review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin. Pharmacother. 10, 2897–2904 [DOI] [PubMed] [Google Scholar]
- 43.Ellis A.L., Wieseler J., Brown K., Blackwood C., Ramos K., Starnes C., Maier S.F., Watkins L.R., and Falci S.P. (2008). Characterization of exaggerated pain behavior and glial activation in a novel rat model of spinal cord injury. Poster from 38th Annual Meeting of the Society for Neuroscience, Washington, DC, November15–19, 2008. [Google Scholar]
- 44.Cho Y., Crichlow G.V., Vermeire J.J., Leng L., Du X., Hodsdon M.E., Bucala R., Cappello M., Gross M., Gaeta F., Johnson K., and Lolis E.J. (2010). Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc. Natl. Acad. Sci. U. S. A. 107, 11313–11318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson L.C., Hastings S.F., McPhee I., Clayton R.A., Darroch C.E., Mackenzie A., Mackenzie F.L., Nagasawa M., Stevens P.A., and Mackenzie S.J. (2006). The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. Eur. J. Pharmacol. 538, 39–42 [DOI] [PubMed] [Google Scholar]
- 46.Wang F., Xu S., Shen X., Guo X., Peng Y., and Yang J. (2011). Spinal macrophage migration inhibitory factor is a major contributor to rodent neuropathic pain-like hypersensitivity. Anesthesiology 114, 643–659 [DOI] [PubMed] [Google Scholar]
- 47.Bland S.T., Schallert T., Strong R., Aronowski J., Grotta J.C., and Feeney D.M. (2000). Early exclusive use of the affected forelimb after moderate transient focal ischemia in rats: functional and anatomic outcome. Stroke 31, 1144–1152 [DOI] [PubMed] [Google Scholar]
- 48.Bland S.T., Pillai R.N., Aronowski J., Grotta J.C., and Schallert T. (2001). Early overuse and disuse of the affected forelimb after moderately severe intraluminal suture occlusion of the middle cerebral artery in rats. Behav. Brain Res. 126, 33–41 [DOI] [PubMed] [Google Scholar]
- 49.Nitz A.J., Dobner J.J., and Matulionis D.H. (1986). Pneumatic tourniquet application and nerve integrity: motor function and electrophysiology. Exp. Neurol. 94, 264–279 [DOI] [PubMed] [Google Scholar]
- 50.Schallert T., Fleming S.M., Leasure J.L., Tillerson J.L., and Bland S.T. (2000). CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39, 777–787 [DOI] [PubMed] [Google Scholar]
- 51.Woodlee M.T., Asseo-Garcia A.M., Zhao X., Liu S.J., Jones T.A., and Schallert T. (2005). Testing forelimb placing “across the midline” reveals distinct, lesion-dependent patterns of recovery in rats. Exp. Neurol. 191, 310–317 [DOI] [PubMed] [Google Scholar]
- 52.Sanberg P.R., Bunsey M.D., Giordano M., and Norman A.B. (1988). The catalepsy test: its ups and downs. Behav. Neurosci. 102, 748–759 [DOI] [PubMed] [Google Scholar]
- 53.Schallert T., De Ryck M., Whishaw I.Q., Ramirez V.D., and Teitelbaum P. (1979). Excessive bracing reactions and their control by atropine and L-DOPA in an animal analog of Parkinsonism. Exp. Neurol. 64, 33–43 [DOI] [PubMed] [Google Scholar]
- 54.Morrissey T.K., Pellis S.M., Pellis V.C., and Teitelbaum P. (1989). Seemingly paradoxical jumping in cataleptic haloperidol-treated rats is triggered by postural instability. Behav. Brain Res. 35, 195–207 [DOI] [PubMed] [Google Scholar]
- 55.Pellis S.M., Pellis V.C., and Teitelbaum P. (1991). Air righting without the cervical righting reflex in adult rats. Behav. Brain Res. 45, 185–188 [DOI] [PubMed] [Google Scholar]
- 56.Pellis S.M., Whishaw I.Q., and Pellis V.C. (1991). Visual modulation of vestibularly-triggered air-righting in rats involves the superior colliculus. Behav. Brain Res. 46, 151–156 [DOI] [PubMed] [Google Scholar]
