

Abstract
Purpose.
The purpose of this study was to analyze the neuroprotective effect of an α7 nAChR agonist, PNU-282987, using an in vivo model of glaucoma in Long Evans rats.
Methods.
One eye in each animal was surgically manipulated to induce glaucoma in control untreated animals and in animals that were treated with intravitreal injections of PNU-282987. To induce glaucoma-like conditions, 0.05 mL of 2 M NaCl was injected into the episcleral veins of right eyes in each rat to create scar tissue and increase intraocular pressure. The left eye in each rat acted as an internal control. One month following NaCl injection, rats were euthanized, retinas were removed, flatmounted, fixed, and nuclei were stained with cresyl violet or RGCs were immunostained with an antibody against Thy 1.1 or against Brn3a. Stained nuclei in the RGC layer and labeled RGCs in NaCl-injected retinas were counted and compared with cell counts from untreated retinas in the same animal.
Results.
NaCl injections into the episcleral veins caused a significant loss of cells by an average of 27.35% (±2.12 SEM) in the RGC layer within 1 month after NaCl injection, which corresponded to a significant loss of RGCs. This loss of RGCs was eliminated if 5 μL of 100 μM PNU-282987 was injected into the right eye an hour before NaCl injection.
Conclusions.
The results from this study support the hypothesis that the α7 agonist, PNU-282987, has a neuroprotective effect in the rat retina. PNU-282987 may be a viable candidate for future therapeutic treatments of glaucoma.
Keywords: neuroprotection, glaucoma, retinal ganglion cells
In this study, the neuroprotective effect of an alpha7 nAChR agonist, PNU-282987, was used in an in vivo rat model of glaucoma. Intravitreal injections of PNU-282987 prevented loss of RGCs normally associated with NaCl injections designed to induce glaucoma-like conditions.
Introduction
Glaucoma is a degenerative retinal disease and is characterized by optic neuropathy and cupping of the optic disk, degeneration of retinal ganglion cells (RGCs), and eventual visual field loss. In several in vitro models of glaucoma, death of RGCs is induced by excessive neurotransmitters that trigger excitotoxicity and apoptosis.1–6 Recently, a great deal of research has explored agents and mechanisms that provide neuroprotection against excitotoxicity.7–9 In previous studies from this lab, acetylcholine (ACh) and nicotine have been found to be neuroprotective in the retina and act to prevent glutamate-induced excitotoxicity in an isolated cultured pig and rat RGC preparation.10,11 Other pharmacological studies using pig- and rat-cultured RGCs have demonstrated that activation of specific α7 nAChRs are linked to this neuroprotective effect.11,12 However, it is unclear if activation of α7 nAChRs have any significant neuroprotective role against RGC loss normally associated with glaucoma. To address this issue, the α7 nicotinic agonist, PNU-282987, was analyzed in an in vivo rat model of glaucoma to test the hypothesis that introduction of the α7 nicotinic agonist, PNU-282987, can significantly reduce the loss of cells in the RGC layer that is normally associated with glaucoma.
Previous studies from our lab have implicated α7 nAChRs on RGCs as a potential target for neuroprotection in the retina and RGCs against glutamate-induced excititoxicity.10,12–14 In these in vitro studies, RGCs from pig or rat11 were isolated from all other retinal tissue and cultured to demonstrate glutamate-induced excitotoxicity and ACh and nicotine-induced neuroprotection. Evidence was provided that excitotoxicity, induced with 500 μM glutamate, was significantly reduced in a dose-dependent manner if relatively low concentrations of ACh or nicotine were applied to isolated cells before glutamate. When the α7 receptor nicotinic antagonists, α-Bgt or MLA, were applied before ACh or nicotine, the neuroprotective effect of ACh and nicotine was significantly reduced.
Further in vitro studies using the α7 nAChR agonist PNU-282987 on isolated rat RGCs support the hypothesis that neuroprotection against glutamate-induced excitotoxicity is mediated through α7 nAChRs.11 Binding studies with rat chimera cells and electrophysiology studies in rats have demonstrated that PNU-282987 is a potent specific agonist for α7 nAChRs.15 These binding studies have shown that PNU-282987 can displace MLA, an α7 antagonist. Using rat hippocampal neurons, application of the PNU compounds cause currents through α7-nAChR channels in a dose-dependent matter. This effect was blocked with MLA.15 As a result of these previous studies, PNU-282987 was used in this study to understand the role of the α7-nACh receptor as a neuroprotective agent against loss of RGCs in a glaucomatous rat model without worrying about cross-reactivity or cross-binding with other nAChRs that may make the analysis and interpretation of data difficult.
The method used to induce glaucoma in Long Evans rats is based on a method of injecting 2 M hypertonic saline into the episcleral veins of the eye.16 The 2 M salt solution causes scarring in the trabecular meshwork and decreases the outflow of aqueous humor. This results in a gradual increase of intraocular pressure and loss of RGCs to mimic glaucoma-like conditions in the retina.16 In this study, this model of glaucoma was used to demonstrate the neuroprotective effect of PNU-282987 to prevent the loss of RGCs normally associated with the model. The results from this study open up possibilities for a new treatment for glaucoma that does not solely deal with treating IOP, which is the sole target for all current glaucoma treatments. Using α7-nAChR agonists in conjunction with already existing treatments could provide an additional therapeutic benefit or could lead to preventative care in patients at high risk for developing glaucoma.
Materials and Methods
Adult male and female Long Evans rats aged 3 months (breeding colony, WMU) were used for these in vivo experiments as this particular outbred strain was also used in previous in vitro studies performed in this lab.11 Each animal was kept in the animal colony prior to experiments. All animals were treated and eventually euthanized according to IACUC protocols. Animal treatment was in adherence to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Inducing Glaucoma
Before injecting NaCl into the episcleral veins to induce glaucoma-like conditions, adult Long Evans rats were anesthetized with 0.1 mL/100 g KAX via intraperitoneal injections until no reflexes were observed. KAX is a combination cocktail consisting of 5 mL ketamine (100 mg/mL; DVM, Dallas, TX); 2.5 mL xylazine (20 mg/mL; Sigma-Aldrich, St. Louis, MO); 1 mL acepromazine (10 mg/mL; Vet Depot, Fort Payne, AL); and 0.5 mL sterile water. A topical anesthetic of procaine hydrochloride was applied to the cornea before any NaCl injection into the episcleral veins. To expose the targeted episcleral veins and to restrict movement of the eye, a hemostat was used to pinch the bottom eyelid, which caused the eye and corresponding episcleral veins to bulge out of the eye socket to expose the episcleral veins. The conjunctiva was incised with fine scissors to further expose the vein, under a dissecting microscope. To inject 50 μL of 2 M hypertonic saline into the right episcleral veins, a 3-mm long glass microneedle, 3 to 5 um in diameter, was attached to a piece of tapered polyethylene tubing. The microneedle was pulled using a vertical electrode puller (NARISHIGE Group; Tokyo, Japan), beveled and inserted into a 23-gauge needle with the tip filed off, and attached to tubing attached to a 1 mL syringe. When the salt was injected into the episcleral veins, the veins blanched as the saline was distributed through the circulatory system. This was taken as confirmation that the injection of 2 M NaCl occurred. Visualization of blanching in the episcleral veins correlated directly with significant loss of cells from the RGC layer after 1 month and an increase of intraocular pressure (IOP). The rats were kept under observation until fully awake before they were transported back to the animal colony. The injections of 2 M hypertonic saline were typically performed in the right eye of most rats. The left eye typically acted as an internal control.
