

Abstract
The oncogenic transcription factor signal transducer and activator of transcription 3 (STAT3) is hyperactivated in most cancers and represents a plausible therapeutic target. In the absence of STAT3-selective small-molecule inhibitors, we sought to develop pSTAT3/1 high-content imaging (HCS) assays to screen for selective inhibitors of STAT3 pathway activation in head and neck squamous cell carcinomas (HNSCC) tumor cell lines. Based on the expression of the interleukin-6 (IL-6)Rα and gp130 subunits of the IL-6 receptor complex and STAT3, we selected the Cal33 HNSCC cell line as our model. After developing image acquisition and analysis procedures, we rigorously investigated the cytokine activation responses to optimize the dynamic ranges of both assays and demonstrated that the pan-Janus kinase inhibitor pyridone 6 nonselectively inhibited pSTAT3 and pSTAT1 activation with 50% inhibition concentrations of 7.19±4.08 and 16.38±8.45 nM, respectively. The optimized pSTAT3 HCS assay performed very well in a pilot screen of 1,726 compounds from the Library of Pharmacologically Active Compounds and the National Institutes of Health clinical collection sets, and we identified 51 inhibitors of IL-6-induced pSTAT3 activation. However, only three of the primary HCS actives selectively inhibited STAT3 compared with STAT1. Our follow-up studies indicated that the nonselective inhibition of cytokine induced pSTAT3 and pSTAT1 activation by G-alpha stimulatory subunit-coupled G-protein-coupled receptor agonists, and forskolin was likely due to cyclic adenosine monophosphate-mediated up-regulation of suppressors of cytokine signaling 3. Azelastine, an H1 receptor antagonist approved for the treatment of seasonal allergic rhinitis, nonallergic vasomotor rhinitis, and ocular conjunctivitis, was subsequently confirmed as a selective inhibitor of IL-6-induced pSTAT3 activation that also reduced the growth of HNSCC cell lines. These data illustrate the power of a chemical biology approach to lead generation that utilizes fully developed and optimized HCS assays as phenotypic screens to interrogate specific signaling pathways.
Introduction
There is a large body of evidence implicating activation of the signal transducer and activator of transcription 3 (STAT3) signaling pathway in the development, progression, and maintenance of many cancers.1–5 In preclinical models, STAT3 is required by the viral oncogene vSrc to transform cells, and transfection of fibroblasts or normal epithelial cells with a constitutively activated mutant STAT3 is sufficient to transform these cells.6–9 STAT3 is activated by phosphorylation at a single tyrosine residue (Y705) of its C-terminal transactivation domain, and pSTAT3-Y705 levels are elevated in most tumor-derived cell lines.1,2,4,8,10 Human tumor biopsies exhibit elevated pSTAT3-Y705 levels that are frequently associated with a poor clinical prognosis in several common malignancies.1,2,4,8,10,11 Although STAT3 mutations resulting in constitutive activation have not been identified in tumors in vivo, other STAT3 signaling pathway components that produce elevated pSTAT3-Y705 levels are frequently altered in many cancer types; over-expression and amplification of epidermal growth factor receptor (EGFR) expression levels, EGFR mutations that result in constitutive receptor tyrosine kinase (RTK) activation, over-expression of Src family kinases (SFKs), and mutations that hyper-activate Janus kinases (JAKs).4,5,8,10,12–15 Elevated levels of growth factors and cytokines that engage receptor complexes which activate STAT3 signaling have also been detected in the serum and/or tumor microenvironments of patients with a variety of human malignancies.1,5,8,11,16 For example, transforming growth factor alpha (TGFα) or interleukin 6 (IL-6) produced in an autocrine or paracrine manner may lead to inappropriate or sustained activation of the STAT3 pathway, which, in turn, promotes tumor development and/or survival.1,5,8,11,16 Despite the compelling data validating STAT3 as an anti-cancer target and nearly two decades of research, a STAT3-selective targeted anti-cancer therapy has yet to be approved.
The STAT3 signaling pathway is activated downstream of receptor complexes for many extracellular inputs, including members of the ErbB/HER family of growth factor receptors, cytokine receptors, and G-protein-coupled receptors (GPCRs).5,7–10,12–15 In addition to multiple growth factors (EGF, TGFα, PDGF, and CSF1) and cytokines (IL-6, LIF, CT-1, CNTF, IL-10, IL-11, and OSM), intracellular SFKs (Src, Lck, Hck, Lyn, Fyn, and Fgr) either activate STAT3 directly, or do so downstream of the activation of RTKs or GPCRs.5,7–9,12,14,17 In the canonical STAT3 signaling pathway, latent STAT3 in the cytoplasm is recruited to specific phospho-tyrosine (pY) docking sites on the receptor subunits of cell surface receptor complexes that are activated by their cognate ligands.2,4,5,8,12,13,18 The recruitment and docking of STAT3 is mediated through the interactions of the Src homology 2 (SH-2) domain of STAT3 with receptor-pY sites, which ultimately lead to phosphorylation of the tyrosine-705 residue of STAT3.2,4,5,8,12,13,19 STAT3-Y705 phosphorylation can be mediated by the intrinsic RTK activity of an activated growth factor receptor, a JAK associated with an activated cytokine receptor, or by an activated SFK.2,4,5,8,12,13 Reciprocal interactions between SH-2 domains of pSTAT3 monomers and the pY705 residue of another pSTAT3 partner lead to dimerization and translocation into the nucleus, where pSTAT3 dimers bind specific DNA response elements in the promoters of target genes to regulate transcription.2,4,5,8,12,13 STAT3 target genes inhibit apoptosis, promote cell proliferation and survival, stimulate angiogenesis and metastasis, and hinder anti-tumor immune responses.1–3,8,20 Although STAT3 and STAT1 are highly homologous and interact with very similar DNA binding sites, STAT1 target genes activate cell cycle arrest, promote apoptosis, and enhance anti-tumor immunity.17,20,21 Under physiological conditions, STAT3 and STAT1 are regulated in a reciprocal manner to produce opposing effects, and in the cancer context, activated STAT3 is an oncogene while activated STAT1 behaves as a tumor suppressor.4,20,21 Therefore, a selective inhibitor of STAT3 pathway activation that does not interfere with STAT1 signaling would seem to be a highly desirable activity profile for an anti-cancer drug.4,20
Several strategies that are used to block STAT3 signaling have been pursued: prevention of STAT3 activation, blockade of STAT3–SH-2 interactions, inhibition of STAT3 translocation and/or entry through the nuclear pore complex, and disruption of DNA binding. The Food and Drug Administration (FDA) has approved the use of EGFR monoclonal antibodies (mAbs; cetuximab and panitumumab) for treating colorectal cancer and head and neck squamous cell carcinomas (HNSCC).5,7,8,22 mAbs to the EGFR antagonizes ligand-receptor interactions to disrupt downstream signaling and may also induce activation of anti-tumor cellular immunity.5,7,8,22 Small-molecule RTK inhibitors (gefitinib, erlotinib, and lapatinib) have been approved by the FDA for pancreatic cancer and non–small-cell lung cancer.5,7,8 Constitutive activation of STAT3 by the persistent activation of highly expressed EGFR levels in squamous carcinoma cells has been linked to the aberrant growth of HNSCC tumors.5,7,8,16 Despite the occurrence of high EGFR expression levels and/or somatic EGFR mutations that constitutively activate the EGFR, the clinical data indicate that many patients are refractory to EGFR inhibitor treatment.7,22 The emergence of secondary mutations in the EGFR that prevent/alter either mAb binding or small-molecule RTK inhibition, activating mutations of the Ras pathway, epithelial–mesenchymal transition, and activation of alternative downstream signaling pathways are potential mechanisms for the development of resistance to EGFR-targeted therapies.22 Although several small-molecule JAK inhibitors have been tested in preclinical tumor xenograft models or have progressed into clinical cancer trials,15,23,24 to date, the FDA has only approved JAK inhibitors for the treatment of myelofibrosis (ruxolitinib) and rheumatoid arthritis (tofacitinib). Dasatinib is a dual Src and BCR/ABL and tyrosine kinase inhibitor that has been approved for use in patients with chronic myelogenous leukemia (CML) after imatinib treatment, and for Philadelphia chromosome-positive acute lymphoblastic leukemia.4,7 Several small-molecule SFK inhibitors (dasatinib, saracatinib, and bosutinib) are in clinical cancer trials for a variety of solid tumors, but thus far, the efficacy of these drugs has not been impressive.4,7,25 Several groups have pursued small-molecule inhibitors that disrupt STAT3–SH-2 binding interactions through peptide- and peptidomimetic-inspired rational design, by in silico computational approaches, and by in vitro high throughput screening.2–4,8,26 Although these small molecules inhibit STAT3 recruitment by activated receptors and/or block the formation of transcriptionally active pSTAT3-Y705 dimers,2–4,8,26 to date, none of these molecules have progressed into clinical trials. The search for small molecules that either block STAT3 translocation or transit through the nuclear pore complex are emerging strategies which have not yet yielded compounds that block STAT3 transcriptional activity.4,27,28 Double-stranded oligodeoxynucleotide (dsODN) decoys that mimic cis-elements within the regulatory regions of STAT3 target genes block DNA binding, down-regulate expression of STAT-3 target genes, restrict cell proliferation, enhance apoptosis, and reduce tumor growth in mouse xenograft models.2–4,8,21,29–31 An intramuscular injection of a STAT3 dsODN decoy produced no observable adverse effects, while STAT3 target gene expression was reduced at the site of injection.30 In a phase 0 pharmacodynamic clinical study, a single intra-tumor injection of a STAT3 dsODN decoy in HNSCC patients abrogated the expression of STAT3 target genes in the tumor.31
Head and neck cancer is the eighth leading cause of cancer worldwide with an incidence of ∼36,000/year in the United States.32–34 Recurrence, metastasis, and second primary cancers account for the 6,900 deaths that are estimated to occur annually from this invasive cancer of the head and neck.32–34 Elevated levels of constitutively activated STAT3 are frequently observed in HNSCC tumor samples, and a number of mouse xenograft models that respond to STAT3 inhibition have been developed with HNSCC tumor cell lines.4,5,7,8,11,29,35 EGFR-independent constitutive activation of STAT3 can be mediated by the autocrine/paracrine activation of the IL-6 receptor complex that confers proliferative and survival advantages to HNSCC cells and may also contribute to the resistance to EGFR-targeted therapies.16 We describe here the development and validation of an IL-6–induced pSTAT3 high-content imaging (HCS) assay to screen in a mechanistic unbiased manner for inhibitors of STAT3 pathway activation in HNSCC cell lines along with an interferon gamma (IFNγ)–induced pSTAT1 pathway activation counter screen conducted in the same cells. It is our expectation that a chemical biology screening campaign conducted in HNSCC cell lines using these phenotypic HCS assays will identify selective inhibitors of STAT3 pathway activation with improved therapeutic potential.
Materials and Methods
Reagents
Methanol, formaldehyde, Triton X-100, Tween 20, Hoechst 33342, azelastine, isoetharine, etomidate, forskolin, and (−)-isoproterenol were purchased from Sigma-Aldrich (St. Louis, MO). Dimethyl sulfoxide (DMSO) (99.9% high-performance liquid chromatography-grade, under argon) was from Alfa Aesar (Ward Hill, MA). Recombinant human IL-6 and IFNγ were purchased from R&D Systems, Inc. (Minneapolis, MN). The mouse monoclonal anti-pSTAT3 Y705 (Cat# 612357) and mouse monoclonal anti-pSTAT1-Y701 (Cat# 612132) primary antibodies were purchased from Becton Dickenson Biosciences (San Diego, CA). The secondary antibody goat anti mouse-IgG conjugated with Alexa Fluor 488 was purchased from Life Technologies (Grand Island, NY). Dulbecco's Mg2+ and Ca2+ free phosphate-buffered saline (PBS) was purchased from Corning (Tewksbury, MA).
Cells and Tissue Culture
Four HNSCC cell lines Cal33,36,37 686LN,38 FaDu,39 and OSC1940 were utilized in these studies. All four HNSCC cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) with 2 mM l-glutamine (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, CA), 100 μM nonessential amino acids (Life Technologies), 100 μM sodium pyruvate (Life Technologies), and 100 U/mL penicillin and streptomycin (Life Technologies).
Western Blotting Analysis of STAT3 Signaling Pathway and IL-6 Receptor Complex Components in HNSCC Cell Lines
To investigate endogenous STAT3 signaling pathway and IL-6 receptor complex components, we seeded HNSCC cells (686LN, Cal33, OSC19, and FaDu) in a 10-cm dish (1×106 cells) and after 24 h in culture, cells were harvested to obtain cell lysates. Forty micrograms of protein/lane were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose, and then probed with rabbit anti-phospho-STAT3-Y705 mAb and rabbit STAT3 polyclonal antibody (Cell Signaling, Danvers, MA), rabbit polyclonal IL-6Rα antibody (Santa Cruz Biotechnology, Dallas, TX), and rabbit polyclonal gp130 antibody (Cell Signaling). β-Tubulin was used as a loading control.
To investigate IL-6-induced activation of pSTAT3-Y705 in the Cal33 HNSCC cell line, we seeded Cal33 cells (3×105 cells/well) in a six-well plate. After 24 h in culture, the cells were serum starved for 24 h and then stimulated in the presence or absence of IL-6 (50 ng/mL) for 15 min. Cells were harvested to obtain lysates, and 40 μg of protein/lane were separated by 10% SDS-PAGE, transferred to nitrocellulose, and probed with anti-phospho-STAT3-Y705 mAb and rabbit STAT3 polyclonal antibody. β-Tubulin was used as a loading control.
Compound Libraries
The 1,280 compound Library of Pharmacologically Active Compounds (LOPAC) purchased from Sigma-Aldrich was supplied at a 10 mM concentration in DMSO arrayed into 96-well microtiter master plates. The 446 compound National Institutes of Health clinical collection (NIH CC) purchased from the NIH was supplied by Evotec (San Francisco, CA) at a 10 mM concentration in DMSO arrayed into 96-well microtiter master plates. The LOPAC and NIH CC compounds were assigned unique University of Pittsburgh Drug Discovery Institute (UPDDI) substance identity numbers and were handled and stored as previously described.41–43 384-well master plates containing 100 μL (LOPAC) or 50 μL (NIH CC) of 10 mM compounds in DMSO were prepared from four 96-well LOPAC or NIH CC master plates mapped into the quadrants of a single 384-well plate using the Bravo automated liquid handling platform (Agilent Technologies, Santa Clara, CA) outfitted with a 96-well transfer head. Daughter plates containing 2 μL of 10 mM compounds in DMSO were prepared and replicated from the 384-well LOPAC and NIH CC master plates using the Bravo outfitted with a 384-well transfer head. Aluminum adhesive plate seals were applied with an Abgene Seal-IT 100 (Rochester, NY) plate sealer, and plates were stored at −20°C in a Matrical MatriMinistore™ (Spokane, WA) automated compound storage and retrieval system. For screening, LOPAC and NIH CC daughter plates were withdrawn from −20°C storage, thawed to ambient temperature, centrifuged at 1–2 min at 50 g, and the plate seals were removed before the transfer of 98 μL of serum-free tissue culture medium (SFM) into wells using the BioTek Microflo liquid handler (BioTek, Winooski, VT) to generate a 200-μM intermediate stock concentration of library compounds (2.0% DMSO). The diluted compounds were mixed by repeated aspiration and dispensation using a 384-well P30 dispensing head on the Evolution-P3 (EP3) liquid handling platform (Perkin Elmer, Waltham, MA), and then, 5 μL of diluted compounds were transferred to the wells of assay plates. In the LOPAC and NIH CC primary screen, compounds were individually tested at a final concentration of 20 μM (0.2% DMSO). After the diluted compounds had been transferred from the daughter plates to the primary HCS assay plates, aluminum adhesive plate seals were applied and the diluted daughter plates were stored frozen at −20°C.