- 57.Schallert T. (2006). Behavioral tests for preclinical intervention assessment. NeuroRx 3, 497–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fanselow M.S. (1986). Associative vs topographical accounts of the immediate shock-freezing deficit in rats: implications for the response selection rules governing species-specific defensive reactions. Learn. Motiv. 17, 16–39 [Google Scholar]
- 59.Rudy J.W., and O'Reilly R.C. (2001). Conjunctive representations, the hippocampus, and contextual fear conditioning. Cogn. Affect Behav. Neurosci. 1, 66–82 [DOI] [PubMed] [Google Scholar]
- 60.Landeira-Fernandez J., DeCola J.P., Kim J.J., and Fanselow M.S. (2006). Immediate shock deficit in fear conditioning: effects of shock manipulations. Behav. Neurosci. 120, 873–879 [DOI] [PubMed] [Google Scholar]
- 61.Rosen J.B., and Schulkin J. (1998). From normal fear to pathological anxiety. Psychol. Rev. 105, 325–350 [DOI] [PubMed] [Google Scholar]
- 62.Rosen J.B. (2004). The neurobiology of conditioned and unconditioned fear: a neurobehavioral system analysis of the amygdala. Behav. Cogn. Neurosci. Rev. 3, 23–41 [DOI] [PubMed] [Google Scholar]
- 63.Loram L.C., Harrison J.A., Sloane E.M., Hutchinson M.R., Sholar P., Taylor F.R., Berkelhammer D., Coats B.D., Poole S., Milligan E.D., Maier S.F., Rieger J., and Watkins L.R. (2009). Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: a novel therapy for neuropathic pain. J. Neurosci. 29, 14015–14025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cole J.T., Yarnell A., Kean W.S., Gold E., Lewis B., Ren M., McMullen D.C., Jacobowitz D.M., Pollard H.B., O'Neill J.T., Grunberg N.E., Dalgard C.L., Frank J.A., and Watson W.D. (2011). Craniotomy: true sham for traumatic brain injury, or a sham of a sham? J. Neurotrauma 28, 359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olesen S.P. (1987). Leakiness of rat brain microvessels to fluorescent probes following craniotomy. Acta Physiol. Scand. 130, 63–68 [DOI] [PubMed] [Google Scholar]
- 66.Siopi E., Llufriu-Daben G., Fanucchi F., Plotkine M., Marchand-Leroux C., and Jafarian-Tehrani M. (2012). Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: the effect of minocycline. Neurosci. Lett. 511, 110–115 [DOI] [PubMed] [Google Scholar]
- 67.Lee H.F., Lee T.S., and Kou Y.R. (2012). Anti-inflammatory and neuroprotective effects of triptolide on traumatic brain injury in rats. Respir. Physiol. Neurobiol. 182, 1–8 [DOI] [PubMed] [Google Scholar]
- 68.Kovesdi E., Kamnaksh A., Wingo D., Ahmed F., Grunberg N.E., Long J.B., Kasper C.E,. and Agoston D.V. (2012). Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front. Neurol. 3, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lopez N.E., Gaston L., Lopez K.R., Coimbra R.C., Hageny A., Putnam J., Eliceiri B., Coimbra R., and Bansal V. (2012). Early ghrelin treatment attenuates disruption of the blood brain barrier and apoptosis after traumatic brain injury through a UCP-2 mechanism. Brain Res. 1489, 140–148 [DOI] [PubMed] [Google Scholar]
- 70.Namas R., Ghuma A., Hermus L., Zamora R., Okonkwo D.O., Billiar T.R., and Vodovotz Y. (2009). The acute inflammatory response in trauma/hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J. Med. 4, 97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gallucci S., and Matzinger P. (2001). Danger signals: SOS to the immune system. Curr. Opin. Immunol. 13, 114–119 [DOI] [PubMed] [Google Scholar]
- 72.Oppenheim J.J., and Yang D. (2005). Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 17, 359–365 [DOI] [PubMed] [Google Scholar]
- 73.Oppenheim J.J., Tewary P., de la Rosa G., and Yang D. (2007). Alarmins initiate host defense. Adv. Exp. Med. Biol. 601, 185–194 [DOI] [PubMed] [Google Scholar]
- 74.Bianchi M.E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 [DOI] [PubMed] [Google Scholar]