IOP Measurements Using Tonometer
IOP measurements were obtained before NaCl injections to obtain a baseline and after NaCl injections to verify an increase of IOP as a result of hypertonic injection. IOP measurements were obtained from awake behaving rats using a tonometer (Tono-Pen XL; Mentor O & O, Inc., Norwell, MA) according to instructions outlined by Morrison et al.16 Rat eyes were anesthetized with 1 drop of 0.5% proparacaine hydrochloride into each eye before using the tonometer (Mentor O & O, Inc.). Rats were loosely held in the experimenter's hand during this procedure and were rewarded with a piece of cereal (Cheerios; General Mills, Minneapolis, MN) after obtaining measurements. IOP measurements were obtained each day for 1 week before NaCl injection and five times each week after NaCl injection until the animals were killed.
Intravitreal Injection of Pharmacological Agents
For these studies, drugs were injected intravitreally into the eye's vitreous cavity in volumes of 5 μL using a syringe (Hamilton Company, Reno, NV). The injection of various concentrations of pharmacological agents included the α7 nicotinic agonist PNU-282987 (Sigma-Aldrich), or the α7 antagonist MLA (Tocris Bioscience, Minneapolis, MN). These sterile agents were injected into the right eyes of animals before NaCl injection that normally induced glaucoma-like conditions. Based on results obtained from preliminary studies and from in vitro results, intravitreal injections of PNU-282987 were applied 1 hour before NaCl injection to obtain maximal effects. For inhibition studies, injections of MLA were applied 1 hour before intravitreal injection of PNU-282987 and 2 hours before NaCl injection into the episcleral veins.
Labeling Cells in the RGC Layer
One month following intravitreal treatment and/or NaCl injection into episcleral veins, rats were euthanized by CO2 asphyxiation and the retinas were removed after forming an eyecup and after introducing a notch into the retina that was used to maintain retinal orientation. Four incisions every 90° were made into the retinas in order to flatmount the retinas onto small sylgard plates using cactus needles. This produced four different retinal quadrants. One quadrant represented the dorsal retina containing the highest density of RGCs in the visual streak, while the other quadrants represented ventral, nasal, and temporal regions of the retina (Fig. 1). The pinned out retinas were then fixed with 10% formalin overnight at 4°C. Once fixed, some retinas were rinsed with PBS and transferred to a pig skin gelatin-coated glass slide and cresyl violet was used to stain cell nuclei according to standard procedures.
Other retinas were processed to directly label the glycoprotein Thy 1.1, found only in the plasma membrane of RGCs in the retina,17 or by using the RGC specific nuclear marker Brn3a.18,19 Once the retinas were flatmounted and fixed overnight, the following day the retinas were rinsed with PBS, incubated in 2% BSA in PBS containing 0.02% saponin for 30 minutes at room temperature to block non-specific binding, then incubated in mouse anti-rat monoclonal antibody against Thy 1.1 (1:300; Millipore Corp., Billerica, MA) or in rabbit anti-mouse polyclonal antibody against Brn3a (1:800; Millipore Corp.) for 1 week at 4°C in PBS containing 0.02% saponin. After 1 week, the retinas were rinsed three times with PBS, then incubated in AlexaFluor 594 (Life Technologies, Grand Island, NY) secondary goat anti-mouse antibody (1:300) to visualize the Thy 1.1 glycoprotein, or in AlexaFluor 594 secondary donkey anti-rabbit (1:300; Life Technologies) for visualization of Brn3a. Secondary antibodies were applied for 5 days at 4°C. Following incubation in secondary antibody, the retinas were transferred to glass slides for viewing and imaging.
In control studies, experiments were conducted to display specificity of the antibodies used. In some negative control experiments, large RGCs were processed with the primary antibodies omitted, while other experiments substituted nonimmune mouse immunoglobulin (dilution: 0.1–1.0 μg/mL) for the antibodies. In other experiments, preabsorption controls were performed where the primary antibody and antigens were added together before applying to tissue. No significant epifluorescence was observed under any of these conditions.
Once mounted, the stained tissues were imaged throughout the ganglion cell layer in each of the four defined retinal quadrants using 1-μm increments using the capabilities of a Zeiss confocal microscope with a rhodamine filter using the optic nerve head and the orientation of the retina. In each quadrant, one series of images were obtained from the retina 4 mm from the optic nerve head (ONH). For some studies, a second series of images were obtained 2 mm from the ONH. The cell bodies in the RGC layer stained with cresyl violet were opaque and could easily be counted. The process produced good contrast between cell nuclei and background, allowing for easy quantification. For RGCs that were immunostained with a fluorescently labeled antibody against Thy 1.1, fluorescence was observed in the plasma membrane of the RGCs as well as in the RGC axons. Anti-Brn3a stained RGC nuclei.
When analyzing RGCs labeled with anti-Thy 1.1, quantification of RGCs from each quadrant was formulated using a 100-μm2 frame. The position of this frame was placed to avoid axon fascicle interference. Preliminary studies used multiple placements of the 100-μm2 frame to determine how reproducible the quantification method was. There were significant differences in RGC counts if only two or three frames were used to quantify RGCs. However, if four, six or eight frames were evenly distributed around the ONH 4 mm from the ONH, the average RGC counts were significantly similar. As a result, a series of images were obtained from each of the four retinal quadrants 4 mm or 2 mm from the ONH for RGC quantification and counts were averaged. For quantification of RGC nuclei labeled with anti-Brn3a, axon fascicles were not labeled and each quadrant could be analyzed using a 2-mm2 frame in the four retinal quadrants 4 mm from the ONH. RGCs were counted using image analysis software (MetaMorph; Molecular Devices, LLC, Sunnyvale, CA), averaged, and compared with internal controls.
To validate this counting method, a blind study was performed using six different student participants. Students were asked to blind count the number of RGCs labeled with the antibody against Thy 1.1 or with the antibody against Brn3a using the procedure described above. Each student counted similar numbers of RGCs (n = 20 retinas). There was no significant difference in RGC counts when students counted cells labeled with anti-Thy 1.1 or when cells were labeled with anti-Brn3a.