To confirm the active compounds identified in the primary screen, we withdrew the LOPAC and NIH CC daughter plates containing the 200-μM intermediate concentration of library compounds (2.0% DMSO) from −20°C storage, thawed them to ambient temperature, centrifuged them for 1–2 min at 50 g, and removed the plate seals. A work list was generated to direct the Multiprobe Janus liquid-handling platform (Perkin Elmer) to cherry pick 75 μL from the wells of the diluted daughter plates that were flagged active in the primary screen and to reformat these into a 384-well plate hit confirmation daughter plate. Five microliter of primary HCS active compounds were then transferred into triplicate wells of assay plates using a 384-well P30 dispensing head on the EP3 liquid-handling platform (Perkin Elmer).
For the determination of the 50% inhibition concentrations (IC50), 10-point twofold serial dilutions of test compounds in 100% DMSO were performed using a 384-well P30 dispensing head on the EP3 liquid-handling platform. Daughter plates containing 2 μL of the serially diluted compounds in DMSO were prepared and replicated from the 384-well serial dilution master plates using the Bravo outfitted with a 384-well transfer head. Aluminum adhesive plate seals were applied, and plates were stored at −20°C. For testing in the bioassays, daughter plates were withdrawn from −20°C storage, thawed to ambient temperature, centrifuged 1–2 min at 50 g, and the plate seals were removed before the transfer of 38 μL of SFM into wells using the BioTek Microflo liquid handler (BioTek) to generate an intermediate stock concentration of library compounds ranging from 1.95 to 500 μM (5.0% DMSO). The diluted compounds were mixed by repeated aspiration and dispensation using a 384-well P30 dispensing head on the EP3, and then, 5 μL of diluted compounds were transferred to the wells of assay plates to provide a final concentration response ranging from 0.195 to 50 μM (0.5% DMSO).
Automated pSTAT3 and pSTAT1 HCS Assay Protocols
The HCS assay protocol for IL-6–induced pSTAT3-Y705 activiation in HNSCC cells is summarized in Table 1. Cal33 HNSCC cells were seeded at 2,000 cells/well in 45 μL of tissue culture media into uncoated, black-walled, clear-bottomed (Greiner Bio-one Cat# 781091) 384-well assay plates on the Zoom dispenser liquid handler (Titertek, Huntsville, AL) and cultured overnight at 37°C, 5% CO2, and 95% humidity. Serum containing media was then exchanged for SFM on the Bravo liquid handler equipped with a 384-well transfer head. For 24 h compound exposure screens, 5 μL of compounds diluted in SFM were transferred to assay plates (20 μM, 0.2% DMSO final) on the Bravo liquid handler equipped with a 384-well transfer head, and the treated assay plates were cultured for 24 h in an incubator at 37°C, 5% CO2, and 95% humidity. For 1 and 3 h compound exposure screens, 5 μL of compounds diluted in SFM were transferred to assay plates (20 μM, 0.2% DMSO final) after the assay plates had been cultured in SFM for 23 and 21 h, respectively. Compound wells and maximum (Max) control wells were treated with 50 ng/mL IL-6 on the Bravo liquid handler equipped with a 384-well transfer head and minimum (Min) control wells received DMSO (0.2% final). The assay plate was then centrifuged at 50 g for 1–2 min and returned to the incubator at 37°, 5% CO2, and 95% humidity. After 15 min, the cells were fixed in prewarmed (37°C) 7.4% paraformaldehyde containing 2 μg/mL Hoechst 33342 using the Elx405 plate washer dispenser (BioTek). After 30 min at room temperature, fixed cell monolayers were washed 1×with PBS, and then, the PBS was replaced on the Elx405 plate washer dispenser. The PBS was aspirated, and then, fixed cells were permeabilized by the addition of 95% ice-cold methanol using the Elx405 plate washer dispenser. After 30 min on ice, the 95% methanol was removed using the Elx405 plate washer dispenser; permeabilized cell monolayers were washed 1×with Tween 20 blocking buffer; and then, the Tween 20 blocking buffer was replaced on the Elx405 plate washer dispenser. Permeabilized cells were incubated in Tween 20 blocking buffer at room temperature for 15 min, and then, a 1:100 dilution of mouse anti-pSTAT3-Y705 or mouse anti-pSTAT1-Y701 primary antibody was added to 384-well assay plates using the Microflo bulk reagent dispenser and incubated for 1 h in the dark at room temperature. Cell monolayers were then washed 1×with Tween 20 blocking buffer, and then, the Tween 20 blocking buffer was replaced on the Elx405 plate washer dispenser. Goat anti-mouse IgG secondary antibody conjugated to Alexa-488 was then added to 384-well assay plates using the Microflo bulk reagent dispenser and incubated for 1 h in the dark at room temperature. Cell monolayers were then washed 1×with PBS, and then, the PBS was replaced on the Elx405 plate washer dispenser, plates were sealed with aluminum foil seals on the Abgene plate sealer, and images were acquired on the ImageXpress Ultra (IXU) automated imaging platform. Images were then analyzed using Translocation Enhanced (TE) image analysis module.
Table 1.
IL-6–Induced pSTAT3-Y705 Activation High-Content Screening Assay Protocol in Head and Neck Squamous Cell Carcinoma Cells
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Plate cells | 40 μL | 2,000 Cal33 HNSCC cells in DMEM+10% FBS |
| 2 | Incubate cells | 24 h | Tissue culture medium, 37°C, 5% CO2, and 95% humidity |
| 3 | Exchange medium | 40 μL | Exchange tissue culture medium for SFM |
| 4 | Incubate cells | 24 h | SFM, 37°C, 5% CO2, and 95% humidity |
| 5 | Library compounds/DMSO to control wells | 5 μL | 20 μM final concentration in well, 0.2% DMSO |
| 6 | Incubate plates | 1 or 3 h | At 37°C, 5% CO2, and 95% humidity |
| 7 | Add IL-6 to compound treated and maximum controls, media to minimum controls | 5 μL | 50 ng/mL IL-6 final in well of compound treated and maximum controls, media to minimum controls |
| 8 | Incubate plates | 15 min | At 37°C, 5% CO2, and 95% humidity |
| 9 | Fix cells | 60 μL | 7.4% formaldehyde containing 2 μg/mL Hoechest 33342 in Ca2+ and Mg2+ free PBS prewarmed to 37 C |
| 10 | Incubate plates | 10–30 min | Ambient temperature |
| 11 | Aspirate fixative and wash 2×with PBS | 50 μL | Aspirate fixative and wash twice with 50 μL Ca2+ and Mg2+-free PBS, 50 μL PBS in well |
| 12 | Cell permeabilization | 50 μL | Aspirate PBS, add ice-cold 95% methanol, and incubate plates on ice for 30 min |
| 13 | Blocking buffer wash | 50 μL | 0.1% (v/v) Tween 20 in PBS (without Ca2+ and Mg2+) |
| 14 | Blocking buffer incubation | 50 μL | 15 min in 0.1% (v/v) Tween 20 in Ca2+ and Mg2+ -free PBS at ambient temperature |
| 15 | Primary antibody incubation | 25 μL | 1 h with a 1:100 dilution of a mouse monoclonal anti-pSTAT3-Y705 antibody in Ca2+ and Mg2+-free PBS in dark at ambient temperature |
| 16 | Blocking buffer wash | 50 μL | 0.1% (v/v) Tween 20 in Ca2+ and Mg2+-free PBS |
| 17 | Blocking buffer incubation | 50 μL | 15 min in 0.1% (v/v) Tween 20 in Ca2+ and Mg2+-free PBS at ambient temperature |
| 18 | Secondary antibody incubation | 25 μL | 1 h of incubation with a 1:1,000 dilution of goat anti-mouse IgG antibody conjugated to Alexa-488 in Ca2+ and Mg2+-free PBS in dark at ambient temperature |
| 19 | PBS wash 2× | 50 μL | Ca2+ and Mg2+-free PBS, 50 μL PBS in well |
| 20 | Seal plates | 1× | Sealed with adhesive aluminum plate seals |
| 21 | Acquire images | 20×/0.4NA objective | Images of the Hoechst (Ch 1) and pSTAT3-Y705 (Ch 2) were sequentially acquired on the IXU platform using the 405 and 488 nm excitation laser lines, a Quad filter cube set, and individual photomultiplier tubes for each channel |
| 22 | Assay readout | Average inner intensity pSTAT3-Y705 | Images were analyzed using the TE image analysis module using the Average Inner Intensity parameter to quantify the nuclear pSTAT3-Y705 signal |
Step Notes
1. 384-well, black-walled, clear-bottom plates, Greiner Bio-one Cat# 781091, Zoom liquid handler (Titertek), or BioTek Microflo (BioTek).
2. Incubate Cal33 HNSCC cells for 24 h at 37°C, 5% CO2, and 95% humidity in DMEM+2 mM l-glutamine+10% FBS+100 μM nonessential amino acids+100 μM sodium pyruvate+100 U/mL penicillin and streptomycin.
3. Serum-containing media exchanged for serum-free media using a Bravo (Agilent Technologies, Inc.) or an Evolution P3 (Perkin Elmer) automated liquid handler outfitted with a 384-well transfer head.
4. Incubate Cal33 HNSCC cells in serum-free media for 24 h at 37°C, 5% CO2, and 95% humidity.
5. Twenty micromolar compounds added to wells in columns 3–22 using a Bravo (Agilent Technologies, Inc.) or an Evolution P3 (Perkin Elmer) automated liquid handler outfitted with a 384-well transfer head.
6. Incubate treated Cal33 HNSCC cells 1–3 h at 37°C, 5% CO2, and 95% humidity.
7. IL-6 (50 ng/mL final in well) added to maximum controls and compound wells, media to minimum control wells using a Bravo (Agilent Technologies, Inc.) or an Evolution P3 (Perkin Elmer) automated liquid handler outfitted with a 384-well transfer head.
8. Incubate Cal33 HNSCC cells±IL-6 15 min at 37°C, 5% CO2, and 95% humidity.
9. Aspiration of media and fixative addition automated on BioTek ELx405 (BioTek) plate washer.
10. Ten to thirty minutes of incubation at ambient temperature to fix cells and stain nuclei with Hoechst.
11. Aspiration of fixative and PBS wash steps automated on BioTek ELx405 (BioTek) plate washer.
12. Ice-cold 95% methanol permeabilization buffer added with the Microflo bulk reagent dispenser (BioTek).
13. Aspiration of permeabilization buffer and blocking buffer wash and addition steps automated on BioTek ELx405 (BioTek) plate washer.
14. Incubate for 15 min in blocking buffer at ambient temperature.
15. Blocking buffer aspirated on BioTek ELx405 (BioTek) plate washer and primary antibody added with the Microflo bulk reagent dispenser (BioTek) and incubated for 1 h in dark at ambient temperature.
16. Aspiration of primary antibody and blocking buffer wash and addition steps automated on BioTek ELx405 (BioTek) plate washer.
17. Incubate for 15 min in blocking buffer at ambient temperature.
18. Blocking buffer aspirated on BioTek ELx405 (BioTek) plate washer and secondary antibody added with the Microflo bulk reagent dispenser (BioTek) and incubated for 45 min in dark at ambient temperature.
19. Aspiration of secondary antibody and PBS wash and addition steps automated on BioTek ELx405 (BioTek) plate washer.
20. Plates sealed with adhesive aluminum plate seals using the Abgene Seal-IT 100 plate sealer (Abgene).
21. Plates loaded into the IXU confocal HCS platform (Molecular Devices LLC) for scanning using a Catalyst robotic plate handler (Thermo Fisher Scientific, Waltham, MA).
22. Images analyzed using the Translocation Enhanced Image analysis module of MetaXpress (Molecular Devices LLC).
Ch 1, channel 1; Ch 2, channel 2; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; HCS, high-content screening; HNSCC, head and neck squamous cell carcinomas; IL-6, interleukin-6; IXU, ImageXpress Ultra; SFM, serum free tissue culture medium; TE, translocation enhanced; PBS, phosphate-buffered saline; DMEM, Dulbecco's modified Eagle's medium.
Image Acquisition on the ImageXpress Confocal Automated Imaging Platform
The IXU platform (Molecular Devices LLC, Sunnyvale, CA) is a fully integrated point-scanning confocal automated imaging platform configured with four independent solid-state lasers providing four excitation wavelengths of 405, 488, 561, and 635 nm. The IXU was equipped with a Quad filter cube providing emission ranges of 417–477, 496–580, 553–613, and 645–725 nm and four independent photomultiplier tubes (PMTs), with each dedicated to a single detection wavelength. The IXU utilizes a dedicated high-speed infra-red laser auto-focus system, has a four-position automated objective changer with air objectives (10×, 20×, 40×, and 60×), and the detection pinhole diameter of the confocal optics was configurable in the software. For the pSTAT3 and pSTAT1 HCS assays, the IXU was set up to acquire two images using a 20×/0.45NA ELWD objective in each of two fluorescent channels that were acquired sequentially. For the studies described here, the Hoechst channel laser autofocus Z-offset was −6.98 μm, the 405 laser was set at 10% power, and the PMT gain was 550. The pSTAT3-Y705 FITC channel Z-offset from W1 (the Hoechst channel) was 12.96 μm, the 488 laser was set at 10% power, and the PMT gain was 625. On average, the IXU scanned a single 384-well plate, two images per channel, in 90 min using these settings.
Image Analysis Using the TE Module
Hoechst 33342 was used to stain and identify the nucleus, and this fluorescent signal from channel 1 (Ch 1) was used by the TE image analysis module to define an “inner” nuclear mask and an “outer” cytoplasm mask in channel 2 (Ch 2). Objects in Ch 1 that exhibited the appropriate fluorescent intensities above background and size (width, length, and area) characteristics were identified and classified by the image segmentation as nuclei. For Hoechst-stained Cal33 cells, the following settings typically proved effective: objects defined as nuclei had to exhibit fluorescent intensities >1,500 gray levels over background, and they had to have an approximate width of 10 μm with a minimum area of 20 μm2, and not to exceed a maximum area of 1,000 μm2. Objects that met these criteria were used to create nuclear masks for each cell. The nuclear mask was eroded by 1 μm from the edge of the detected nucleus to reduce cytoplasmic contamination within the nuclear area, and the reduced “inner” mask was used to quantify the amount of target Ch 2 fluorescence (pSTAT3-Y705 or pSTAT1-Y701) within the nucleus. The “outer” cytoplasm mask was then established at 1 μm from the edge of the detected nucleus and the width of the outer mask was set at 3 μm, to cover as much of the cytoplasm region as possible without going outside the cell boundary. The “outer” mask was used to quantify the amount of target Ch 2 fluorescence (pSTAT3-Y705 or pSTAT1-Y701) within the cytoplasm. The TE image analysis module outputs quantitative data, such as the average fluorescent intensities of the Hoechst-stained objects in Ch 1, the selected object or cell count in Ch 1, and the integrated and average fluorescent intensities of the Ch 2 signal in the nucleus (inner) or cytoplasm (outer) regions as an overall well average value, or on an individual cell basis. The translocation of an object from the cytoplasm to the nucleus was quantified using the TE image analysis module, which provides a mean average inner:outer (Nuc:Cyt) intensity ratio as an indicator of the relative distribution of an object within the cytoplasm and nucleus, and is a measure of translocation between these compartments.