- 75.Matzinger P. (2002). An innate sense of danger. Ann. N. Y. Acad. Sci. 961, 341–342 [DOI] [PubMed] [Google Scholar]
- 76.Olson J.K., and Miller S.D. (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 173, 3916–3924 [DOI] [PubMed] [Google Scholar]
- 77.Hill S.J., Barbarese E., and McIntosh T.K. (1996). Regional heterogeneity in the response of astrocytes following traumatic brain injury in the adult rat. J. Neuropathol. Exp. Neurol. 55, 1221–1229 [DOI] [PubMed] [Google Scholar]
- 78.Nonaka M., Chen X.H., Pierce J.E., Leoni M.J., McIntosh T.K., Wolf J.A., and Smith D.H. (1999). Prolonged activation of NF-kappaB following traumatic brain injury in rats. J. Neurotrauma 16, 1023–1034 [DOI] [PubMed] [Google Scholar]
- 79.Gueorguieva I., Clark S.R., McMahon C.J., Scarth S., Rothwell N.J., Tyrrell P.J., Hopkins S.J., and Rowland M. (2008). Pharmacokinetic modelling of interleukin-1 receptor antagonist in plasma and cerebrospinal fluid of patients following subarachnoid haemorrhage. Br. J. Clin. Pharmacol. 65, 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grady M.S., Charleston J.S., Maris D., Witgen B.M., and Lifshitz J. (2003). Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J. Neurotrauma 20, 929–941 [DOI] [PubMed] [Google Scholar]
- 81.Clausen F., Hanell A., Bjork M., Hillered L., Mir A.K., Gram H., and Marklund N. (2009). Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 30, 385–396 [DOI] [PubMed] [Google Scholar]
- 82.Yu I., Inaji M., Maeda J., Okauchi T., Nariai T., Ohno K., Higuchi M., and Suhara T. (2010). Glial cell-mediated deterioration and repair of the nervous system after traumatic brain injury in a rat model as assessed by positron emission tomography. J. Neurotrauma 27, 1463–1475 [DOI] [PubMed] [Google Scholar]
- 83.D'Ambrosio R., Fairbanks J.P., Fender J.S., Born D.E., Doyle D.L., and Miller J.W. (2004). Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 127, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Graeber M.B., and Kreutzberg G.W. (1988). Delayed astrocyte reaction following facial nerve axotomy. J. Neurocytol. 17, 209–220 [DOI] [PubMed] [Google Scholar]
- 85.McCann M.J., O'Callaghan J.P., Martin P.M., Bertram T., and Streit W.J. (1996). Differential activation of microglia and astrocytes following trimethyl tin-induced neurodegeneration. Neuroscience 72, 273–281 [DOI] [PubMed] [Google Scholar]
- 86.Hanisch U.K. (2002). Microglia as a source and target of cytokines. Glia 40, 140–155 [DOI] [PubMed] [Google Scholar]
- 87.Iravani M.M., Leung C.C., Sadeghian M., Haddon C.O., Rose S., and Jenner P. (2005). The acute and the long-term effects of nigral lipopolysaccharide administration on dopaminergic dysfunction and glial cell activation. Eur. J. Neurosci. 22, 317–330 [DOI] [PubMed] [Google Scholar]
- 88.Herber D.L., Maloney J.L., Roth L.M., Freeman M.J., Morgan D., and Gordon M.N. (2006). Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia 53, 382–391 [DOI] [PubMed] [Google Scholar]
- 89.Hanisch U.K., and Kettenmann H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394 [DOI] [PubMed] [Google Scholar]
- 90.Griffin W.S., Sheng J.G., Royston M.C., Gentleman S.M., McKenzie J.E., Graham D.I., Roberts G.W., and Mrak R.E. (1998). Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 8, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rauch S.L., Savage C.R., Alpert N.M., Fischman A.J., and Jenike M.A. (1997). The functional neuroanatomy of anxiety: a study of three disorders using positron emission tomography and symptom provocation. Biol. Psychiatry 42, 446–452 [DOI] [PubMed] [Google Scholar]
- 92.Simmons A., Strigo I., Matthews S.C., Paulus M.P., and Stein M.B. (2006). Anticipation of aversive visual stimuli is associated with increased insula activation in anxiety-prone subjects. Biol. Psychiatry 60, 402–409 [DOI] [PubMed] [Google Scholar]