LC/MS/MS Analysis
Liquid chromatography-quadrupole mass spectrometry (LC/MS/MS) was performed on retina removed from euthanized Long Evans rats at various time points following intravitreal injections of three different concentrations of PNU-282987. Specifically, retinas were removed from euthanized Long Evans rats 1, 2, 4, 8, and 12 hours after injecting 5 μL of 10 μM, 100 μM, or 1 mM PNU-282987 into the rat's vitreous cavity. These time intervals corresponded to those that were done in the rabbit retina with the same compound (Linn DM, et al. IOVS 2011:ARVO E-Abstract 3237). Removed retinas were rinsed to remove any residual PNU-282987, weighed, and sent to the Southwest Michigan Innovation Center (Kalamazoo, MI) for LC/MSMS detection of PNU-282987. Analysis was performed using standard procedures on a mass spectrometer (Micromass Quattro Micro triple quadrupole mass spectrometer; Waters Corp., Milford, MA) using positive ion electrospray ionization. A capillary HPLC (CapLC System; Waters Corp.) was configured for online SPE.
Data Analysis
All cell counts were compared with the internal control counts for each experiment. Student's t-tests were used for single comparisons, and one-way ANOVA was used for multiple comparisons to statistically measure cellular loss with Tukey's post hoc tests using statistical software (Prism GraphPad version 4.0; GraphPad Software Inc., San Diego, CA) and a statistics package (Minitab; Pennsylvania State University, University Park, Pennsylvania). For normalized data, statistical analysis was performed using Kruskall-Wallis nonparametric analysis of variance with post hoc comparisons (Dunn's test). A P value <0.05 represented significance. Graphs were plotted with statistical software (GraphPad Software, Inc.).
Results
Flatmounted Retina
Figure 1 demonstrates where images were obtained from flatmounted retinas. The retina displayed in Figure 1 was removed from a rat eyecup and positioned in a sylgard dish with the RGC layer facing up. The retina was then orientated with the visual streak in the dorsal quadrant and four quadrants were produced. Images were obtained 2 mm (white boxes) and 4 mm (yellow boxes) from the optic nerve head in the dorsal, nasal, temporal, and ventral quadrants. Great care was taken to maintain the orientation of the retina throughout the imaging process.
Figure 1.

Flatmount retina. One month following NaCl injection, the retina was removed and flatmounted onto a sylgard dish using cactus needles with the RGC layer facing up. The yellow boxes represent the areas of the retina where RGCs were counted (4 mm from the ONH), while the white boxes represent a distance of 2 mm from the ONH. The dotted lines represent retinal orientation maintained throughout the experiments. d, dorsal; n, nasal; t, temporal; v, ventral.
Cell Loss in the RGC Layer
The image in Figure 2 was obtained after a flatmounted retina was processed with cresyl violet. Under the Zeiss confocal microscope, all nuclei in cresyl violet–stained cells appeared opaque. The image in Figure 2A was obtained from the left eye of a rat, 4 mm from the ONH in the nasal quadrant. This eye was untreated and acted as an internal control. The image in Figure 2B was obtained from the right eye of the same rat, 4 mm from the ONH from the same region of the retina one month after 50 μL of 2M NaCl was injected into the animal's episcleral veins. As seen in Figure 2B, there are fewer stained nuclei in the RGC layer compared with the internal control (Fig. 2A).
Figure 2.

Visualization of cell loss from RGC layer. The images in Figure 2 were taken 4 mm from the ONH and represent cells in the RGC layer stained with cresyl violet. (A) Image obtained from the left control eye. (B) Image obtained 1 month after an injection of hypertonic saline. Both images were taken from the same rat, from the equivalent retinal region and from the same depth in the RGC layer. Single-headed arrows indicate stained nuclei in the RGC layer, while double-headed arrows point to defined axon tracks.
The number of stained cells in the RGC layer were counted at 2- and 4-mm distances from the ONH in each of the four quadrants and compared with internal controls. The bar graphs in Figure 3 summarize the results of these experiments. Each bar graph represents the average number of stained cell bodies counted 4 mm from ONH and 2 mm from ONH throughout the RGC layer. The part of the retina 4 mm from the ONH had an average cell loss of 27.35% (±2.12 SEM; n = 12) from the RGC layer 1 month after injecting 2 M NaCl hypertonic saline (Fig. 3, left bar), compared with the retina 2 mm from the ONH, which demonstrated an average of 20.01% (±5.21 SEM; n = 12) cell loss (Fig. 3, right bar). As more cell loss in the periphery is typically associated with glaucoma, all other experiments counted cells 4 mm from the ONH.
Figure 3.
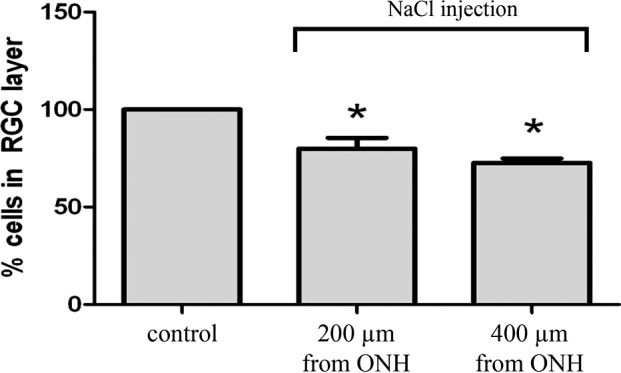
Percent cell loss associated with glaucoma-inducing procedure. This graph depicts the percent survival of cells in the RGC layer at 200 and 4 mm from the ONH 1 month after the episcleral veins were injected with hypertonic saline. Each bar was produced using between 6 and 12 rat eyes. Asterisk represents significance from internal untreated controls. Error bars represent SEM.
The bar graphs in Figure 4 demonstrate that there is significant cell loss from the RGC layer 1 month following NaCl injection in Long Evans rats. Each bar in the figure represents the average cell survival compared with the internal control retinas when animals were euthanized 1 and 2 weeks, and 1, 2, and 4 months following the hypertonic injections. At the end of each time interval, the animals were euthanized and the retinas were removed, flatmounted, fixed, and stained with cresyl violet. As can be seen by the summarized results in Figure 4, significant loss of cells 4 mm from the ONH in the RGC layer occurred 1 month following the NaCl injection and there was no additional significant difference in the loss of cells at 2 and 4 months compared to 1 month results. Though longer studies were done at 5 and 6 months, (data not shown), at 1 month, hypertonic injections of saline into the episcleral vein always caused a significant amount of cell loss in the RGC layer, ranging between 18% and 27% if blanching occurred throughout the episcleral veins following the injection. Based on these results, all rats in subsequent glaucoma-inducing experiments were killed 1 month following NaCl injection and cell counts were obtained 4 mm from the ONH.
Figure 4.