HNSCC Cell Line Growth Inhibition Assays
The four HNSCC cell lines Cal33, 686LN, FaDu, and OSC19 were cultured in an incubator at 37°C in 5% CO2 and 95% humidity in DMEM supplemented with 10% FBS, nonessential amino acids, sodium pyruvate, penicillin, and streptomycin. On day 1, each cell line was harvested, counted, and seeded into two 384-well assay plates, a time zero (T0) and a time 72 h (T72) plate. Cal33, 686LN, FADU, and OSC19 HNSCC cells were seeded at 1,000 cells/well in 45 μL of tissue culture media into uncoated white opaque 384-well assay plates (Greiner Bio-one Cat# 781080/1B) on the Zoom dispenser liquid handler (Titertek) or the BioTek Microflo liquid handler (BioTek) and cultured overnight at 37°C, 5% CO2, and 95% humidity. On day 2, 25 μL of the Cell Titer Glo (CTG; Promega Corporation, Madison, WI) detection reagent was dispensed into the wells of the T0 assay plate on the BioTek Microflo liquid handler, and the luminescence signal was captured on the Envision (Perkin Elmer) microtiter plate reader. In addition, on day 2, 5 μL of compounds diluted in SFM were transferred into the test wells of the T72 384-well assay plates on the Bravo liquid handler that was equipped with a 384-well transfer head, and the compound-treated assay plates were cultured for 72 h in an incubator at 37°C, 5% CO2, and 95% humidity. Control wells received DMSO alone. On day 5, 25 μL of the CTG detection reagent was dispensed into the wells of the T72 assay plate on the BioTek Microflo liquid handler, and the luminescence signal was captured on the Envision microtiter plate reader platform.
Data Processing, Visualization, Statistical Analysis, and Curve Fitting
HCS data processing for the LOPAC and NIH CC screens and the HNSCC growth inhibition assays were performed using ActivityBase™ (IDBS, Guildford, United Kingdom) and CytoMiner (UPDDI). Processed data and HCS multi-parameter features were visualized using Spotfire™ DecisionSite™ (Somerville, MA) software.41–43 An ActivityBase primary HTS template was created that automatically calculated % inhibition along with plate control signal-to-background ratios (S:B) and Z′-factor coefficients. For the LOPAC and NIH CC screens, we utilized the mean average inner intensity values (pSTAT3-Y705 or pSTAT1-Y701) of the 0.2% DMSO minimum plate control wells (n=32) and the mean average inner intensity values of the 50-ng/mL IL-6 (pSTAT3-Y705) or 30 ng/mL IFNγ (pSTAT1-Y701) maximum plate control wells (n=32) to normalize the mean average inner intensity values (pSTAT3-Y705 or pSTAT1-Y701) of the compound data and to represent 0% and 100% activation/translocation of pSTAT3 or pSTAT1 to the nucleus, respectively.
For the HNSCC growth inhibition assays, we used the DMSO control data from the T0 and T72 assay plates to assess the dynamic range of the T0 to T72 cell growth, and to calculate S:B ratios and Z′-factor coefficient statistics for the assay signal window (T0 to T72). To normalize the 72-h compound exposure HNSCC growth inhibition data, the signals from the compound treated wells were processed and expressed as % of the T72 DMSO plate controls.
We also constructed an ActivityBase concentration-response template to calculate percent inhibition along with plate control S:B ratios and Z′-factor coefficients for quality control purposes.41–43 IC50 values were calculated using an XLfit four-parameter logistic model, also called the Sigmoidal Dose–Response Model: y=(A+((B−A)/(1+((C/x)^D)))), where y was the percent activation and x was the corresponding compound concentration. The fitted C parameter was the IC50 and defined as the concentration giving a response half way between the fitted top (B) and bottom (A) of the curve. For normalized data, the A and B parameters were locked as 0 and 100, respectively. For non-normalized concentration response data, we used GraphPad Prism 5 software to plot and fit data to curves using the Sigmoidal dose response variable slope equation:
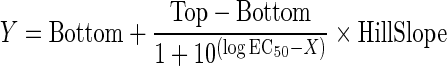 |
Results
Selection of the HNSCC Cell Model
We first investigated the endogenous expression levels of total and phosphorylated STAT3 and IL-6 receptor complex components in a panel of four HNSCC cell lines by western blotting analysis using specific antibodies for these proteins (Fig. 1). Although all four HNSCC cell lines exhibited comparable total STAT3 signals, the endogenous levels of pSTAT3-Y705 were elevated in Cal33 and 686LN cells compared with FADU and OSC19 cells (Fig. 1A). The four HNSCC cell lines also expressed the IL-6Rα (gp80) and gp130 signaling subunits of the IL-6 receptor complex (Fig. 1B), but to varying levels. While OSC19 cells exhibited barely detectable levels of the IL-6Rα subunit, Cal33 and 686LN cells expressed readily detectable levels of IL-6Rα, which were, in turn, higher than the levels observed in FADU cells (Fig. 1B). For the IL-6Rα subunit, the rank order of expression was Cal33=686LN>FADU>OSC19. Similarly, while all HNSCC cell lines exhibited detectable levels of the gp130 signaling subunit, the gp130 signals in Cal33 and OSC19 cells were stronger than in 686LN and FADU cells (Fig. 1B). For the gp130 signaling subunit, the rank order of expression was Cal33=OSC19>686LN=FADU. Since the Cal33 HNSCC cell line exhibited readily detectable expression levels of IL-6Rα, gp130, and STAT3, we examined the effects of IL-6 treatment on the activation of the STAT3 signaling pathway (Fig. 1C). Treatment of Cal33 cells with 50 ng/mL of IL-6 for 15 min dramatically elevated the pSTAT3-Y705 signal without altering total STAT3 levels (Fig. 1C). Based on the results, we selected the Cal33 cell line as our HNSCC model for the development of an HCS assay to screen for selective STAT3 pathway inhibitors.
Fig. 1.
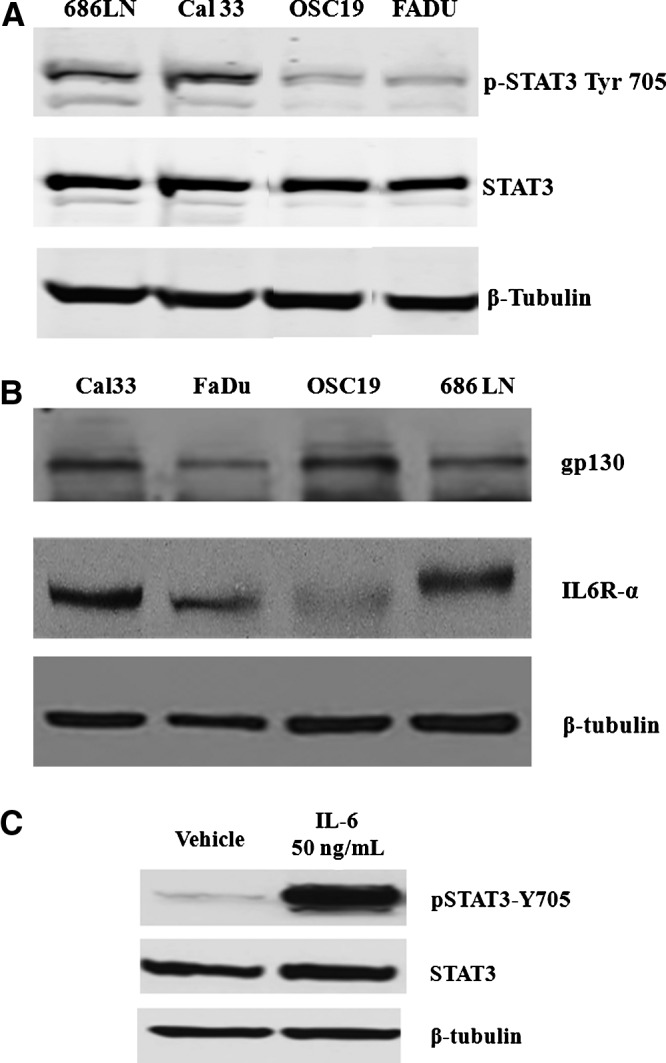
Western blotting analysis of STAT3 signaling pathway and IL-6 receptor complex components in HNSCC cell lines and activation by IL-6. (A) Endogenous levels of STAT3 and pSTAT3-Y705 in the 686LN, Cal33, FADU, and OSC19 HNSCC cell lines. HNSCC cells (686LN, Cal33, OSC19, and FaDu) were seeded in a 10-cm dish (1×106 cells) and after 24 h in culture, cells were harvested to obtain cell lysates. Forty micrograms of protein/lane were separated by 10% SDS-PAGE, transferred to nitrocellulose, and probed with rabbit anti-phospho-STAT3-Y705 mAb and rabbit STAT3 polyclonal antibody (Cell Signaling). β-Tubulin (Abcam, Cambridge, MA) was used as a loading control. (B) Endogenous levels of IL-6Rα and gp130 in the 686LN, Cal33, FADU, and OSC19 HNSCC cell lines. Cal33, FaDu, OSC19, and 686LN cells were seeded in a 10-cm dish (1×106 cells) and after 24 h in culture, cells were harvested to obtain cell lysates. Forty micrograms of protein/lane were separated by 10% SDS-PAGE, transferred to nitrocellulose, and probed with rabbit polyclonal IL-6Rα antibody (Santa Cruz Biotechnology, Dallas, TX) and rabbit polyclonal gp130 antibody (Cell Signaling). β-Tubulin was used as a loading control. (C) Activation of pSTAT3-Y705 in the Cal33 HNSCC cell line by IL-6. Cal33 cells (3×105 cells/well) were seeded into a six-well plate. After 24 h in culture, the cells were serum starved and after an additional 24 h in culture cells, cells were stimulated in the presence or absence of IL-6 (50 ng/mL) for 15 min. Cells were harvested to obtain lysates, and 40 μg of protein/lane were separated by 10% SDS-PAGE, transferred to nitrocellulose, and probed with anti-phospho-STAT3-Y705 mAb and rabbit STAT3 polyclonal antibody. β-Tubulin was used as a loading control. The experiment was repeated thrice. HNSCC, head and neck squamous cell carcinomas; IL-6, interleukin 6; mAb, monoclonal antibody; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; STAT3, signal transducer and activator of transcription 3.
Configuring an HCS Assay for pSTAT3 Pathway Activation
Cal33 cells that had been seeded into 384-well plates in serum-containing medium were cultured overnight and then serum starved for 24 h before a 15-min treatment±50 ng/mL IL-6. The cells were then fixed in 3.7% paraformaldehyde containing Hoechst, permeabilized with methanol, and stained with a monoclonal anti-pSTAT3-Y705 primary antibody and an FITC-conjugated anti-mouse secondary antibody. We then utilized the IXU confocal automated imaging platform to sequentially acquire images for the Hoechst (Ch 1) and FITC (Ch 2) fluorescent channels using a 20×/0.45NA ELWD objective. The Ch 1 laser autofocus Z-offset was −6.98 μm, the 405 laser was set at 10% power, and the PMT gain was 550. The Ch 2 Z-offset from Ch 1 was 12.96 μm, the 488 laser was set at 10% power, and the PMT gain was 625. Fluorescent images of Hoechst-stained nuclei from Cal33 cells acquired on the IXU were unaffected by treatment with IL-6 (Fig. 2A). In contrast, the indirect immuno-fluorescent pSTAT3-Y705 staining was dramatically higher in images acquired of Cal33 cells treated with IL-6 compared with those of un-stimulated cells (Fig. 2A). The enhanced pSTAT3-Y705 staining in IL-6-treated Cal33 cells is consistent with the elevated pSTAT3-Y705 western blotting signals observed in IL-6-treated Cal33 cells (Fig. 1C). A color composite overlay of the Ch 1 and Ch 2 fluorescent images indicates that the increased immuno-fluorescent pSTAT3-Y705 staining is predominantly localized within the nuclei of IL-6-treated Cal33 cells (Fig. 2A). To extract and analyze quantitative data from these digital images, we used the TE image analysis module of the MetaXpress software on the IXU platform (Fig. 2B–H). The Hoechst 33342 DNA staining in Ch 1 was used to identify the nuclei of Cal33 cells (Fig. 2B), and the TE image analysis module used this fluorescent signal to define “inner” nuclear and “outer” cytoplasm masks in Ch 2 (Fig. 2D). Objects in Ch 1 that exhibited the appropriate fluorescent intensities above background, and morphology (width, length, and area) characteristics were identified and classified by the TE image segmentation as nuclei (Fig. 2B). For Hoechst-stained Cal33 cells, the following TE image analysis module settings typically proved effective; objects defined as nuclei exhibited fluorescent intensities >1,500 gray levels over background, and had an approximate width of 10 μm with a minimum area of 20 μm2, and did not exceed a maximum area of 1,000 μm2. Objects that met these criteria were used to create nuclear masks for each cell in Ch 1. The Ch 1 nuclear mask was then eroded 1 μm from the edge of the detected nucleus to reduce cytoplasmic contamination within the nuclear area, and the reduced Ch 2 “inner” mask (Fig. 2D, light green and red) was established to quantify the amount of target pSTAT3-Y705 fluorescence (Fig. 2C) within the nucleus. A Ch 2 “outer” cytoplasm mask was then established at 1 μm from the edge of the Ch 1 detected nucleus, and the width of the outer mask was set at 3 μm, to cover as much of the cytoplasm region as possible without going outside the cell boundary (Fig. 2D, dark green and red). The Ch 2 “outer” mask was used to quantify the amount of target pSTAT3-Y705 fluorescence (Fig. 2C) within this region of the cytoplasm. The TE image analysis module outputs quantitative individual cell or well-averaged data, including the average fluorescent intensities of the Hoechst-stained objects in Ch 1 (data not shown), the selected object or cell count per image in Ch 1 (Fig. 2E), the integrated (data not shown), and average fluorescent intensities of the Ch 2 signal in the cytoplasm (Fig. 2F) or nucleus (Fig. 2G) regions. The TE image analysis module also provides a mean average inner:outer (Nuc:Cyt) intensity ratio as an indicator of the relative distribution of an object within the cytoplasm and nucleus, and a measure of translocation between these compartments (Fig. 2H). While exposure to 50 ng/mL of IL-6 for 15 min had no significant effect on the cell counts per image (Fig. 2E), the average fluorescent intensities of pSTAT3-Y705 in the cytoplasm (Fig. 2F) and nucleus (Fig. 2G) regions were significantly higher in IL-6-treated Cal33 cells compared with untreated cells. Although IL-6 induced a 30% increase in the average fluorescent intensity of pSTAT3-Y705 in the cytoplasm of Cal33 cells (Fig. 2F), the corresponding increase in the average fluorescent intensity of pSTAT3-Y705 in the nucleus was much more pronounced, typically five to sevenfold higher than in untreated cells (Fig. 2G). Consistent with these data, the mean average inner:outer intensity ratio of IL-6-treated Cal33 cells was three to fourfold higher than in untreated cells (Fig. 2H). Although both the average “inner” fluorescent intensity of pSTAT3-Y705 in the nucleus and the mean average inner:outer intensity ratio parameters provided usable assay signal windows (Fig. 2G, H), we selected the more robust dynamic range of the average “inner” intensity of pSTAT3-Y705 from Ch 2 as the primary indicator of STAT3 pathway activation. Very similar data were generated for the configuration of a high-content STAT1 pathway activation assay in Cal33 cells using IFNγ as the stimulus and a monoclonal anti-pSTAT1-Y701 primary antibody as the detection reagent (data not shown).
Fig. 2.