- 93.Stein M.B., Simmons A.N., Feinstein J.S., and Paulus M.P. (2007). Increased amygdala and insula activation during emotion processing in anxiety-prone subjects. Am. J. Psychiatry 164, 318–327 [DOI] [PubMed] [Google Scholar]
- 94.Shin L.M., and Liberzon I. (2010). The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology 35, 169–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carlson J.M., Greenberg T., Rubin D., and Mujica-Parodi L.R. (2011). Feeling anxious: anticipatory amygdalo-insular response predicts the feeling of anxious anticipation. Soc. Cogn. Affect. Neurosci. 6, 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davidson R.J. (2002). Anxiety and affective style: role of prefrontal cortex and amygdala. Biol. Psychiatry 51, 68–80 [DOI] [PubMed] [Google Scholar]
- 97.Shin L.M., Rauch S.L., and Pitman R.K. (2006). Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann. N. Y. Acad. Sci. 1071, 67–79 [DOI] [PubMed] [Google Scholar]
- 98.Milad M.R., Pitman R.K., Ellis C.B., Gold A.L., Shin L.M., Lasko N.B., Zeidan M.A., Handwerger K., Orr S.P., and Rauch S.L. (2009). Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol. Psychiatry 66, 1075–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bremner J.D., Randall P., Scott T.M., Bronen R.A., Seibyl J.P., Southwick S.M., Delaney R.C., McCarthy G., Charney D.S., and Innis R.B. (1995). MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am. J. Psychiatry 152, 973–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sapolsky R.M. (2000). Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch. Gen. Psychiatry 57, 925–935 [DOI] [PubMed] [Google Scholar]
- 101.Elder G.A., Dorr N.P., De Gasperi R., Gama Sosa M.A., Shaughness M.C., Maudlin-Jeronimo E., Hall A.A., McCarron R.M., and Ahlers S.T. (2012). Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J. Neurotrauma 29, 2564–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davis M. (2006). Neural systems involved in fear and anxiety measured with fear-potentiated startle. Am. Psychol. 61, 741–756 [DOI] [PubMed] [Google Scholar]
- 103.Samuels E.R., and Szabadi E. (2008). Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr. Neuropharmacol. 6, 235–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hailer N.P. (2008). Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog. Neurobiol. 84, 211–233 [DOI] [PubMed] [Google Scholar]
- 105.Alessandri B., Rice A.C., Levasseur J., DeFord M., Hamm R.J., and Bullock M.R. (2002). Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J. Neurotrauma 19, 829–841 [DOI] [PubMed] [Google Scholar]
- 106.Campbell J.N., Churn S.B., and Register D. (2011). Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J. Neurotrauma 29, 201–217 [DOI] [PubMed] [Google Scholar]
- 107.Yatsiv I., Grigoriadis N., Simeonidou C., Stahel P.F., Schmidt O.I., Alexandrovitch A.G., Tsenter J., and Shohami E. (2005). Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. FASEB J. 19, 1701–1703 [DOI] [PubMed] [Google Scholar]
- 108.Xiong Y., Mahmood A., Meng Y., Zhang Y., Qu C., Schallert T., and Chopp M. (2010). Delayed administration of erythropoietin reducing hippocampal cell loss, enhancing angiogenesis and neurogenesis, and improving functional outcome following traumatic brain injury in rats: comparison of treatment with single and triple dose. J. Neurosurg. 113, 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Byrnes K.R., Loane D.J., Stoica B.A., Zhang J., and Faden A.I. (2012). Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J. Neuroinflammation 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]