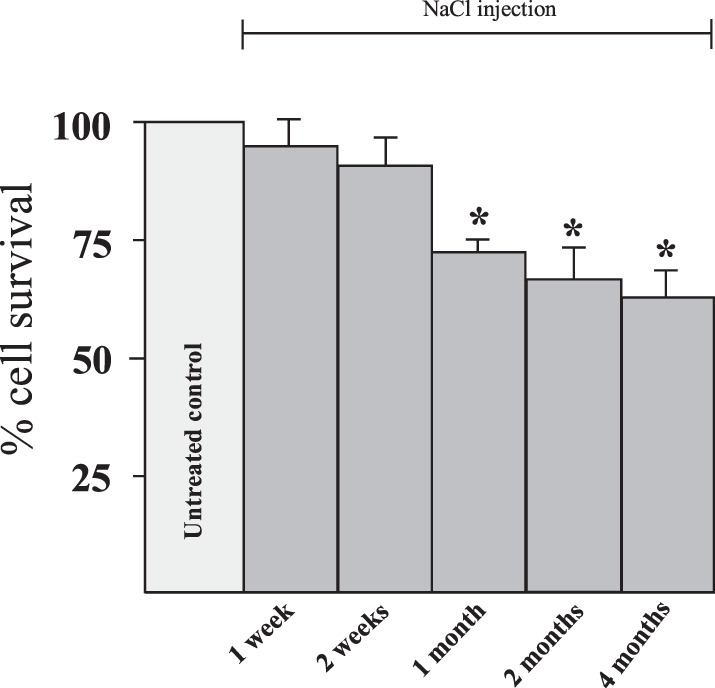
Time-dependent effects following hypertonic injections. Animals were euthanized at different time points after NaCl injection and the loss of cells in the RGC layer were quantified in a time-dependent manner. Each bar graph represents the average number of cell bodies stained with cresyl violet that were counted in the RGC layer after inducing glaucoma-like conditions with hypertonic saline injections. Asterisk indicates significance from the untreated control. Each bar graph was generated from between 6 to 12 animals. Error bars represent SEM.
Labeling RGCs In Vivo
Although careful consideration was given to cell counts from specific regions in the RGC layer in vivo, cresyl violet stains all cell bodies in the RGC layer. As displaced amacrine cells are also found in the retinal ganglion cell layer,20,21 it is unlikely that cell counts obtained with cresyl violet only represent the RGC population. To determine what percentage of cells in the RGC layer are represented by RGCs in these experiments, some retinas were immunostained with an antibody against Thy 1.1. Experimental rat eyes injected with hypertonic saline were euthanized after 1 month and the retinas were removed, pinned on sylgard plates, and fixed. The flatmounted retinas were then incubated with a monoclonal antibody against Thy 1.1 and secondarily labeled with AlexaFluor 594 goat anti-mouse IgG. Figure 5 represents images obtained 4 mm from the ONH from the temporal quadrant after RGCs were stained with the Thy 1.1 antibody. Figure 5A is an image obtained from an untreated control retina. An average of 8.22% (±2.21) of cells in images obtained 4 mm from the ONH in the RGC layer did not label with the primary antibody (n = 25).
Figure 5.
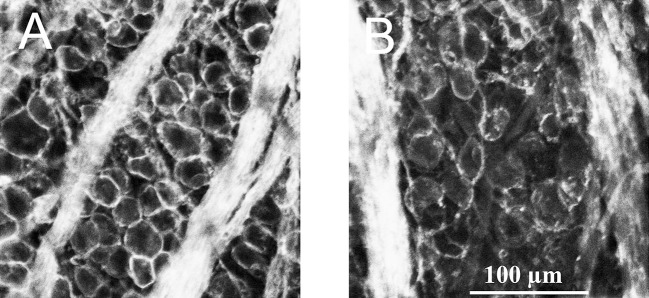
Fluorescent labeling of RGCs. (A) Represents a left eye image of RGCs immunostained with an antibody against Thy 1.1 under control untreated conditions 4 mm from the ONH. (B) Represents immunostained RGCs from the right eye of the same rat, from an equivalent position, 1 month after NaCl injection into the episcleral vein.
Figure 5B was obtained from the same animal's right eye from the same temporal retinal location 1 month after NaCl injection. As shown in this image, the hypertonic saline injection resulted in fewer RGCs and less defined axon tracks compared with the internal control. There was an average significant loss of 28.12% (±3.2, n = 8) of RGCs compared with the internal control. Based on cresyl violet and anti-Thy 1.1 results, the loss of cells in the RGC layer due to hypertonic injections of NaCl into the rat's episcleral veins are hypothesized to be primarily RGCs.
Although it is likely that Thy 1.1-labeled RGCs in the RGC layer, other studies have demonstrated that Thy 1.1 expression can be downregulated by RGC alterations without actual RGC death.22,23 To address this issue, glaucoma-inducing experiments were repeated using the specific RGC nuclear marker Brn3a18,19 to determine whether results obtained using the RGC nuclear marker coincide with results obtained using Thy 1.1. The Brn3 family of POU-domain transcription factors has been shown to play important roles in differentiation, survival, and axonal elongation24 and have emerged as a reliable marker for RGCs in mice and rats.25,26
Figure 6 illustrates a typical example obtained when the nuclear marker, Brn3a, was used to label RGCs. In this example, the left eye was untreated and the right eye was injected with 2 M hypertonic saline to induce glaucoma-like conditions. One month following NaCl injection, the animal was killed, retinas were removed while maintaining eye orientation and flatmounted for processing with anti-Brn3a. Figure 6 (left panel) represents an image obtained from an untreated control retina 4 mm from the ONH from the nasal quadrant, while Figure 6 (right panel) represents an image obtained from the same region in the right eye 1 month after NaCl injection. As shown in this image, the hypertonic saline injection resulted in significantly fewer RGCs. When labeled with an antibody against Brn3a, there was an average significant loss of 25.82% (±5.1, n = 5) of RGCs compared with the internal control. This percent loss is statistically similar to the 28.12% (±3.2) loss of RGCs measured when anti-Thy 1.1 was used to label RGCs and supports the hypothesis that both Thy 1.1 and Brn3a labeled a similar number of RGCs in the rat retina.
Figure 6.

Labeling of RGCs with anti-Brn3a. Left panel represents a left eye image of RGCs immunostained with an antibody against Brn3a under control untreated conditions 4 mm from the ONH. Right panel represents immunostained RGCs from the right eye of the same rat, from an equivalent position, 1 month after NaCl injection into the episcleral vein. Arrows are pointed to Brn3a-labeled cells.
An α7 nAChR Agonist Prevents Loss of RGCs
Previous studies from this laboratory using an in vitro excitotoxic model on isolated rat RGCs have demonstrated that the α7 nAChR agonist, PNU-282987, prevents the loss of RGCs against glutamate-induced excitotoxity.11 The next experiments were designed to determine if PNU-282987 provided neuroprotection against loss of RGCs in the in vivo rat model. For these studies, various concentrations of PNU-282987 were injected into the vitreous cavity of anesthetized rats after standard LC/MSMS procedures were conducted to verify that PNU-282987 could be detected in the retina after intravitreal injection. For the LC/MSMS studies, retinas were removed from euthanized Long Evans rats 1, 2, 4, 8, and 12 hours after intravitreal injections containing 5 μL of 10 μM, 100 μM, or 1 mM PNU-282987. Removed retinas were rinsed of any residual PNU-282987, weighed, and analyzed for LC/MSMS detection of PNU-282987.