Quantifying IL-6-induced pSTAT3 activation in HNSCC cells by high-content analysis. (A) Grayscale and color composite images of Cal33 cells±IL-6 treatment. Grayscale images of Hoechst 33342-stained nuclei (Ch 1) and pSTAT3-Y705 staining (Ch 2) and color composite images of nonstimulated and IL-6-treated (50 ng/mL, 15 min) Cal33 HNSCC cells. (B) Enlarged grayscale images of Cal33 Hoechst-stained nuclei. Enlarged grayscale images of Hoechst 33342-stained nuclei (Ch 1) of nonstimulated and IL-6 treated (50 ng/mL, 15 min) Cal33 HNSCC cells. (C) Enlarged grayscale images of Cal33 ofpSTAT3-Y705 staining. Enlarged grayscale images of pSTAT3-Y705 staining (Ch 2) from nonstimulated and IL-6-treated (50 ng/mL, 15 min) Cal33 HNSCC cells. (D) Nucleus and cytoplasm masks generated by the TE image analysis module segmentation. Corresponding nucleus (inner, dark green or red) and cytoplasm (outer, light green or red) masks generated by the TE image analysis module segmentation. (E–H) Quantitative data extracted from the digital images of the Hoechst-stained nuclei and pSTAT3-Y705 staining in nonstimulated and IL-6-treated Cal33 HNSCC cells by the TE image analysis module; (E) cell counts per image, (F) average pSTAT3-Y705 intensity in the outer cytoplasm mask area, and (G) average pSTAT3-Y705 intensity in the inner nuclear mask area, (H) average inner:outer (Nuc:Cyt) pSTAT3-Y705 intensity ratio. First, 2,000 Cal33 HNSSC cells were seeded into the wells of 384-well microtiter plates in DMEM containing 10% FBS and cultured overnight in an incubator at 37°C in 5% CO2 and 95% humidity. The cell culture medium was then aspirated, monolayers were washed once with SFM, fresh SFM was added to the wells, and the plates were returned to the incubator at 37°C in 5% CO2 and 95% humidity for an additional 24 h. Cal33 cells were treated with SFM (□) or 50 ng/mL of IL-6 (■) for 15 min at 37°C in 5% CO2 and 95% humidity and were then fixed and stained with Hoechst and a mouse monoclonal anti-p-STAT3-Y705 antibody as described in Materials and Methods (Table 1). Images of two fields of view for two fluorescent channels, Hoechst (Ch 1) and pSTAT3-Y705-FITC (Ch 2), were acquired on the IXU confocal HCS platform using a 20×/0.45NA objective, and the quantitative data presented were extracted from the digital images using the TE image analysis module as described in Materials and Methods. The images presented are representative of similar images obtained in numerous independent experiments, and the data represent the mean±SD of triplicate determinations from one of these independent experiments. Ch 1, channel 1; Ch 2, channel 2; FBS, fetal bovine serum; HCS, high-content imaging; IXU, ImageXpress Ultra; SFM, serum-free tissue culture medium; TE, translocation enhanced; DMEM, Dulbecco's modified Eagle's medium.
Development and Optimization of HCS Assays for pSTAT3 and pSTAT1 Pathway Activation
Since our primary interest is to identify selective inhibitors of STAT3 pathway activation, we proceeded to co-develop and optimize a STAT1 pathway activation assay as a selectivity counter screen (Fig. 3). HNSCC cell lines have previously been shown to synthesize and secrete growth factor and cytokines into culture medium that can activate the STAT3 pathway in an autocrine or paracrine manner.5,8,11,16 In an attempt to control for potential paracrine or autocrine activation of the STAT3 and STAT1 signaling pathways, we incorporated a medium exchange step into our protocols that involved multiple aspiration and wash steps along with a 24-h serum starvation period before cytokine treatment. Since media aspiration and multiple wash steps along with an extended serum starvation period have the potential to significantly disrupt cell integrity and can be challenging to automate, we directly compared this process with one in which the cells were cultured undisturbed in medium containing 10% FBS before cytokine administration to see whether any of the medium exchange and serum starvation steps could be eliminated (Fig. 3A, D). Compared with Cal33 cells processed by the medium exchange and serum starvation protocol, undisturbed cells cultured in medium containing 10% FBS for 48 h exhibited both a higher background un-induced pSTAT3-Y705 signal and a reduced activation response to IL-6 (Fig. 3A). The corresponding IL-6-induced pSTAT3 assay signal window collapsed from >5-fold for the medium exchange and serum starvation protocol to around 2-fold in the “undisturbed” protocol. In contrast, the impact of these two protocols on IFNγ-induced pSTAT1 activation was much less pronounced (Fig. 3D). For pSTAT1 activation, Cal33 cells cultured in medium containing 10% FBS for 48 h did not exhibit a higher background un-induced pSTAT1-Y701 signal, although their activation response to IFNγ was slightly reduced and more variable than in cells that had undergone the medium exchange and serum starvation protocol (Fig. 3D). Given our primary focus on screening for selective inhibitors of STAT3 pathway activation, we elected to incorporate the media exchange and serum starvation steps into both the pSTAT3 and pSTAT1 HCS protocols.
Fig. 3.


Development and optimization of the pSTAT3-Y705 and pSTAT1-Y701 HCS assays. Treated Cal33 HNSSC cells were fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 (A–D) antibody or a mouse monoclonal anti-p-STAT1-Y701 (E–H) as described in Materials and Methods (Table 1). Images of two fields of view for two fluorescent channels, Hoechst (Ch 1) and either pSTAT3-Y705-FITC or pSTAT1-Y701-FITC (Ch 2), were acquired on the IXU confocal HCS platform using a 20×/0.45NA objective, and the quantitative data presented were extracted from the digital images using the TE image analysis module as described in Materials and Methods. Serum-free media exchange, (A) IL-6 and pSTAT3-Y705 and (E) IFNγ and pSTAT1-Y701 HCS. First, 2,000 Cal33 HNSSC cells were seeded into the wells of duplicate 384-well microtiter plates in DMEM containing 10% FBS and cultured overnight in an incubator at 37°C in 5% CO2 and 95% humidity. In one of the duplicate plates, the cell culture medium was then aspirated, monolayers were washed once with SFM, fresh SFM was added to the wells, and the plates were returned to the incubator at 37°C in 5% CO2 and 95% humidity for an additional 24 h. The other plate was left in the incubator untouched. After 24 h, both the serum-containing and serum-free plates of Cal33 cells were then treated with SFM, 50 ng/mL of IL-6, or 30 ng/mL IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity and were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=6) average inner (nuclear) intensities of the p-STAT3-Y705 or p-STAT1-Y701 signals from six wells treated with medium or cytokine in cells cultured in serum containing (filled bars) conditions throughout, or serum starved for 24 h (hashed bars) before cytokine treatment are presented. Representative experimental data from one of three independent experiments are shown. Cytokine concentration responses, (B) IL-6 and pSTAT3-Y705 and (F) pSTAT1-Y701 HCS. First, 2,000 Cal33 HNSSC cells that had been serum starved for 24 h as described earlier were treated with the indicated concentrations of IL-6 or IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity, and the treated cells were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=4) average inner (nuclear) intensities of the p-STAT3-Y705 (B, •) or p-STAT1-Y701 (F, •) signals from four wells treated with each cytokine concentration are presented. Representative experimental data from one of four independent experiments are shown. Cytokine time courses, (C) IL-6 and pSTAT3-Y705 and (G) IFNγ and pSTAT1-Y701 HCS. First, 2,000 Cal33 HNSSC cells that had been serum starved for 24 h as described earlier were treated with 50 ng/mL of IL-6 or 30 ng/mL IFNγ for the indicated time periods at 37°C in 5% CO2 and 95% humidity, and the treated cells were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=8) average inner (nuclear) intensities of the p-STAT3-Y705 (C, •) or p-STAT1-Y701 (G, •) signals from eight wells treated with each cytokine concentration are presented. Representative experimental data from one of four independent experiments are shown. Cal33 HNSSC cell seeding density, (D) IL-6 and pSTAT3-Y705 and (H) IFNγ and pSTAT1-Y701 HCS. Cal33 HNSSC cells that had been seeded into the wells of 384-well microtiter plates at the indicated cell seeding densities in serum-containing medium for 24 h and then serum starved for 24 h as described earlier were then treated with medium, 50 ng/mL of IL-6, or 30 ng/mL IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity, and were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=6) average inner (nuclear) intensities of the p-STAT3-Y705 (D, ◯ medium, • IL-6) or p-STAT1-Y701 (H, ◯ medium, ■ IFNγ) signals from six wells treated with each cytokine are presented. Representative experimental data from one of three independent experiments are shown. IFNγ, interferon gamma.
To evaluate the cytokine concentration-dependent activation of the pSTAT3 and pSTAT1 HCS assays, we exposed serum-starved Cal33 cells to the indicated concentrations of IL-6 (Fig. 3B) or IFNγ (Fig. 3E) for 15 and 45 min, respectively. For IL-6-induced activation of pSTAT3, the average pSTAT3-Y705 “inner” intensity response increased linearly through 20 ng/mL IL-6 and thereafter, the magnitude of the increase in signal with higher IL-6 concentrations appeared to decline until a maximum response was approached in the 50–60-ng/mL range (Fig. 3B). For IFNγ activation of pSTAT1, the average pSTAT1-Y701 “inner” intensity response increased linearly through 2.5 ng/mL IFNγ and thereafter, the magnitude of the increase in signal with higher IFNγ concentrations appeared to decline until a maximum response was reached at 20 ng/mL, which was maintained through 40 ng/mL (Fig. 3B). To evaluate the time course of cytokine-dependent activation of the pSTAT3 and pSTAT1 HCS assays, we exposed serum-starved Cal33 cells to 50 ng/mL IL-6 (Fig. 3C), or 30 ng/mL IFNγ (Fig. 3F) for the indicated time periods. For IL-6-induced activation of pSTAT3, the average pSTAT3-Y705 “inner” intensity response increased through 10 min of cytokine exposure to reach a maximum plateau that was maintained through 20 min, and thereafter declined steadily through 30 and 45 min even with continuous cytokine exposure (Fig. 3C). For IFNγ activation of pSTAT1, the average pSTAT1-Y701 “inner” intensity response dramatically increased through 15 min of cytokine exposure and thereafter exhibited a much slower rate of increase that was maintained through 60 min of continuous cytokine exposure (Fig. 3G). On the basis of these data, we selected a 15-min exposure to 50 ng/mL IL-6 and a 30-min exposure to 30 ng/mL IFNγ, respectively, for all further pSTAT3 and pSTAT1 HCS assay development experiments. Using these optimized conditions, we evaluated the effects of Cal33 cell seeding density on the dynamic ranges of the pSTAT3 and pSTAT1 HCS assays (Fig. 3D, H). The 384-well pSTAT3 HCS assay exhibited a robust and reproducible separation between the untreated background signals and IL-6-induced responses (Fig. 3D). At seeding densities of 1,000, 1,500, and 2,000 cells/well, the IL-6-induced pSTAT3 activation S:B ratios were 6.25, 5.66, and 5.53, respectively, but declined to <5-fold with higher seeding densities. The 384-well pSTAT1 HCS assay also exhibited a reproducible separation between the untreated background signals and IFNγ-induced responses (Fig. 3H), but had a larger dynamic range than the pSTAT3 assay. Indeed, all of the seeding densities tested exhibited IFNγ-induced pSTAT1 activation S:B ratios >10-fold. To reduce the cell culture burden and be consistent between the two assays, we selected a 384-well plate seeding density of 2,000 cells/well for both.
Validation of the HCS Assay for pSTAT3 Pathway Activation
Since our compound libraries are dissolved in DMSO, we needed to determine the DMSO tolerance of the pSTAT3 and pSTAT1 HCS assays (Fig. 4A, C). At DMSO concentrations greater than 1.25%, both the IL-6-induced pSTAT3 (Fig. 4A) and IFNγ-induced pSTAT1 (Fig. 4C) activation responses were dramatically inhibited in a concentration-dependent manner. For the primary compound screen at 20 μM, the final DMSO concentration was 0.2%, and for concentration response assays beginning at a maximal concentration of 50 μM, the final DMSO concentration was 0.5%. To validate that the pSTAT3 and pSTAT1 HCS assays could correctly identify inhibitors of these signaling pathways, we exposed serum-starved Cal33 cells to the indicated concentrations of the JAK inhibitor pyridone 6 for 3 h before the addition of 50 ng/mL IL-6 (Fig. 4B) or 30 ng/mL IFNγ (Fig. 4D) for 15 and 45 min, respectively. Pretreatment of Cal33 cells with the pan-JAK inhibitor pyridone 644 inhibited both the IL-6-induced pSTAT3 (Fig. 4B) and IFNγ-induced pSTAT1 (Fig. 4D) activation responses in a concentration-dependent manner. Pyridone 6 nonselectively inhibited the IL-6-induced pSTAT3 and IFNγ-induced pSTAT1 activation responses with IC50s of 7.193±4.082 nM and 16.382±8.453 nM, respectively (Fig. 4B, D).
Fig. 4.

DMSO tolerance and JAK inhibitor concentration responses of the pSTAT3-Y705 and pSTAT1-Y701 HCS assays. DMSO tolerance of the (A) IL-6 and pSTAT3-Y705 and (C) IFNγ and pSTAT1-Y701 HCS assays. First, 2,000 Cal33 HNSSC cells that had been serum starved for 24 h as described earlier were exposed to the indicated concentrations of DMSO for 3 h. Next, the DMSO-exposed cells were treated with either medium, 50 ng/mL of IL-6, or 30 ng/mL IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity, and were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=3) average inner (nuclear) intensities of the p-STAT3-Y705 (A, ○ medium, • IL-6) or p-STAT1-Y701 (C, ○ medium, • IFNγ) signals from triplicate wells treated with medium or cytokine are presented. Representative experimental data from one of three independent experiments are shown. Pyridone 6 concentration responses of the (B) IL-6 and pSTAT3-Y705 and (D) IFNγ and pSTAT1-Y701 HCS assays. First, 2,000 Cal33 HNSSC cells that had been serum-starved for 24 h as described earlier were exposed to the indicated concentrations of the JAK inhibitor pyridone 6 for 3 h. Next, the cells were treated with either 50 ng/mL of IL-6 or 30 ng/mL IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity, and were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=3) average inner (nuclear) intensities of the p-STAT3-Y705 (B) or p-STAT1-Y701 (C) signals from triplicate wells in three independent experiments are presented: Experiment 1 (•), Experiment 2 (○), and Experiment 3 (▪). DMSO, dimethyl sulfoxide; JAK, Janus kinase.
To validate the compatibility of the pSTAT3 HCS assay with screening, we ran our 3-day assay signal window and DMSO validation tests.45 The 3-day assay signal window and Z-factor determination consisted of three independent experiments of two full 384-well plates each of the minimum (Min, 0.2% DMSO) and maximum (Max, 50 ng/mL IL-6 in 0.2% DMSO) plate controls conducted on three separate days (Table 2). The Max and Min plate controls performed very reproducibly, and their respective average pSTAT3-Y705 “inner” intensity population responses were well separated from each other with S:B ratios of 6.1-, 6.3-, and 7.8-fold on days 1, 2, and 3, respectively (Table 2). Overall, the IL-6-induced pSTAT3 activation assay exhibited intra-plate Z-factor coefficients on all 3 days ranging between 0.38 and 0.72, with inter-plate Z-factors of 0.61, 0.69, and 0.35 on days 1, 2, and 3, respectively (Table 2). The day 1–2 Z-factor coefficient was 0.61, while the day 2–3 Z-factor coefficient was negative because the responses on the two maximum plates from day 2 were appreciably higher than on day 3, such that the corresponding SDs and CVs for the day-to-day comparison exceeded acceptable limits. Based on the statistical indices of the pSTAT3 HCS assay (Table 2), we selected a plate controls-based % inhibition data processing method for the DMSO validation test and set a preliminary active criterion of ≥50% inhibition of IL-6-induced pSTAT3 activation. The 3-day five-plate DMSO validation test mimics 3 days of automated screening operations, and all fifteen 384-well plates exhibited S:B ratios >5-fold with only 3 of the 15 plates producing Z′-factor coefficients <0.5 (Table 3). All three plates were from day 1, and with Z′-factor coefficients of 0.21, 0.29, and 0.41 and S:B ratios >5-fold, these assay plates still passed our QC criteria. The % inhibition data from the 4,800 wells of the 15×384-well DMSO plates closely approximated a normal distribution (data not shown), and only 11 wells exhibited ≥50% inhibition of IL-6-induced pSTAT3 activation producing an estimated false-positive rate of 0.23%. An analysis of variance (ANOVA) in the DMSO validation data revealed no significant row/column effects or other positional biases (data not shown). Based on the statistical indices of the pSTAT3 HCS assay45 (Tables 2 and 3), we elected to continue with a plate controls-based % inhibition data processing method and a preliminary active criterion of ≥50% inhibition of IL-6-induced pSTAT3 activation. For quality control review purposes, we failed plates that produced both an S:B ratio <5-fold and a Z′-factor coefficient <0.25.
Table 2.
Three-Day Assay Signal Window and Z-Factor Coefficient Determination for the IL-6–Induced pSTAT3-Y705 Activation High-Content Screening Assay
| Class/Day | Plate | Max/Min | Mean | SD | CV | PL to PL | Z-factor | S:B ratio |
|---|---|---|---|---|---|---|---|---|
| Intraplate | ||||||||
| 1 | 1 | Max | 10,690.4 | 1,102.6 | 10.3 | 1–3 | 0.59 | 5.73 |
| 2 | Max | 11,082.8 | 947.4 | 8.6 | 1–4 | 0.6 | 6.23 | |
| 3 | Min | 1,866.0 | 103.3 | 5.5 | 2–3 | 0.66 | 5.94 | |
| 4 | Min | 1,714.6 | 86.5 | 5.1 | 2–4 | 0.67 | 6.46 | |
| All plates | All | 0.61 | 6.08 | |||||
| 2 | 1 | Max | 12,108.7 | 782.5 | 6.5 | 1–3 | 0.73 | 6.33 |
| 2 | Max | 11,697.7 | 920.4 | 7.9 | 1–4 | 0.72 | 6.39 | |
| 3 | Min | 1,911.6 | 130.9 | 6.9 | 2–3 | 0.68 | 6.12 | |
| 4 | Min | 1,894.3 | 161.3 | 8.5 | 2–4 | 0.67 | 6.18 | |
| All plates | All | 0.69 | 6.26 | |||||
| 3 | 1 | Max | 7,038.9 | 1,079.8 | 15.3 | 1–3 | 0.38 | 6.93 |
| 2 | Max | 8,078.0 | 1,196.8 | 14.8 | 1–4 | 0.4 | 7.62 | |
| 3 | Min | 1,015.9 | 174.7 | 17.2 | 2–3 | 0.42 | 7.95 | |
| 4 | Min | 923.3 | 144.2 | 15.6 | 2–4 | 0.44 | 8.75 | |
| All plates | All | 0.35 | 7.8 | |||||
| Interplate | ||||||||
| 1 | 1 and 2 | Max | 10,886.6 | 1,046.3 | 9.6 | 0.61 | 6.08 | |
| 3 and 4 | Min | 1,790.3 | 121.7 | 6.8 | ||||
| 2 | 1 and 2 | Max | 11,903.2 | 878.5 | 7.4 | 0.69 | 6.26 | |
| 3 and 4 | Min | 1,903.0 | 147.1 | 7.7 | ||||
| 3 | 1 and 2 | Max | 7,558.4 | 1,252.5 | 16.6 | 0.35 | 7.80 | |
| 3 and 4 | Min | 969.6 | 166.7 | 17.2 | ||||
| Day to day | ||||||||
| 1 and 2 | All plates | Max | 11,394.9 | 1,091.5 | 9.6 | 0.61 | 6.17 | |
| Min | 1,846.7 | 146.2 | 7.9 | |||||
| 2 and 3 | All plates | Max | 9,730.8 | 2,427.0 | 24.9 | −0.06 | 6.78 | |
| Min | 1,436.3 | 492.5 | 34.3 | |||||
The average inner intensity pSTAT3-Y705 Ch 2 data presented in the table was generated in three independent experiments of 2×384-well plates each of the maximum (50 ng/mL IL-6 and 0.2% DMSO) and minimum (0.2% DMSO) plate controls conducted on three separate days.
S:B, signal-to-background ratio.
Table 3.
Three-Day 5-Plate Dimethyl Sulfoxide Validation of the IL-6–Induced pSTAT3-Y705 Activation High-Content Screening Assay
| Plate controls average inner intensity pSTAT3-Y705 Ch 2 | Plate performance statistics | |||
|---|---|---|---|---|
| Plate ID | Mean_Max (n=32) | Mean_Min (n=32) | Z′-factor | S:B ratio |
| Day 1 | ||||
| 1 | 8,526.2 | 1,111.5 | 0.53 | 7.67 |
| 2 | 8,444.2 | 1,113.7 | 0.41 | 7.58 |
| 3 | 6,887.6 | 1,104.6 | 0.21 | 6.24 |
| 4 | 8,444.2 | 1,188.2 | 0.53 | 7.11 |
| 5 | 7,282.1 | 1,169.3 | 0.29 | 6.23 |
| Day 2 | ||||
| 1 | 7,797.2 | 1,519.8 | 0.62 | 5.13 |
| 2 | 7,767.1 | 1,372.3 | 0.59 | 5.66 |
| 3 | 7,843.9 | 1,378.5 | 0.69 | 5.69 |
| 4 | 8,401.7 | 1,481.0 | 0.62 | 5.67 |
| 5 | 9,203.9 | 1,558.2 | 0.62 | 5.91 |
| Day 3 | ||||
| 1 | 8,466.3 | 1,388.0 | 0.64 | 6.10 |
| 2 | 7,889.1 | 1,368.2 | 0.53 | 5.77 |
| 3 | 7,893.8 | 1,381.0 | 0.57 | 5.72 |
| 4 | 8,020.5 | 1,351.9 | 0.55 | 5.93 |
| 5 | 9,018.4 | 1,553.3 | 0.67 | 5.81 |
The average inner intensity pSTAT3-Y705 Ch 2 data presented in the table were generated in three independent experiments of 5×384-well DMSO validation plates. These experiments were designed to mimic 3 days of automated screening operations in the pSTAT3-Y705 activation HCS assay, save that DMSO was added to wells instead of compounds. The average inner intensity pSTAT3-Y705 Ch 2 data of the maximum (50 ng/mL IL-6 and 0.2% DMSO, n=32) and minimum (0.2% DMSO, n=32) plate controls from each of the 15×384-well plates are presented.
Screening the LOPAC and NIH CC in the pSTAT3 HCS Assay
To confirm that our optimized pSTAT3 assay would perform well in the presence of compounds, we screened the 1,280-member LOPAC and the 446-member NIH CC after a 24-h compound exposure at 20 μM to identify compounds that could inhibit IL-6-induced pSTAT3-Y705 activation (Fig. 5). Figure 5A is a 4×384-well plate overlay scatter plot of the % inhibition exhibited by the LOPAC set, and Figure 5B is a 2×384-well plate overlay scatter plot of the % inhibition exhibited by the NIH CC set. In addition to 320 compound wells, each 384-well plate had 32×IL-6-induced Max plate control wells and 32×0.2% DMSO Min plate control wells. The LOPAC set plate Z′-factor coefficients were 0.45, 0.56, 0.50, and 0.48, and the corresponding plate S:B ratios were 5.94, 5.82, 5.61, and 5.67 for plates 1, 2, 3, and 4, respectively (Fig. 5A). The NIH CC plate Z′-factor coefficients were 0.51 and 0.57, and the corresponding plate S:B ratios were 5.01 and 6.16 for plates 1, and 2, respectively (Fig. 5B). These data indicated that the pSTAT3 HCS assay performed very well and was compatible with compound library screening. To identify compounds that behaved as outliers compared with the other substances (n=320) tested on the assay plate, we utilized the z-score plate-based statistical scoring method to analyze selected multi-parameter measurements output by the image analysis module as previously described.41–43,46–48 Compounds that met all of the following criteria were designated qualified inhibitors of IL-6-induced pSTAT3 activation (Table 4): ≥50% inhibition of IL-6-induced pSTAT3-Y705 activation, cell counts per image >250, an average fluorescent intensity in Ch 1 z-score >−3 and <3, an integrated “outer” fluorescent intensity Ch 2 signal z-score <3, an average “outer” fluorescent intensity Ch 2 signal z-score <3, and an integrated “inner” fluorescent intensity Ch 2 signal z-score <3. After excluding 189 (11%) cytotoxic compounds and 36 (2.1%) compounds that were fluorescent outliers in one or more of the intensity parameters from the Hoechst and FITC channels, 51 (3%) compounds were designated qualified inhibitors of IL-6-induced pSTAT3-Y705 activation (Table 4).
Fig. 5.

The 384-well plate overlays of % inhibition of IL-6–induced pSTAT3-Y705 activation screen; (A) LOPAC and (B) NIH CC. Six 384-well plates were seeded with 2,000 Cal33 HNSSC cells per well and then serum starved for 24 h as described earlier. Immediately after the serum-free media exchange, diluted compounds were transferred from the 4×384-well LOPAC daughter plates and the 2×384 NIH CC daughter plates into the assay plates to provide a final screening concentration of 20 μM (0.2% DMSO) and then incubated at 37°C, 5% CO2, and 95% humidity for 24 h. Compound-treated wells and Max plate controls then received 50 ng/mL IL-6 in 0.2% DMSO (final), Min controls received 0.2% DMSO (final) in media, and assay plates were then incubated at 37°C, 5% CO2, and 95% humidity for 15 min and then fixed and stained with Hoechst and a mouse monoclonal anti-p-STAT3-Y705 antibody as described in Materials and Methods (Table 1). Images of two fields of view for two fluorescent channels, Hoechst (Ch 1) and pSTAT3-Y705-FITC (Ch 2) were acquired on the IXU confocal HCS platform using a 20×/0.45NA objective, and the average inner “nuclear” intensity of pSTAT3-Y705 was derived from the digital images using the TE image analysis module as described in Materials and Methods. The mean average innerpSTAT3-Y705 intensity values of the 0.2% DMSO Min plate control wells (n=32) and the 50-ng/mL IL-6 Max plate control wells (n=32) were used to normalize the mean average inner intensity pSTAT3-Y705 values of the compound-treated wells and to represent 100% and 0% inhibition of IL-6-induced pSTAT3 activation, respectively. (A) Overlay scatter plot of the % inhibition of the average inner pSTAT3-Y705 intensity data from the 4×384-well assay plates of the 1,280 compound LOPAC screen; Max (•) controls, Min (•) controls, inactive (<50% inhibition) compound treated wells (•), and active (≥50% inhibition) compound-treated wells (•). (B) Overlay scatter plot of the % inhibition of the average inner pSTAT3-Y705 intensity data from the 2×384-well assay plates of the 446 compound NIH CC screen; Max (•) controls, Min (•) controls, inactive (<50% inhibition) compound-treated wells (•), and active (≥50% inhibition) compound-treated wells (•). LOPAC, Library of Pharmacologically Active Compounds; NIH CC, National Institutes of Health Clinical Collection.
Table 4.
Library of Pharmacologically Active Compounds and National Institutes of Health Clinical Collection pSTAT3 Primary High-Content Screening Summary
| Compound library | No. of compounds | % of total |
|---|---|---|
| Compound information | ||
| LOPAC | 1,280 | 74.2 |
| NIH CC | 446 | 25.8 |
| Total | 1,726 | 100 |
| Cytotoxic and fluorescent outlier analysis | ||
| Ch 1: Average cell count/image <250 | 189 | 11.0 |
| Ch 2: Hoechst average nuclear intensity z-score >3 | 8 | 0.5 |
| Ch 2: Integrated inner intensity z-score >3 | 7 | 0.4 |
| Ch 2: Average outer intensity z-score >3 | 15 | 0.9 |
| Ch 2: Integrated outer intensity score >3 | 6 | 0.3 |
| Primary HCS actives at 20 μM | ||
| % Inhibition of IL-6-induced pSTAT3 ≥50, 24 h exp. | 51 | 3.0 |
| Active confirmation at 20 μM | ||
| Mean % inhibition of IL-6-induced pSTAT3 ≥50, 24 h exp. | 24 | 47.1 |
| Mean % inhibition of IL-6-induced pSTAT3 ≥50, 1 h exp. | 35 | 68.6 |
| Mean % inhibition of IFNγ-induced pSTAT1 ≥50, 24 h exp. | 6 | 11.8 |
| Mean % inhibition of IFNγ induced pSTAT1 ≥50, 1 h exp. | 18 | 35.3 |
| Selective inhibitors of IL-6-induced pSTAT3 activation at 20 μM | ||
| Mean % pSTAT3 inhibition ≥50% (24 and 1 h) and mean % pSTAT1 inhibition <30% (24 and 1 h) | 3 | 5.9 |
HCS data represent % inhibition of compounds tested in a single well at 20 μM, and confirmation data represent mean % inhibition±SD (n=3) of compounds tested in triplicate wells at 20 μM, after 1 or 24 h of compound exposures.
IFNγ, interferon-gamma; LOPAC, Library of Pharmacologically Active Compounds; NIH CC, National Institutes of Health Clinical Collection.
To confirm the qualified active compounds identified in the primary pSTAT3 screen, we cherry picked the 51 compounds into a 384-well hit confirmation daughter plate and rescreened them at 20 μM in triplicate wells in the IL-6-induced pSTAT3 assay and also the IFNγ-induced pSTAT1 counter screen (Tables 4 and 5). In addition to the 24-h compound exposure of the primary HCS, actives were also tested in both assays after a 1-h compound exposure period (Table 5). After a 24-h exposure, 24 (47.1%) of the compounds exhibited ≥50% inhibition of IL-6-induced pSTAT3 activation, thereby confirming their activity in the primary screen (Table 5). After1 h compound exposure, however, an additional 15 compounds exhibited ≥50% inhibition of IL-6-induced pSTAT3 activation (Table 5), giving a total of 39 (76.5%) confirmed actives. Six (11.8%) of the compounds exhibited ≥50% inhibition of IFNγ-induced pSTAT1 activation after a 24-h exposure, and an additional 14 inhibited the IFNγ-induced pSTAT1 activation assay by ≥50% after a 1-h exposure (Table 5). Taking the pSTAT1 counter screening data into consideration, only three compounds (3.9%) exhibited ≥50% inhibition of IL-6-induced pSTAT3 activation at both compound exposures along with <30% inhibition of IFNγ-induced pSTAT1 activation at both exposures: azelastine, etomidate and α-methyl-norepinephrine.
Table 5.