Results from the LC/MSMS studies demonstrated that all three doses of PNU-282987 could be detected in the retinas in a dose-dependent manner. The results of these experiments are summarized in Figure 7. The highest amount of PNU-282987 detected in the retina was recorded 2 hours after 100 μM PNU-282987 was injected into the vitreous. Two hours after application, LC/MSMS detected 3.1 ng PNU/g (±0.65) in the retina after 10 μM was injected, 5.25 ng PNU/g (±0.48) after 100 μM was injected and 4.81 ng PNU/g (±0.48) after 1 mM was injected. Each of these values obtained 2 hours after intravitreal injection was significantly different from all other time intervals for each particular concentration. PNU-282987 detection was the lowest at 12 hours.
Figure 7.
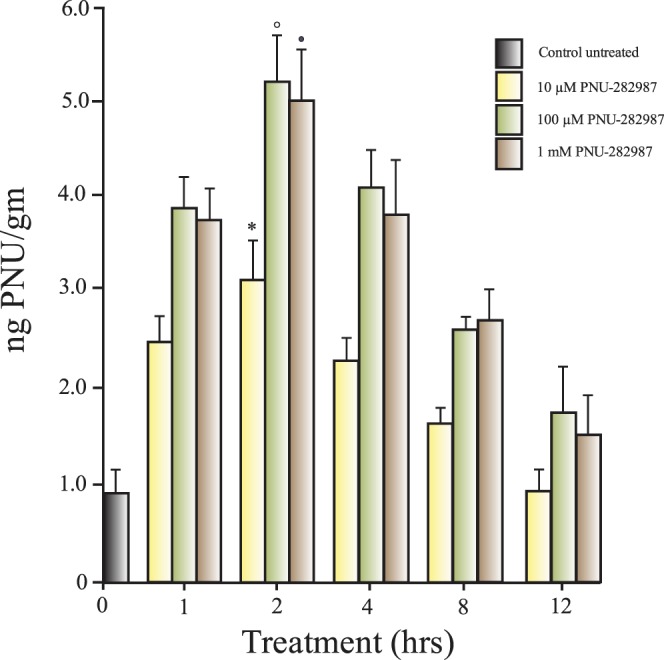
PNU-282987 detection in the rat retina. Retinas were removed from animals after intravitreal injections of three different concentrations of PNU-282987. Retinas were removed after different periods of time following intravitreal injection of the α7 agonist. Each bar represents the amount of PNU-282987 detected (ng PNU/g wet tissue) under each condition. The open circle, solid circle, and star indicate significance from all other time points using the same PNU-282987 concentration. Each bar graph was generated from three retinas. Error bars represent SEM.
After providing evidence that PNU-282987 can be detected in the rat retina after intravitreal injection, the next experiments were designed to determine if PNU-282987 provided neuroprotection against loss of RGCs using the in vivo rat glaucoma model. The right eye of each experimental rat was intravitreally injected with 5 μL of 10 μM, 100 μM, or 1 mM PNU-282987 1 hour before NaCl injection. Previous in vitro studies,10,11 as well as preliminary in vivo time-dependent studies, demonstrated that neuroprotective agents had to be applied 1 to 2 hours before application of excessive glutamate or NaCl injection into the episcleral veins for neuroprotection to occur.
Figure 8 represents sample images obtained from one of these experiments, where 5 μL of 100 μM PNU-282987 was injected into the vitreous humor 1 hour before NaCl injection. Figure 8A represents an image obtained from the untreated left internal control eye 4 mm from the ONH in the temporal quadrant, while Figure 8B represents an image obtained from the right eye from the same rat and from the same retinal region, where PNU-282987 was intravitreally injected before NaCl injection into the episcleral veins. One month following NaCl injection, eyes were harvested, processed for Thy 1.1 labeling, and RGC counts were compared to the internal control eyes. As shown in Figure 8, intravitreal injection of 100 μM PNU-282987 into the vitreous cavity prevented the loss of RGCs that normally occur due to the NaCl injection. Studies performed using Brn3a to label RGC nuclei demonstrated similar results. If retinas were injected with 100 μM PNU-282987 before hypertonic injection into the episcleral veins to induce glaucoma-like conditions, the typical loss of RGC nuclei associated with the hypertonic saline injections were significantly reduced by an average of 87.3% (±5.6; n = 3). This is significantly similar to the neuroprotective effect recorded when PNU-282987 was intravitreally injected and cells were labeled with anti-Thy 1.1.
Figure 8.
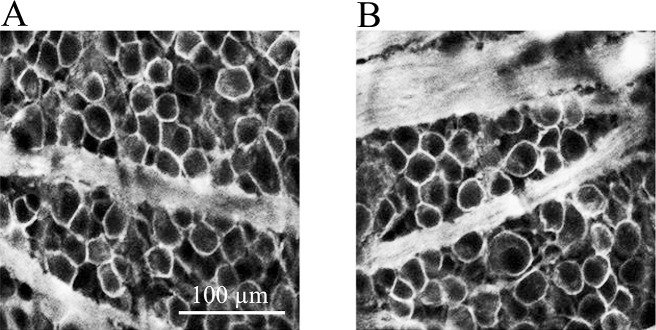
Neuroprotective effect of PNU-282987. (A) Represents a left eye image of RGCs immunostained with an antibody against Thy 1.1. under control untreated conditions, 4 mm from the ONH. (B) To obtain the image, the right eye was intravitreally injected with 100 μM PNU-282987 1 hour before NaCl injection into the episcleral vein. The image shown in (B) was obtained from the same rat that produced (A), from an equivalent retinal location, 1 month after NaCl injection.
NaCl Injections Increased IOP
As shown in the Table, besides a loss of RGCs, NaCl injection into the episcleral veins also increased intraocular pressure. IOP measurements were obtained from awake rats before and after NaCl injection into the episcleral veins using the tonometer (Mentor O & O, Inc.) according to the methods outlined by Morrison et al.16 Before NaCl injection, IOP measurements fluctuated between 10 and 13 mm Hg, averaging 12.3 mm Hg (±1.9, n = 8). However, within 2 weeks following NaCl injection, the IOP began to increase. One month after NaCl injection, there was a significant increase in the average IOP measurement compared with the average IOP measurement obtained from the internal controls. One month following NaCl injection, the IOP measurements ranged from 18 to 23 mm Hg, averaging 21.22 mm Hg (±3.21).
Table.
Mean IOP Measurements Obtained Before and After Surgery to Induce Glaucoma-Like Conditions
|
Left Control Eyes, n
= 8 |
Right Experimental Eyes, n
= 8 |
|
| Baseline before surgery | 12.6 (±1.7) | 12.3 (±1.9) |
| 1 wk postsurgery | 13.8 (±2.2) | 14.1 (±2.9) |
| 2 wk postsurgery | 14.3 (±2.5) | 15.6 (±1.7) |
| 3 wk postsurgery | 12.6 (±1.8) | 17.2 (±1.5)* |
| 4 wk postsurgery | 13.6 (±2.1) | 21.4 (±3.1)* |
Represents significance from internal control.
In control experiments, IOP measurements were obtained from untreated retinas and then 100 μM PNU-282987 was intravitreally injected without performing the NaCl injection insult. This was performed to demonstrate that it was not the PNU-282987 intravitreal injection that caused an increase of IOP. In six different retinas, there was no significant increase of IOP from baseline levels after intravitreal injection of PBS (n = 3) or 100 μM PNU-282987 (n = 3) if NaCl injections into episcleral veins did not occur.