Confirmation of the Library of Pharmacologically Active Compounds and National Institutes of Health Clinical Collection Primary pSTAT3 High-Content Screening Actives
| Compound information | % Inhibition of IL-6-induced pSTAT3-Y705 | % Inhibition of IFNγ–induced pSTAT1-Y701 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SID | SL | Name | HCS | Mean 24 h | SD | Mean 1 h | SD | Mean 24 h | SD | Mean 1 h | SD |
| 46386661 | N | Azelastine | 82.6 | 90.4 | 4.8 | 96.2 | 14.5 | 24.2 | 18.1 | 29.6 | 41.4 |
| 53777461 | L | Dihydroouabain | 97.8 | 84.2 | 6.2 | 4.7 | 16.9 | 81.3 | 8.0 | −55.5 | 19.0 |
| 53777973 | L | NF449 octasodium salt | 85.2 | 72.6 | 5.2 | 96.9 | 2.7 | 90.3 | 7.8 | 96.1 | 1.4 |
| 46386563 | N | Desloratadine | 72.9 | 72.0 | 1.5 | 111.3 | 1.0 | 27.4 | 22.2 | 73.5 | 8.5 |
| 53778225 | L | SB 205384 | 85.0 | 69.8 | 7.0 | 56.6 | 2.8 | 89.2 | 1.4 | 25.4 | 1.1 |
| 53778026 | L | Phenylephrine hydrochloride | 51.6 | 69.5 | 4.7 | 85.4 | 1.3 | 40.2 | 4.6 | 75.9 | 5.3 |
| 53778265 | L | Trazodone hydrochloride | 53.5 | 68.2 | 4.5 | 72.0 | 3.9 | 35.7 | 12.4 | 50.5 | 15.2 |
| 46387007 | N | R(+)-SCH-23390 HCl | 63.4 | 66.4 | 5.5 | 77.1 | 2.5 | 30.5 | 23.8 | 65.6 | 3.1 |
| 53778017 | L | Nylidrin hydrochloride | 59.8 | 66.1 | 8.7 | 72.4 | 3.6 | 20.7 | 12.0 | 45.7 | 16.9 |
| 53778204 | L | Albuterol hemisulfate | 60.2 | 63.5 | 2.3 | 102.8 | 2.1 | 20.9 | 9.0 | 94.0 | 1.7 |
| 53777777 | L | Isotharine mesylate | 52.1 | 59.1 | 9.5 | 23.0 | 2.3 | −11.0 | 5.5 | −29.6 | 13.2 |
| 53777593 | L | Forskolin | 75.2 | 57.2 | 4.8 | 91.3 | 1.8 | 3.8 | 9.6 | 84.4 | 3.5 |
| 46386983 | N | Etomidate | 61.3 | 56.7 | 7.1 | 65.8 | 8.1 | 17.1 | 22.3 | 13.4 | 7.8 |
| 53777746 | L | R(−)-Isoproterenol (+)-bitartrate | 64.4 | 56.1 | 1.8 | 52.8 | 4.8 | 27.0 | 5.2 | 44.6 | 12.2 |
| 53777650 | L | Fenoterol hydrobromide | 61.5 | 56.0 | 10.0 | 107.5 | 1.7 | 7.5 | 16.2 | 97.0 | 1.2 |
| 46386731 | N | Cefpodoxime proxetil | 55.1 | 54.6 | 1.1 | 99.0 | 3.3 | 13.1 | 18.6 | 94.0 | 2.7 |
| 53778255 | L | Tulobuterol hydrochloride | 57.5 | 53.0 | 11.4 | 60.6 | 5.6 | 22.6 | 8.2 | 57.5 | 7.7 |
| 53778281 | L | Tyrphostin AG 879 | 59.1 | 52.4 | 6.9 | 27.8 | 10.6 | 53.5 | 10.6 | 52.4 | 20.6 |
| 53777778 | L | (−)-Isoproterenol hydrochloride | 69.4 | 52.2 | 9.3 | 66.2 | 7.2 | 25.2 | 22.2 | 52.2 | 16.6 |
| 53777840 | L | Metaproterenol hemisulfate | 58.5 | 51.3 | 0.7 | 102.6 | 3.0 | 8.1 | 26.1 | 93.1 | 0.7 |
| 53777465 | L | (−)-α-methylnorepinephrine | 55.8 | 51.2 | 9.1 | 68.1 | 3.3 | 27.4 | 16.0 | 12.7 | 6.2 |
| 53778191 | L | (±)-Synephrine | 61.5 | 50.7 | 7.8 | 59.6 | 7.4 | 12.3 | 22.3 | 50.8 | 16.3 |
| 53777817 | L | (±)-Isoproterenol hydrochloride | 63.4 | 50.3 | 2.6 | 63.9 | 3.3 | 17.8 | 20.7 | 32.1 | 33.1 |
| 53778152 | L | Cortexolone maleate | 57.1 | 50.3 | 6.2 | 46.5 | 3.5 | 5.6 | 21.3 | −35.5 | 30.4 |
| 53777595 | L | N-(3,3-diphenylpropyl)glycinamide | 61.0 | 49.6 | 11.6 | 62.6 | 2.7 | 9.3 | 15.4 | 34.3 | 5.8 |
| 53778147 | L | Ritodrine hydrochloride | 63.1 | 49.0 | 2.6 | 69.7 | 12.7 | −3.1 | 17.6 | 48.0 | 7.9 |
| 53778120 | L | Propantheline bromide | 57.0 | 47.7 | 9.9 | 53.4 | 3.0 | 11.7 | 26.7 | 35.6 | 8.2 |
| 46386828 | N | Balsalazida | 58.5 | 47.1 | 3.0 | 53.1 | 4.0 | 10.0 | 29.3 | 15.4 | 25.4 |
| 53778232 | L | Terbutaline hemisulfate | 61.4 | 46.3 | 6.1 | 97.6 | 2.6 | −12.4 | 16.9 | 78.4 | 8.3 |
| 53778036 | L | Perphenazine | 73.3 | 46.2 | 2.8 | 87.3 | 7.7 | 50.9 | 8.5 | 68.5 | 0.7 |
| 53777521 | L | 6,7-ADTN hydrobromide | 52.8 | 45.7 | 6.3 | 50.4 | 9.4 | 19.1 | 19.6 | 32.8 | 3.6 |
| 53777200 | L | L(−)-Norepinephrine bitartrate | 55.8 | 40.5 | 10.9 | 61.9 | 2.2 | 34.3 | 23.7 | 39.1 | 10.9 |
| 46387005 | N | Salmeterol | 63.2 | 38.5 | 4.8 | 110.4 | 1.4 | 2.8 | 26.3 | 90.0 | 1.9 |
| 53777665 | L | Guanidinyl-naltrindole di-trifluoroacetate | 57.8 | 36.8 | 12.2 | 52.7 | 5.2 | −13.6 | 10.0 | −15.3 | 7.6 |
| 53777160 | L | Aurintricarboxylic acid | 86.3 | 36.5 | 5.4 | 105.0 | 0.8 | 99.4 | 0.2 | 98.6 | 2.5 |
| 53777487 | L | 8-Cyclopentyl-1,3-dipropylxanthine | 52.1 | 33.8 | 10.6 | 60.6 | 4.6 | 23.8 | 15.0 | 26.6 | 18.3 |
| 53777634 | L | cis-(Z)-flupenthixol dihydrochloride | 79.0 | 30.8 | 10.1 | 72.3 | 5.3 | 13.3 | 13.2 | 15.5 | 23.4 |
| 53777608 | L | (−)-Epinephrine bitartrate | 62.1 | 24.7 | 11.4 | 58.0 | 7.4 | 1.6 | 16.5 | 11.1 | 15.7 |
| 53777159 | L | (±)-Norepinephrine (+)bitartrate | 69.5 | 24.4 | 7.6 | 62.1 | 2.2 | 43.4 | 12.2 | 45.6 | 11.0 |
HCS data represent % inhibition of a single well, confirmation data represent mean % inhibition±SD (n=3), 1 and 24 h of compound exposure data presented.
SID, PubChem substance identifier, SL, source library, L, LOPAC, N, NIH CC.
Hit Characterization
Nineteen (48.7%) of the 39 compounds that inhibited IL-6-induced pSTAT3 activation and/or IFNγ-induced pSTAT1 activation are G-alpha stimulatory subunit (Gs)-coupled GPCR agonists (Table 5). Agonists of Gs-coupled GPCRs elevate cellular cyclic adenosine monophosphate (cAMP) levels.49 In addition, forskolin, another of the 39 confirmed actives (Table 5), elevates cellular cAMP levels by direct activation of adenylate cyclase.50 To determine whether forskolin or the beta-adrenoreceptor (β-AR) agonist isoproterenol could inhibit IL-6-induced pSTAT3 activation and/or IFNγ-induced pSTAT1 activation, serum-starved Cal33 cells were exposed to 20 μM forskolin or isoproterenol for either 3 or 24 h and then treated with either medium, 50 ng/mL of IL-6, or 30 ng/mL IFNγ for 15 min (Fig. 6). Pre-exposure of Cal33 cells to 20 μM forskolin or isoproterenol for 3 h significantly inhibited the activation of both pathways by their respective cytokines (Fig. 6A, B). While a 24-h exposure to 20 μM forskolin was almost as effective as a 3-h exposure, a 24-h exposure to isoproterenol was significantly less effective than a 3-h exposure (Fig. 6A, B).
Fig. 6.

Nonselective inhibition of pSTAT3 and pSTAT1 activation by forskolin and isoproterenol. First, 2,000 Cal33 HNSSC cells that had been serum starved for 24 h as described earlier were exposed to the indicated concentrations of forskolin or isoproterenol for either 3 or 24 h and then treated with either medium, 50 ng/mL of IL-6, or 30 ng/mL IFNγ for 15 min. Next, the cells were fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). (A) Inhibition of IL-6-induced pSTAT3 activation. The mean±SD (n=3) average inner (nuclear) intensities of the p-STAT3-Y705 in medium and 50 ng/mL IL-6 treated controls, or in IL-6-treated wells pre-exposed to either 20 μM forskolin or 20 μM isoproterenol for 3 h (□) or 24 h (▪). Representative experimental data from one of three independent experiments are shown. (B) Inhibition of IFNγ-induced pSTAT1 activation. The mean±SD (n=3) average inner (nuclear) intensities of the p-STAT1-Y701 in medium and 30 ng/mL γ-treated controls, or in IFNγ-treated wells pre-exposed to either 20 μM forskolin or 20 μM isoproterenol for 3 h (□) or 24 h (▪). Representative experimental data from one of three independent experiments are shown.
To confirm the selective inhibition of the STAT3 pathway by azelastine and etomidate, we purchased powdered samples and conducted 10-point concentration response experiments in the pSTAT3 and pSTAT1 HCS assays starting at a maximum of 50 μM (Fig. 7). The newly purchased sample of etomidate failed to significantly inhibit the activation of either cytokine pathway (Fig. 7). In contrast, pre-exposure of Cal33 cells to azelastine for 3 h selectively inhibited IL-6-induced pSTAT3 activation in a concentration-dependent manner (Fig. 7A, C), without significantly affecting IFNγ-induced pSTAT1 activation (Fig. 7B, D). We repeated the azelastine concentration response experiments in the pSTAT3 (Fig. 8A) and pSTAT1 (Fig. 8B) HCS assays and in growth inhibition assays conducted in four HNSCC cell lines: Cal33, 686LN, FaDu, and OSC19 (Fig. 8C). Azelastine selectively inhibited the IL-6-induced pSTAT3 activation response with an IC50 of 26.56±10.42 μM (Figs. 7A and 8A, D). In marked contrast, azelastine did not significantly inhibit IFNγ-induced pSTAT1 activation, even at 50 μM of the highest concentration tested (Figs. 7B and 8B, D). Exposure of HNSCC cell lines to azelastine for 72 h inhibited their growth in a concentration-dependent manner (Fig. 8C). Azelastine inhibited the growth of the four cell lines with IC50s of 13.62±7.5, 38.82±5.15, 33.18±11.8, and 23.77±2.15 μM for Cal33, 686LN, FaDu, and OSC19 cells, respectively (Fig. 8C, D). The chemical structure of azelastine hydrochloride, ((±)-1-(2H)-phthalazinone,4-[(4-chlorophenyl)methyl]-2-(hexahydro-1methyl-1Hazepin-4-yl)-monhydrochloride), is presented along with a summary of its IC50 values in the IL-6-induced pSTAT3 activation assay, the IFNγ-induced pSTAT1 activation assay, the HNSCC growth inhibition assays, and the results of a cross-target query of the biological activity of SID 46386661 in the PubChem database (Fig. 8D). A cross-target query of PubChem indicated that azelastine had been tested in 454 bioassays, but there were only 12 (2.6%) active flags recorded in the database.
Fig. 7.

Concentration-dependent effects of primary HCS hits on (A) IL-6-induced pSTAT3 activation, (B) IFNγ-induced pSTAT1 activation, (C) composite images of Hoechst and pSTAT3-Y705-stained Cal33 cells in controls and 50 μM Azelastin-treated wells, and (D) composite images of Hoechst and pSTAT1-Y701-stained Cal33 cells in controls and 50 μM Azelastin-treated wells. First, 2,000 Cal33 HNSSC cells that had been serum starved for 24 h as described earlier were exposed to the indicated concentrations of azelastine or etomidate for 3 h. Next, the cells were treated with either 50 ng/mL of IL-6 or 30 ng/mL of IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity, and were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=3) average inner (nuclear) intensities of the p-STAT3-Y705 (A) or p-STAT1-Y701 (B) signals from triplicate wells for each compound concentration are presented; azelastine (•) or etomidate (○). Representative experimental data from one of three independent experiments are shown. Representative composite images from control wells and 50 μM-treated Cal33 HNSSC cells that were fixed and stained with Hoechst and either (C) a mouse monoclonal anti-p-STAT3-Y705 antibody or (D) a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). Images of two fields of view for two fluorescent channels, Hoechst (Ch 1) and either pSTAT3-Y705-FITC or pSTAT1-Y701-FITC (Ch 2) were acquired on the IXU confocal HCS platform using a 20×/0.45NA objective.
Fig. 8.

Concentration-dependent effects of azelastine on (A) IL-6 induced pSTAT3 activation, (B) IFNγ-induced pSTAT1 activation, (C) HNSCC cell growth inhibition, and (D) azelastine chemical structure and biological activity summary. (A) Inhibition of IL-6-induced pSTAT3 activation, and (B) inhibition of IFNγ-induced pSTAT1 activation. First, 2,000 Cal33 HNSSC cells that had been serum starved for 24 h as described earlier were exposed to the indicated concentrations of azelastine for 3 h. Next, those cells were treated with either 50 ng/mL of IL-6 or 30 ng/mL of IFNγ for 15 min at 37°C in 5% CO2 and 95% humidity, and were then fixed and stained with Hoechst and either a mouse monoclonal anti-p-STAT3-Y705 antibody or a mouse monoclonal anti-p-STAT1-Y701 antibody as described in Materials and Methods (Table 1). The mean±SD (n=3) average inner (nuclear) intensities of the p-STAT3-Y705 (A) or p-STAT1-Y701 (B) signals from triplicate wells for each compound concentration are presented. Representative experimental data from one of three independent experiments are shown.(C) HNSCC growth inhibition. First, 1,000 Cal33, 686LN, FaDu, or OSC19 HNSCC cells were seeded into 384-well assay plates and incubated at 37°C in 5% CO2 and 95% humidity for 24 h. After 24 h, the indicated concentrations of azelastine were transferred into the test wells of the 384-well assay plates that were then incubated at 37°C in 5% CO2 and 95% humidity for an additional 72 h. Control wells received DMSO alone. After 72 h of exposure to azelastine, Cell Titer Glo detection reagent was dispensed into the wells of the assay plates, and the luminescent signal was captured on the Envision microtiter plate reader. The mean±SD (n=3) growth inhibition data from triplicate wells for each concentration of azelastine are presented as the % of the DMSO plate controls; Cal33 (•), 686LN (○), FaDu (▪), and OSC19 (□). Representative experimental data from one of three independent experiments are shown. (D) Azelastine chemical structure and biological activity summary of the chemical structure of azelastine, (RS)-4-[(4-chlorophenyl)methyl]-2-(1-methylazepan-4-yl)-phthalazin-1-one, is presented along with a summary of the IC50 values for azelastine in the IL-6-induced pSTAT3 activation assay, the IFNγ-induced pSTAT1 activation assay, and the HNSCC growth inhibition assays, and the results of a cross-target query of the biological activity of SID 46386661 in the PubChem database. IC50, 50% inhibition concentration.
Discussion
Based on the IL-6 receptor complex and STAT3 protein expression data (Fig. 1), we selected the Cal33 cell line as our HNSCC model for the development of the pSTAT3 and pSTAT1 HCS assays. The Cal33 HNSCC cell line was established from a biopsy that had been extracted before therapy from a moderately differentiated squamous cell carcinoma of the tongue in a 69-year-old male patient.37 Cal33 cells are of epithelial origin that display chromosomal rearrangements and copy number changes which mirror the cytogenetic status of primary HNSCC.36 To configure the pSTAT3 and pSTAT1 assays for the IXU-automated HCS platform, we developed sample preparation protocols, image acquisition procedures, and image analysis settings such that only minor adjustments to the laser power and/or PMT gain were required to ensure that the Hoechst stained nuclei and fluorescent pSTAT3/pSTAT1 staining did not saturate the PMT detectors. Similarly, after we had established and optimized the parameter settings of the TE image analysis module (Fig. 2), these settings were saved to a protocol that was utilized to analyze all of the images acquired in this study.
We illustrated the assay development process by documenting the variables that directly impacted the dynamic ranges of the pSTAT3 and pSTAT1 HCS assays (Fig. 3). HNSCC cell lines have previously been shown to synthesize and secrete TGFα and IL-6 into culture medium that can constitutively activate the STAT3 pathway in an autocrine or paracrine manner.7,8,11,16 To control for potential autocrine/paracrine activation of pSTAT3 and pSTAT1, we developed an automated medium exchange and serum starvation protocol. The IL-6-induced pSTAT3 assay signal window collapsed from >5-fold using this protocol to around 2-fold in Cal33 cells cultured “undisturbed” in serum-containing medium, because un-stimulated pSTAT3-Y705 levels were higher and the response to IL-6 was smaller in serum- containing medium (Fig. 3A). In contrast, the IFNγ-induced pSTAT1-Y701 responses of Cal33 cells were largely unaffected by these culture protocols (Fig. 3E). These data are consistent with previous reports that IL-6 is a major autocrine/paracrine factor secreted by HNSCC cells.16 To maintain consistency, we incorporated the medium exchange and serum starvation steps into both the pSTAT3 and pSTAT1 HCS protocols. The activation of both pSTAT3 and pSTAT1 was shown to be dependent on the concentrations of their respective cytokines (Fig. 3B, F) and on the length of cytokine exposure (Fig. 3C, G). A 15-min exposure to IL-6 50 ng/mL and a 30-min exposure to IFNγ 30 ng/mL were selected as the optimal conditions for the pSTAT3 and pSTAT1 HCS assays, respectively. To reduce the cell culture burden, we selected a 384-well plate seeding density of 2,000 cells/well for both assays (Fig. 3D, H). Utilizing these optimized assay conditions, the dynamic range of the IL-6-induced activation of pSTAT3-Y705 was typically >5-fold, and typically >10-fold for IFNγ-induced activation of pSTAT1-Y701.
The activation of both pSTAT3 and pSTAT1 was inhibited by DMSO concentrations greater than 1.25% (Fig. 4A, C). DMSO has two major effects on HCS assays.41–43,46–48,51 At DMSO concentrations ≥5%, there is a significant cell loss due to cytoxicity and/or reduced cell adherence, and at concentrations >1% but <5%, cells change from a well-spread and well-attached morphology to a more rounded loosely attached morphology that interferes with the ability of the image analysis module to segment images into distinct cytoplasm and nuclear regions.41–43,46–48,51 The DMSO tolerance data for the pSTAT3 and pSTAT1 HCS assays indicated that we could conduct a primary compound (20 μM) screen at a final DMSO concentration of 0.2%, and confirm actives in concentration response assays at a final DMSO concentration of 0.5%, without significantly impacting the dynamic ranges of either assay (Fig. 4A, C). The pan-JAK inhibitor pyridone 644 inhibited both IL-6-induced pSTAT3-Y705 and IFNγ-induced pSTAT1-Y701 activation with IC50s of 7.19±4.08 and 16.38±8.45 nM, respectively (Fig. 4B, D). Two additional JAK inhibitors, AZD 1,480 and JSI-124 (cucurbitacin I), also inhibited IL-6 and IFNγ-induced STAT pathway activation nonselectively (data not shown). The lack of selectivity of the JAK inhibitors is not surprising, as both cytokines activate canonical JAK/STAT signaling pathways.12,13,52 For the IL-6 signaling pathway, IL-6 first binds to the IL-6Rα subunit, and the IL-6-IL-6Rα complex then recruits the gp130 signaling subunit, the gp130 subunit, and associated JAKs become rapidly activated by phosphorylation. Nonphosphorylated STAT3 in the cytoplasm is recruited to gp130 pY motifs through interactions with the STAT3 SH domain, the gp130-associated JAK phosphorylates STAT3 at Y705, and pSTAT3-Y705 dimerizes and translocates to the nucleus, where they bind to specific DNA sequences and activate STAT3 target gene transcription.12,13 In the IFNγ signaling pathway, IFNγ dimers bind to the IFNγR1 subunit, which then engage the IFNγR2 subunit, causing JAK1 and JAK2 to cross-phosphorylate each other and the receptor subunits. Nonphosphorylated parallel STAT1 homodimers recruited to the receptors are phosphorylated by the JAKs at Y701, the pSTAT1-Y701 homodimers reorient into an anti-parallel configuration, translocate to the nucleus, where they bind to gamma-activated sequence sites in the promoters of STAT1 target genes and activate transcription.52 Several small-molecule JAK inhibitors have been tested in preclinical tumor xenograft models or have been progressed into clinical trials for cancer: AG490, LS-104, ICNB18424, and CEP701.4,15,23,24 To date, however, JAK inhibitors have only been approved for the treatment of myelofibrosis (ruxolitinib) and rheumatoid arthritis (tofacitinib). JAK inhibitors have also been associated with a mild myelosuppression and a mild but frequent GI toxicity.4,15,23,24 It is our hypothesis that a selective inhibitor of activation of the oncogenic STAT3 pathway which does not interfere with activation of the tumor-suppressive STAT1 pathway would be a more desirable anti-cancer drug profile. Since JAK inhibitors nonselectively inhibit the activation of both STAT pathways, we would de-prioritize even a novel JAK inhibitor. To identify nonselective STAT pathway inhibitors, we, therefore, incorporated the IFNγ-induced pSTAT1 activation counter screen early in our HCS testing paradigm.
We conducted the 3-day assay signal window and DMSO validation tests to evaluate the robustness and reproducibility of the pSTAT3 assay signal window, to determine Z-factor coefficients, to establish quality control review parameters, and to select the most appropriate data processing method and active criterion for screening.45 The pSTAT3 HCS assay preformed robustly and reproducibly in both tests; the assay signal window S:B ratio was >5-fold for all plates, the Z′-factor coefficients were >0.5 for 20 of the 27 plates tested, and only 1 of the 7 plate Z′-factor coefficients was <0.25 (Tables 2 and 3). For quality control review purposes, we established the following criteria for failed plates: pSTAT3 plates that produced both an S:B ratio <5-fold and a Z′-factor coefficient <0.25 would be failed and scheduled for retesting. Eleven of the 4,800 wells in the 3×5-plate DMSO validation test exhibited ≥50% inhibition of IL-6-induced pSTAT3 activation producing an estimated false-positive rate of 0.23%. These false positives were likely due to clogged tips that failed to deliver cells or primary or secondary antibodies during the automated process. Based on the combined statistical indices from both tests,45 we selected a plate controls-based % inhibition data processing method for the screen and set an active criterion of ≥50% inhibition of IL-6-induced pSTAT3 activation.
The pSTAT3 HCS assay performed very well in the LOPAC and NIH CC screen conducted at 20 μM with a 24-h compound exposure (Fig. 5). We selected a 24-h compound exposure for the pilot screens, because pyridone 6, one of the three JAK inhibitors used to validate the assay, was more potent after 24 h than at 1 h (data not shown). All of the assay plates passed our quality control criteria and after eliminating 189 (11%) cytotoxic and 36 (2.1%) fluorescent outlier compounds, 51 (3%) compounds inhibited IL-6-induced pSTAT3 activation by ≥50% (Table 4). The 51 HCS active compounds were cherry picked and rescreened in triplicate at 20 μM in both the IL-6-induced pSTAT3 and IFNγ-induced pSTAT1 assays that were exposed to compounds for either 1 h or 24 h before cytokine administration (Tables 4 and 5). Although 39 compounds (76.5%) were confirmed to inhibit IL-6-induced pSTAT3 activation by ≥50%, only three compounds (3.9%) also exhibited <30% inhibition of IFNγ-induced pSTAT1 activation at both compound exposures (Table 5): azelastine, etomidate and α-methyl-norepinephrine. Twenty (51%) of the 39 compounds that were confirmed to inhibit pSTAT3 and/or pSTAT1 activation elevate cellular cAMP levels either because they are Gs-coupled GPCR agonists49 (17 β-AR agonists, and 2 dopamine receptor 1 agonists) or they directly activate adenylate cyclase50 (e.g., forskolin; Table 5). Pre-exposure of Cal33 cells to 20 μM forskolin for 3 or 24 h inhibited the subsequent activation of both the pSTAT3 and pSTAT1 pathways by their respective cytokines (Fig. 6A, B). In contrast, exposure to the β-AR agonist isoproterenol for 24 h was less effective at inhibiting both pathways than a 3-h exposure (Fig. 6A, B). Eighteen of the Gs-coupled GPCR agonists that inhibited the pSTAT3 assay were active after 1 h of exposure compared with only 15 after 24 h of exposure (Table 5). In the pSTAT1 assay, 16 inhibited activation after 1 h of exposure compared with only 2 after 24 h of exposure (Table 5). After prolonged agonist exposures, GPCRs become desensitized, most commonly due to receptor phosphorylation and internalization.49,53,54 The apparent loss in efficacy of Gs-coupled GPCR agonists at inhibiting pSTAT3 and/or pSTAT1 activation after 24 h compound exposures would be consistent with receptor desensitization. The suppressors of cytokine signaling (SOCS) family of proteins are endogenous regulators of cytokine activation of JAK/STAT signaling pathways.55 SOCS3 blocks STAT3 and STAT1 signaling by one of three mechanisms: by binding to and directly inhibiting JAKs, by competing with the STATs for pY-binding sites on activated receptor chains, or by targeting bound signaling proteins for proteasomal degradation.55 In addition to being a STAT3 target gene and classical feedback inhibitor of STAT3 activation,55 SOCS3 expression levels are also regulated by STAT independent mechanisms.50,56–58 Agents that elevate intracellular cAMP levels up-regulate SOCS3 levels and inhibit STAT3 activation.50,56–58 Forskolin treatment of HUVEC cells elevated intracellular cAMP levels that led to the induction of SOCS3 and inhibition of IL-6-induced pSTAT3 activation.58 Forskolin treatment of the MDA-MB-231 breast cancer cell line elevated intracellular cAMP levels that inhibited leptin-induced pSTAT3 activation, an effect which was mediated through protein kinase A (PKA) activation.50 T47D breast cancer cells that were treated with forskolin or prostaglandin E2 (PGE2), an agonist of the Gs-coupled PGE2-receptor, activated the cAMP/PKA pathway and up-regulated SOCS3 expression which blocked lipopolysaccharide induced STAT3 activation.56Treatment of primary rat hepatocytes or transfected HepG2 cell with the α1B-adrenergic receptor agonist phenylephrine inhibited IL-6-induced STAT3 pathway activation.57 It is likely, therefore, that the nonselective inhibition of cytokine-induced JAK/STAT pathway activation by Gs-coupled GPCR agonists and forskolin was mediated by cAMP-dependent up-regulation of SOCS3. Since α-methyl-norepinephrine is an agonist of Gs-coupled adrenoreceptors, we did not pursue this compound further. In addition, a dry powder sample of etomidate that was purchased from commercial sources failed to inhibit either cytokine pathway in concentration response assays (Fig. 7).
In contrast, a dry powder sample of azelastine selectively inhibited the IL-6-induced pSTAT3-Y705 activation response with an IC50 of 26.56±10.42 μM (Figs. 7A, C and 8A, D) without affecting IFNγ-induced pSTAT1-Y705 activation, even at the highest concentration tested, 50 μM (Figs. 7B, D and 8B, D). Exposure of actively proliferating HNSCC cell lines to azelastine for 72 h inhibited their growth with IC50s of 13.62±7.5, 38.82±5.15, 33.18±11.8, and 23.77±2.15 μM for Cal33, 686LN, FaDu, and OSC19 cells, respectively (Fig. 8C, D). Intranasal formulations of azelastine hydrochloride ((±)-1-(2H)-phthalazinone,4-[(4-chlorophenyl)methyl]-2-(hexahydro-1methyl-1Hazepin-4-yl)-monhydrochloride) have been approved in the United States for the treatment of seasonal allergic rhinitis or nonallergic vasomotor rhinitis, and ophthalmic formulations for ocular conjunctivitis.59,60 Outside the United States, oral formulations are available for the treatment of allergic rhinitis, asthma, and urticaria.59,60 Azelastine is very well tolerated, and the most frequently reported adverse events that are either mild or moderate in the majority of cases are bitter taste, somnolence, and nasal burning.59,60 Azelastine is oxidatively metabolized by the cytochrome P450 (CYP450) system to an active metabolite desmethylazelastine and two inactive carboxylic-acid metabolites.59,60 Azelastine and desmethylazelastine were shown to bind to plasma protein at ∼88% and 97%, respectively.59,60 Intranasal azelastine has a systemic bioavailability of 40%, and Cmax levels are achieved in 2–3 h, while oral azelastine has an elimination half life of 22 h.59,60
Azelastine is a high-affinity histamine H1-receptor antagonist (Ki 6.4 nM) with some affinity for H2 receptors that blocks histamine release in mast cells and basophils, and inhibits the response of airway H1 receptors to histamine.59–61 In addition, in vitro studies have shown that azelastine causes a significant inhibition of the synthesis and release of leukotrienes (LTC4 and LTD4) from lung tissues, polymorphonuclear leukocytes, and eosinophils with IC50s in the 2–50-μM range, and blocks the leukotriene-induced contraction of guinea pig ileum with IC50s of 1–2 μM range.59,60,62 It has, therefore, been suggested that azelastine may also inhibit phospholipase A2 and leukotriene C4 synthase activity.59,60 Our cross-target query of the PubChem database revealed that although azelastine had been tested in 454 bioassays, there were only 12 (2.6%) active flags in the database (Fig. 8D), and the only confirmed (concentration dependent) active flag was for an assay to identify inhibitors/substrates of CYP450 3A4, a major liability for drug development. The PubChem data are consistent with published reports that azelastine is oxidatively metabolized by the CYP450 system,59,60 and they also suggest that azelastine has a selective biological activity profile. In vitro treatment of human peripheral blood lymphocytes and monocytes with 10 μM azelastine suppressed the mRNA and protein expression levels of pro-inflammatory cytokines by 33%; tumor necrosis factor alpha (TNFα), IL-1β, granulocyte-macrophage colony-stimulating factor, and IL-6.63 Azelastine treatment (10 μM) also suppressed reactive oxygen intermediate and nitric oxide production, and decreased total cellular protein tyrosine phopsphorylation levels.63 In vivo, azelastine causes decreases in circulating blood levels of IL-4, IL-8, and nitric oxide.59,60 The studies reported here demonstrate that azelastine selectively blocks the IL-6-induced activation of the oncogenic STAT3 signaling pathway in HNSCC cells without affecting the IFNγ-induced activation of the STAT1 tumor suppressor pathway, and that exposure of HNSCC cell lines to azelastine inhibits their growth (Figs. 7 and 8). However, given the relatively modest potency of azelastine at inhibiting STAT3 activation and HNSCC growth (∼25 μM), it is unlikely that it would be a candidate for drug repurposing as a cancer therapeutic.