Figure 9 demonstrates the dose-response curve generated when various concentrations of PNU-282987 between 0.1 and 1000 μM were intravitreally injected into rat eyes before NaCl injections. One month after NaCl injection, significant neuroprotection against RGC loss occurred if 5 μL of 10 μM PNU-282987 was injected into the vitreous cavity, but maximal neuroprotection occurred when 100 μM PNU-282987 was used. The neuroprotective effect using 100 and 1000 μM PNU-282987 was not significantly different. 100 μM PNU-282987 acted to prevent the loss of RGCs by an average of 96.22% (±14.2). The EC50 for the neuroprotective effect was calculated to be 42 μM.
Figure 9.
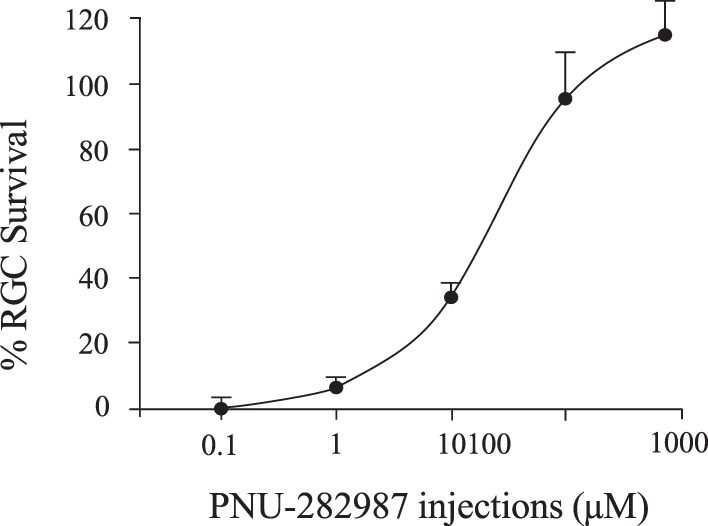
Dose-response effect of PNU-282987. Each data point shown represents the average percent of RGC survival compared with control untreated conditions using different concentrations of PNU-282987. Different concentrations of PNU-282987 were injected into the right eye of experimental rats 1 hour before NaCl injection. Animals were killed 1 month later and the percent of RGC survival was quantified. Data points were curve fit. Error bars represent SEM.
If PNU-282987's neuroprotective effect is mediated through activation of α7 nAChRs on rat RGCs, then α7 nAChR antagonists should block the effect. To test this hypothesis, additional experiments were performed where 5 μL of the α7 nAChR specific antagonist, MLA (10 μM), was injected into the vitreous cavity of right eyes an hour before intravitreal injection of PNU and 2 hours before NaCl injection into the episcleral veins. The summary of these experiments are illustrated in Figure 10. After injection of hypertonic saline into the episcleral veins, there was an average RGC loss of 23.86% (±6.68, n = 28) 4 mm from the ONH. However, when retinas were pretreated with the α7 nAChR agonist PNU-282987, RGC loss was prevented. When MLA was injected prior to the PNU agonist and prior to NaCl injection into the episcleral veins, the neuroprotective effect of PNU was eliminated and a significant loss of RGCs occurred, averaging 20.23% (±6.29, n = 4). In sham experiments, 5 μL of PBS vehicle was injected into the eye before the NaCl injection instead of PNU-282987 to determine if the NaCl injection alone was responsible for increased RGC survival. The sham treatment had no significant effect on RGC survival and there was significant loss of RGCs when PBS was substituted for the α7 nAChR agonist. One month after PBS intravitreal injections and NaCl injection designed to induce glaucoma-like conditions, RGC loss averaged 19.85% (±5.72, n = 4).
Figure 10.
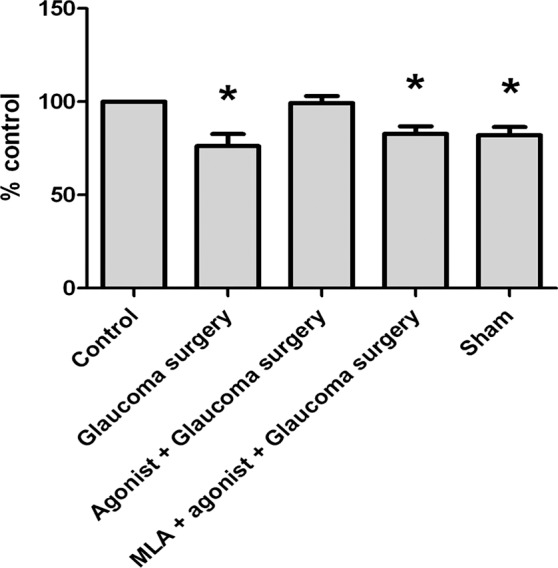
Summary of PNU-282987 experiments. Each bar graph shown represents the average percent change in RGCs compared with each animal's internal untreated control eye. Sham experiments were obtained where intravitreal injections consisted of PBS instead of PNU-282987. Asterisk represents significance from the untreated control. Error bars represent SEM.
The next experiments were designed to directly measure the neuroprotective effects of PNU-282987 on RGC survival in the hypertonic injection glaucoma model. For these experiments, both rat eyes were injected with 2M NaCl to induce loss of RGCs in both retinas, after 5 μL of 100 μM PNU-282987 was injected intravitreally into the right eye only. As seen in Figure 11A, NaCl injection into the left eye resulted in fewer RGCs and loss of defined axon tracks. However, as shown in Figure 11B, PNU-282987 prevented the loss of RGCs associated with the NaCl injection in the right eye. When comparing these two experimental conditions, the retinas pretreated with 100 μM PNU-282987 significantly increased RGC survival by an average of 130.33% (±12.72, n = 6).
Figure 11.
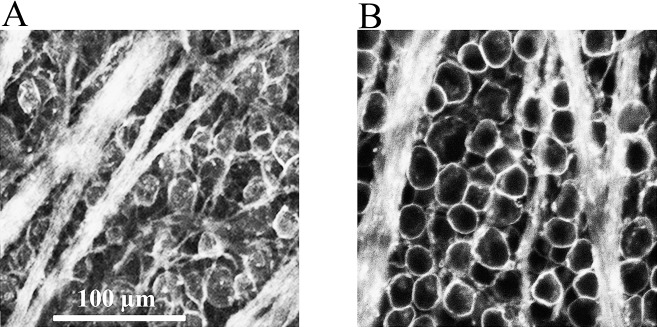
PNU-282987 protects against loss of RGCs. The fluorescent images represented were obtained 1 month after hypertonic injections were made into both eyes. (A) Image (left eye) represents the effect of hypertonic saline injection into the episcleral vein on RGC survival. (B) The image shown was obtained from the same animal, from an equivalent retinal position after 5 μL of 100 μM PNU-282987 was injected into the right eye before the hypertonic injections to induce glaucoma-like conditions. Both images were obtained 1 month following injections to induce glaucoma-like conditions and were obtained 4 mm from the ONH.