In conclusion, we have described the development, optimization, and validation of pSTAT3 and pSTAT1 HCS assays to screen for selective inhibitors of STAT3 pathway activation in HNSCC cell lines. The IL-6-induced pSTAT3 HCS assay has the potential to discover inhibitors that block activation of oncogenic STAT3 signaling at any step between the engagement and activation of the IL-6 receptor complex to entry of pSTAT3-Y705 into the nucleus of Cal33 HNSCC cells. By incorporating the IFNγ-induced pSTAT1 HCS assay as a counter screen early in our testing paradigm, we can identify and de-prioritize nonselective inhibitors of STAT signaling that would also block STAT1 tumor suppressor function. A small pilot screen of 1,726 compounds from the LOPAC and NIH CC sets identified 51 compounds that inhibited pSTAT3 activation, but only 3 of these selectively inhibited STAT3 compared with STAT1 in our confirmation studies. Azelastine, an H1 receptor antagonist which was approved for the treatment of seasonal allergic rhinitis, nonallergic vasomotor rhinitis, and ocular conjunctivitis,59,60 was subsequently confirmed as a selective inhibitor of IL-6-induced pSTAT3 activation that also inhibited the growth of HNSCC cell lines. These data illustrate the power of a chemical biology approach to lead generation that utilizes fully developed and optimized HCS assays as phenotypic screens to interrogate specific signaling pathways. We have completed an HCS campaign of 100,000 compounds in this testing paradigm and are using the pSTAT3 and pSTAT1 HCS assays to support a lead an optimization effort for four selective pSTAT3 inhibitor series identified in the screen (manuscript in preparation).
Abbreviations
- β-AR
beta adrenoreceptor
- cAMP
cyclic adenosine monophosphate
- Ch 1 and 2
channels 1 and 2
- CYP450
cytochrome P450
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- dsODN
double-stranded oligodeoxynucleotide
- EGFR
epidermal growth factor receptor
- EP3
Evolution-P3
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- GPCRs
G-protein–coupled receptors
- Gs
G-alpha stimulatory subunit
- HCS
high content screening
- HNSCC
head and neck squamous cell carcinomas
- IC50
50% inhibition concentration
- IFNγ
interferon gamma
- IL-6
interleukin 6
- IXU
ImageXpress Ultra
- JAK
Janus kinase
- LOPAC
Library of Pharmacologically Active Compounds
- mAbs
monoclonal antibodies
- NIH CC
National Institutes of Health clinical collection
- PBS
phosphate-buffered saline
- PGE2
prostaglandin E2
- PKA
protein kinase A
- PMT
photomultiplier tube
- pSTAT3-Y701 or pSTAT1
STAT1 phosphorylated at tyrosine 701
- pSTAT3-Y705 or pSTAT3
STAT3 phosphorylated at tyrosine 705
- pY
phospho-tyrosine
- RTK
receptor tyrosine kinase
- S:B
signal-to-background ratio
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SFK
Src family kinase
- SFM
serum free tissue culture medium
- SH-2
Src homology 2
- SOCS3
suppressors of cytokine signaling 3
- STAT3
signal transducer and activator of transcription 3
- TE
translocation enhanced
- TGFα
transforming growth factor alpha
Acknowledgments
This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. NExT-CBC Project ID #1015, S08-221 Task Order 6 “STAT3 Pathway Inhibitor HCS” (Grandis, PI), NCI Chemical Biology Consortium, Pittsburgh Specialized Application Center (PSAC) (J.S.L. and P.A.J. co-PIs). The project was also supported in part by funds from the American Cancer Society (Grandis) and a Head and Neck Spore P50 award (Grandis, CA097190).
Disclosure Statement
The authors have no conflicts of interest to declare.
References
- 1.Frank DA: STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett 2007;251:199–210 [DOI] [PubMed] [Google Scholar]
- 2.Germain D, Frank DA: Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res 2007;13:5665–5669 [DOI] [PubMed] [Google Scholar]
- 3.Jing N, Tweardy DJ: Targeting Stat3 in cancer therapy. Anticancer Drugs 2005;16:601–607 [DOI] [PubMed] [Google Scholar]
- 4.Johnston PA, Grandis JR: STAT3 signaling: anticancer strategies and challenges. Mol Interv 2011;11:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quesnelle KM, Boehm AL, Grandis JR: STAT-mediated EGFR signaling in cancer. J Cell Biochem 2007;102:311–319 [DOI] [PubMed] [Google Scholar]
- 6.Bromberg JF, Wrzeszczynska MH, Devgan G, et al.: Stat3 as an oncogene. Cell 1999;98:295–303 [DOI] [PubMed] [Google Scholar]
- 7.Egloff AM, Grandis JR: Improving response rates to EGFR-targeted therapies for head and neck squamous cell carcinoma: Candidate predictive biomarkers and combination treatment with Src inhibitors. J Oncol 2009;2009:896407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leeman RJ, Lui VW, Grandis JR: STAT3 as a therapeutic target in head and neck cancer. Expert Opin Biol Ther March2006;6:231–241 [DOI] [PubMed] [Google Scholar]
- 9.Silva CM: Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004;23:8017–8023 [DOI] [PubMed] [Google Scholar]
- 10.Aggarwal BB, Kunnumakkara AB, Harikumar KB, et al.: Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship?. Ann NY Acad Sci 2009;1171:59–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seethala RR, Gooding WE, Handler PN, et al.: Immunohistochemical analysis of phosphotyrosine signal transducer and activator of transcription 3 and epidermal growth factor receptor autocrine signaling pathways in head and neck cancers and metastatic lymph nodes. Clin Cancer Res 2008;14:1303–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L: Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 1998;334:297–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F: Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 2003;374:1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ram PT, Iyengar R: G protein coupled receptor signaling through the Src and Stat3 pathway: role in proliferation and transformation. Oncogene 2001;20:1601–1606 [DOI] [PubMed] [Google Scholar]
- 15.Wilks AF: The JAK kinases; not just another kinase drug discovery target. Sem Cell Dev Biol 2008;19:319–328 [DOI] [PubMed] [Google Scholar]
- 16.Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS: Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res 2003;63:2948–2956 [PubMed] [Google Scholar]
- 17.Murray P: The JAK-STAT signaling pathway: input and output integration. J Immunol 2007;178:2632–2629 [DOI] [PubMed] [Google Scholar]
- 18.Zhong Z, Wen Z, Darnell JE, Jr: Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994;264:95–98 [DOI] [PubMed] [Google Scholar]
- 19.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD: Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995;267:1349–1353 [DOI] [PubMed] [Google Scholar]
- 20.Regis G, Pensa S, Boselli D, Novelli F, Poli V: Ups and downs: the STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin Cell Dev Biol 2008;19:351–359 [DOI] [PubMed] [Google Scholar]
- 21.Lui VW, Boehm AL, Koppikar P, et al.: Antiproliferative mechanisms of a transcription factor decoy targeting signal transducer and activator of transcription (STAT) 3: the role of STAT1. Mol Pharmacol 2007;71:1435–1443 [DOI] [PubMed] [Google Scholar]
- 22.Chen LF, Cohen EE, Grandis JR: New strategies in head and neck cancer: understanding resistance to epidermal growth factor receptor inhibitors. Clin Cancer Res 2010;16:2489–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santos FP, Kantarjian HM, Jain N, et al.: Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010;115:1131–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verstovsek S, Kantarjian H, Mesa RA, et al. : Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis N Engl J Med 2010;363:1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sen B, Saigal B, Parikh N, Gallick G, Johnson FM: Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res 2009;69:1958–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fletcher S, Drewry JA, Shahani VM, Page BD, Gunning PT: Molecular disruption of oncogenic signal transducer and activator of transcription 3 (STAT3) protein. Biochem Cell Biol 2009;87:825–833 [DOI] [PubMed] [Google Scholar]
- 27.Herrmann A, Vogt M, Mönnigmann M, et al.: Nucleocytoplasmic shuttling of persistently activated STAT3. J Cell Sci 2007;120:3249–3261 [DOI] [PubMed] [Google Scholar]
- 28.Liu L, McBride KM, Reich NC: STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sci USA 2005;102:8150–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boehm AL, Sen M, Seethala R, et al. : Combined targeting of epidermal growth factor receptor, signal transducer and activator of transcription-3, and Bcl-X(L) enhances antitumor effects in squamous cell carcinoma of the head and neck. Mol Pharmacol 2008;73:1632–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen M, Tosca PJ, Zwayer C, et al.: Lack of toxicity of a STAT3 decoy oligonucleotide. Cancer Chemother Pharmacol 2009;63:983–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sen M, Thomas SM, Kim S, et al.: First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov 2012;2:694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brockstein B: Management of recurrent head and neck cancer: recent progress and future directions. Drugs 2011;71:1551–1559 [DOI] [PubMed] [Google Scholar]
- 33.Goerner M, Seiwert TY, Sudhoff H: Molecular targeted therapies in head and neck cancer—an update of recent developments. Head Neck Oncol 2010;2:8–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stransky N, Egloff AM, Tward AD, et al.: The mutational landscape of head and neck squamous cell carcinoma. Science 2012;333:1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leeman-Neill RJ, Wheeler SE, Singh SV, et al.: Guggulsterone enhances head and neck cancer therapies via inhibition of signal transducer and activator of transcription-3. Carcinogenesis 2009;30:1848–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauer V, Hieber L, Schaeffner Q, et al.: Establishment and molecular cytogenetic characterization of a cell culture model of head and neck squamous cell carcinoma (HNSCC). Genes 2010;1:338–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gioanni J, Fischel JL, Lambert JC, et al.: Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: establishment, characterization and response to cytotoxic treatment. Eur J Cancer Clin Oncol 1988;24:1445–1455 [DOI] [PubMed] [Google Scholar]
- 38.Sturgis E, Sacks PG, Masui H, Mendelsohn J, Schantz SP: Effects of antiepidermal growth factor receptor antibody 528 on the proliferation and differentiation of head and neck cancer. Otolaryngol Head Neck Surg 1994;111:633–643 [DOI] [PubMed] [Google Scholar]
- 39.Rangan S: A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer 1972;29:117–121 [DOI] [PubMed] [Google Scholar]
- 40.Khan M, Yasuda M, Higashino F, et al.: nm23-H1 suppresses invasion of oral squamous cell carcinoma-derived cell lines without modifying matrix metalloproteinase-2 and matrix metalloproteinase-9 expression. Am J Pathol 2001;158:1785–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudgeon D, Shinde SN, Shun TY, et al.: Characterization and optimization of a novel protein-protein interaction biosensor HCS assay to identify disruptors of the interactions between p53 and hDM2. Assay Drug Dev Technol 2010;8:437–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dudgeon D, Shinde SN, Hua Y, et al.: Implementation of a 220,000 compound HCS campaign to identify disruptors of the interaction between p53 and hDM2, and characterization of the confirmed hits. J Biomol Screen 2010;15:152–174 [DOI] [PubMed] [Google Scholar]
- 43.Johnston PA, Shinde SN, Hua Y, Shun TY, Lazo JS, Day BW: Development and validation of a high-content screening assay to identify inhibitors of cytoplasmic Dynein-mediated transport of glucocorticoid receptor to the nucleus. Assay Drug Dev Technol 2012;10:432–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedranzini L, Dechow T, Berishaj M, et al.: Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res 2006;66:9714–9721 [DOI] [PubMed] [Google Scholar]
- 45.Shun TY, Lazo JS, Sharlow ER, Johnston PA: Identifying actives from HTS data sets: practical approaches for the selection of an appropriate HTS data-processing method and quality control review. J Biomol Screen 2011;16:1–14 [DOI] [PubMed] [Google Scholar]
- 46.Nickischer D, Laethem C, Trask OJ Jr, et al.: Development and implementation of three mitogen-activated protein kinase (MAPK) signaling pathway imaging assays to provide MAPK module selectivity profiling for kinase inhibitors: MK2-EGFP translocation, c-Jun, and ERK activation. Methods Enzymol 2006;414:389–418 [DOI] [PubMed] [Google Scholar]
- 47.Trask OJ, Jr, Baker A, Williams RG, et al.: Assay development and case history of a 32K-biased library high-content MK2-EGFP translocation screen to identify p38 mitogen-activated protein kinase inhibitors on the ArrayScan 3.1 imaging platform. Methods Enzymol 2006;414:419–439 [DOI] [PubMed] [Google Scholar]
- 48.Williams RG, Kandasamy R, Nickischer D, et al. : Generation and characterization of a stable MK2-EGFP cell line and subsequent development of a high-content imaging assay on the Cellomics ArrayScan platform to screen for p38 mitogen-activated protein kinase inhibitors. Methods Enzymol 2006;414:364–389 [DOI] [PubMed] [Google Scholar]
- 49.Vasudevan N, Mohan ML, Goswami SK, Naga Prasad SV: Regulation of β-adrenergic receptor function: an emphasis on receptor resensitization. Cell Cycle 2011;10:3684–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naviglio S, Di Gesto D, Illiano F, et al.: Leptin potentiates antiproliferative action of cAMP elevation via protein kinase A down-regulation in breast cancer cells. J Cell Physiol 2010;225:801–809 [DOI] [PubMed] [Google Scholar]
- 51.Trask O, Nickischer D, Burton A, et al.: High-throughput automated confocal microscopy imaging screen of a kinase-focused library to identify p38 mitogen-activated protein kinase inhibitors using the GE InCell 3000 analyzer. Methods Mol Biol 2009;565:159–186 [DOI] [PubMed] [Google Scholar]
- 52.Zaida M, Merlino G: The two faces of Interferon-gamma in Cancer. Clin Cancer Res 2011;17:6118–6124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Charlton S: Agonist efficacy and receptor desensitization: from partial truths to a fuller picture. Br J Pharmacol 2009;158:165–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelly E, Bailey CP, Henderson G: Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol 2008;153:5379–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Croker BA, Kiu H, Nicholson SE: SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol 2008;19:414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barclay J, Anderson ST, Waters MJ, Curlewis JD: Characterization of the SOCS3 promoter response to prostaglandin E2 in T47D cells. Mol Endocrinol 2007;21:2516–2528 [DOI] [PubMed] [Google Scholar]
- 57.Nguyen V, Gao B: Cross-talk between alpha(1B)-adrenergic receptor (alpha(1B)AR) and interleukin-6 (IL-6) signaling pathways. Activation of alpha(1b)AR inhibits il-6-activated STAT3 in hepatic cells by a p42/44 mitogen-activated protein kinase-dependent mechanism. J Biol Chem 1999;274:35492–35498 [DOI] [PubMed] [Google Scholar]
- 58.Woolson HD, Thomson VS, Rutherford C, Yarwood SJ, Palmer TM: Selective inhibition of cytokine-activated extracellular signal-regulated kinase by cyclic AMP via Epac1-dependent induction of suppressor of cytokine signalling-3. Cell Signal 2009;21:1706–1715 [DOI] [PubMed] [Google Scholar]
- 59.Bernstein J: Azelastine hydrochloride: a review of pharmacology, pharmacokinetics, clinical efficacy and tolerability. Curr Med Res Opin 2007;23:2441–2452 [DOI] [PubMed] [Google Scholar]
- 60.Lee C, Corren J: Review of azelastine nasal spray in the treatment of allergic and non-allergic rhinitis. Expert Opin Pharmacother 2007;8:701–709 [DOI] [PubMed] [Google Scholar]
- 61.Kubo N, Shirakawa O, Kuno T, Tanaka C: Antimuscarinic effects of antihistamines: quantitative evaluation by receptor-binding assay. Jpn J Pharmacol 1987;43:277–282 [DOI] [PubMed] [Google Scholar]
- 62.Katayama S, Tsunoda H, Sakuma Y, Kai H, Tanaka I, Katayama K: Effect of azelastine on the release and action of leukotriene C4 and D4. Int Arch Allergy Appl Immunol 1987;83:284–289 [DOI] [PubMed] [Google Scholar]
- 63.Yoneda K, Yamamoto T, Ueta E, Osaki T: Suppression by azelastine hydrochloride of NF-kappa B activation involved in generation of cytokines and nitric oxide. Jpn J Pharmacol 1997;73:145–153 [DOI] [PubMed] [Google Scholar]