Discussion
In this study, the neuroprotective effects of PNU-282987 was analyzed to prevent the loss of RGCs mediated through α7 nAChRs in an in vivo rat model of glaucoma. Results from this study demonstrated that: (1) injection of 2M NaCl into the episcleral veins significantly reduced RGC density after 1 month, 4 mm from the ONH in the retinal ganglion cell layer; and (2) when PNU-282987 was directly injected into the vitreous humor before NaCl injection designed to induce glaucoma-like conditions, it prevented loss of RGCs due to hypertonic injections in a dose-dependent manner. The maximal neuroprotective effect of PNU-282987 occurred when 100 μM PNU-282987 was injected into the vitreous cavity of the eye before NaCl was injected into the episcleral veins and recovered an average of 96% of RGCs normally lost due to the NaCl injection when using Thy 1.1 and recovered a similar 87% when the RGC nuclear marker was used.
Results from this study are in agreement with the effectiveness of the episcleral injection of hypertonic saline to produce glaucoma-like conditions to produce a decrease of RGCs in the rat retina that has been reported in other laboratories.16,27 This consistency demonstrates that the methods used to induce glaucoma in rat retina are appropriate to examine glaucoma-like conditions. Many other studies have also linked glaucoma-like conditions in rodent models with an increase of intraocular pressure.16,28
This study also revealed that there was a larger percentage of RGCs present in the rat ganglion cell layer at 4 mm from the ONH than was previously reported in mice. We found this increase of RGCs to be higher than that reported for mice in all four retinal quadrants, including the dorsal visual streak region. In several mouse studies, several reports indicated that as many as 56% to 59% of neurons in the ganglion cell layer were not RGCs.20,21 However, in the rat, when cells were labeled with antibodies against Thy 1.1 or the nuclear marker Brn3a, nearly 90% of cells were labeled 4 mm from the ONH. This may be due to species differences that occur in rats,29–31 the age of the animals used in this study (3 months),32–34 or the different procedures used for detecting cells in previous studies.29,35,36
The mechanism responsible for the loss of RGCs associated with glaucoma-like conditions is still under investigation. A number of studies have argued that excitotoxicity plays a role at the onset of glaucoma,37 but different contributing mechanisms are also likely to play a role in the progression of the disease such as; oxidative stress by reactive species, vascular dysregulation, cytoskeletal dysfunction, changes in growth factor levels, genetic contributions, or other pathological pathways.37,38 In glaucoma however, it is accepted that the primary risk factor associated with glaucoma is an increase of intraocular pressure, leading to the degeneration of the optic nerve and loss of vision, which is the ultimate result of contributing factors.
Regardless of the mechanism involved in loss of RGCs associated with glaucoma-like conditions, neuroprotection against cell loss has been used as a therapeutic approach for many neurodegenerative diseases. In this study, we analyzed the neuroprotective effect of PNU-282987, an α7 nAChR-specific agonist, against RGC loss that was correlated with NaCl injections into the episcleral veins. Quantification of RGC neuroprotection using PNU-282987 demonstrated a clear dose-dependent effect, which plateaued when 5 μL of 100 μM PNU-282987 was injected into the vitreous cavity.
Activation of α7 nAChRs in the brain have also been linked to neuroprotection against several neurodegenerative diseases.9,39 What are the physiological implications of α7 nAChR mediated neuroprotection in the retina? Starburst amacrine cells are a category of amacrine cells that release ACh onto RGCs.40,41 Pharmacological and immunocytochemical studies have provided evidence of α7 nAChRs on both large and small RGCs.11 As a result, ACh should initiate neuroprotection in RGCs as it can be released from displaced starburst amacrine cells in the RGC layer42 to protect against a glaucomatous insult or from the inner nuclear layer. It's possible that neuroprotection from these cholinergic cells are lost under glaucoma-like conditions due to death of the displaced amacrine cells and/or loss of ACh. However, it has been shown that cholinergic amacrine cells aren't lost in glaucoma-like conditions.43 Therefore, other factors are likely responsible for the loss of neuroprotection normally provided by ACh release. Alternate hypotheses include scenarios where the naturally occurring neuroprotective defense mechanisms provided by ACh are overwhelmed or that ACh release is compromised in a glaucomatous environment and a reduction in ACh release from amacrine cells contribute to the loss of RGCs normally associated with glaucoma. If the release of ACh from starburst amacrine cells is compromised under glaucoma-like conditions, it would unveil a previously unknown neuroprotective function of starburst amacrine cells in the retina. Current studies are underway to investigate ACh release from the rat glaucomatous retina to enhance our understanding of this possibility and to gain an understanding of different neuroprotective mechanisms used in the retina.
The main risk factor associated with glaucoma is an increase in IOP. All current therapies are designed to decrease IOP. Currently the two main treatments for glaucoma are drugs or surgery. Either applied topically or orally, drugs can be administered to decrease the production of aqueous humor or can increase the drainage of fluid to decrease the IOP.44 In other instances, a surgical procedure can produce small holes in the eye to drain the aqueous humor, or a laser can manufacture holes through the trabecular meshwork. Although these treatments work to decrease intraocular pressure, in most cases, decreasing the IOP does not completely stop the progression of the disease. In some cases, patients with normal IOP still develop glaucoma and vision loss, which indicates that something other than increased IOP may be causing the vision loss. Because of this, it may be advantageous to use an alternate treatment that provides neuroprotection against loss of RGCs in the retina in conjunction with current treatments. This would represent a unique and different strategy for treating glaucoma. However, there are limitations attached to any new treatment strategy. For instance, previous studies have demonstrated that ACh, nicotine and PNU-282987 had to be applied before inducing excitotoxicity or before NaCl injections into the episcleral veins for neuroprotection to occur.10,14 As a result, treatments using an α7 nAChR agonist would act as a preventative measure for patients at high risk of glaucoma based on age, genetics or race. A neuroprotective treatment with an α7 nAChR agonist under these limitations could be used regardless of IOP.
In conclusion, these data suggest that introduction of α7 nicotinic agonist, PNU-282987 into the eye, could significantly reduce the loss of RGCs associated with glaucoma. These results strengthen the strategy of developing α7 nAChR agonists as a potential therapeutic treatment for glaucoma.
Acknowledgments
The authors thank Rob Eversole for his confocal imaging expertise and the Southwest Michigan Innovation Center for their LC/MS/MS analysis of retinal samples.
Supported by NIH NEI Grant EY 022795 and a Technology Development Award from Western Michigan University (CLL).
Disclosure: K. Iwamoto, None; P. Birkholz, None; A. Schipper, None; D. Mata, None; D.M. Linn, None; C.L. Linn, None
References
- 1. Vickers JC, Schumer RA, Podos SM, Wang RF, Riederer BM, Morrison JH. Differential vulnerability of neurochemically identified subpopulations of retinal neurons in a monkey model of glaucoma. Brain Res. 1995; 680: 23–35 [DOI] [PubMed] [Google Scholar]
- 2. Brooks DE, Garcia GA, Dreyer EB, Zurakowski D, Franco-Bourland RE. Vitreous body glutamate concentration in dogs with glaucoma. Amer J Vis Res. 1997; 58: 864–867 [PubMed] [Google Scholar]
- 3. Dkhissi O, Chanut E, Wasowicz M, et al. Retinal TUNEL-positive cells and high glutamate levels in vitreous humor of mutant quail with a glaucoma-like disorder. Invest Ophthalmol Vis Sci. 1999; 40: 990–995 [PubMed] [Google Scholar]
- 4. Dong CJ, Guo Y, Agey P, Wheeler L, Hare WA. Alpha2 adrenergic modulation of NMDA receptor function as a major mechanism of RGC protection in experimental glaucoma and retinal excitotoxicity. Invest Ophthalmol Vis Sci. 2008; 49: 4515–4522 [DOI] [PubMed] [Google Scholar]
- 5. Seki M, Soussou W, Manabe S, Lipton SA. Protection of retinal ganglion cells by caspase substrate-binding peptide IQACRG from N-methyl-D-aspartate receptor-mediated excitotoxicity. Invest Ophthalmol Vis Sci. 2010; 51: 1198–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guerin MB, Donovan M, McKernan DP, O'Brien CJ, Cotter TG. Age-dependent rat retinal ganglion cell susceptibility to apoptotic stimuli: implications for glaucoma. Clin Exp Ophthalmol. 2011; 39: 243–251 [DOI] [PubMed] [Google Scholar]
- 7. Smith AJ, Tauskela JS, Stone TW, Smith RA. Preconditioning with 4-aminopyridine protects cerebellar granule neurons against excitotoxicity. Brain Res. 2009; 1294: 165–175 [DOI] [PubMed] [Google Scholar]
- 8. Matteucci A, Cammarota R, Paradisi S, et al. Curcumin protects against NMDA-induced toxicity. Invest Ophthalmol Vis Sci. 2011; 52: 1070–1077 [DOI] [PubMed] [Google Scholar]
- 9. Liu Z, Cai H, Zhang P, Li H, Liu H, Li Z. Activation of ERK1/2 and PI3K/Akt by IGF-1 on GAP-43 expression in DRG neurons with excitotoxicity induced by glutamate in vitro. Cell Mol Neurobiol. 2012; 32: 191–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wehrwein E, Thompson SA, Coulibaly SF, Linn DM, Linn CL. Acetylcholine protection of adult pig retinal ganglion cells from glutamate-induced excitotoxicity. Invest Ophthalmol Vest Sci. 2004; 45: 1531–1543 [DOI] [PubMed] [Google Scholar]
- 11. Iwamoto K, Linn DM, Mata D, Linn CL. Neuroprotection of rat retinal ganglion cells mediated through alpha7 nicotinic acetylcholine receptors. Neuroscience. 2013; 237: 184–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thompson SA, Smith O, Linn DM, Linn CL. Acetylcholine neuroprotection against glutamate-induced excitotoxicity in adult pig retinal ganglion cells is partially mediated through alpha4 nAChRs. Exp Eye Res. 2006; 83: 1135–1145 [DOI] [PubMed] [Google Scholar]
- 13. Asomugha CO, Linn DM, Linn CL. ACh receptors link two signaling pathways to neuroprotection against glutamate-induced excitotoxicity in isolated RGCs. J Neurochem. 2010; 112: 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brandt SK, Weatherly ME, Ware L, Linn DM, Linn CL. Calcium preconditioning triggers neuroprotection in retinal ganglion cells. Neuroscience. 2011; 172: 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005; 48: 905–908 [DOI] [PubMed] [Google Scholar]
- 16. Morrison JC, Moore CG, Deppmeier LM, Gold BG, Meshul CK, Johnson EC. A rat model of chronic pressure-induced optic nerve damage. Exp Eye Res. 1997; 64: 85–96 [DOI] [PubMed] [Google Scholar]
- 17. Barnstable CJ, Dräger UC. Thy-1 antigen: a ganglion cell specific marker in rodent retina. Neuroscience. 1984; 11: 847–855 [DOI] [PubMed] [Google Scholar]
- 18. Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naïve and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009; 50: 3860–3868 [DOI] [PubMed] [Google Scholar]
- 19. Zhang PP, Yang XL, Zhong YM. Cellular localization of P2Y6 receptor in rat retina. Neuroscience. 2012; 220: 62–69 [DOI] [PubMed] [Google Scholar]
- 20. Jeon CJ, Kong JH, Strettoi E, Rockhill R, Stasheff SF, Masland RH. Pattern of synaptic excitation and inhibition upon direction-selective retinal ganglion cells. J Comp Neurol. 2002; 449: 195–205 [DOI] [PubMed] [Google Scholar]
- 21. Pang JJ, Gao F, Wu SM. Light responses and morphology of bNOS-immuoreactive neurons in the mouse retina. J Comp Neurol. 2010; 518: 2456–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlamp CL, Johnson EC, Li Y, Morrison JC, Nickells RW. Changes in Thy1 gene expression associated with damaged retinal ganglion cells. Mol Vis. 2001; 7: 192–201 [PubMed] [Google Scholar]
- 23. Huang W, Fileta J, Guo Y, Grosskreutz CL. Downregulation of Thy1 in retinal ganglion cells in experimental glaucoma. Curr Eye Res. 2006; 31: 265–271 [DOI] [PubMed] [Google Scholar]
- 24. Wang SW, Mu X, Bowers WJ, et al. Brn3b/Brn3c double knockout mice reveal an unsuspected role for Brn3c in retinal ganglion cell axon outgrowth. Development. 2002; 129: 467–477 [DOI] [PubMed] [Google Scholar]
- 25. Bernstein SL, Koo JH, Slater BJ, Guo Y, Margolis FL. Analysis of optic nerve stroke by retinal Bex expression. Mol Vis. 2006; 12: 147–155 [PubMed] [Google Scholar]
- 26. Buckingham BP, Inman DM, Lambert W, et al. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008; 28: 2735–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bouhenni RA, Dunmire J, Sewell A, Edward DP. Animal models of glaucoma. J Biomed Biotechnol. 2012; 11: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morrison JC. Elevated intraocular pressure and optic nerve injury models in the rat. J Glaucoma. 2005; 14: 315–317 [DOI] [PubMed] [Google Scholar]
- 29. Danias J, Shen F, Goldblum D, et al. Cytoarchitecture of the retinal ganglion cells in the rat. Invest Ophthalmol Vis Sci. 2002; 43: 587–594 [PubMed] [Google Scholar]
- 30. Fileta JB, Huang W, Kwon GP, et al. Efficient estimation of retinal ganglion cell number: a stereological approach. J Neurosci Methods. 2008; 170: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlamp CL, Montgomery AD, Mac Nair CE, Schuart C, Willmer DJ, Nickells RW. Evaluation of the percentage of ganglion cells in the ganglion cell layer of the rodent retina. Mol Vis. 2013; 19: 1387–1396 [PMC free article] [PubMed] [Google Scholar]
- 32. Danias J, Lee KC, Zamora M-F, et al. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57/BL6 mice. Invest Ophthalmol Vis Sci. 2003; 44: 5151–5162 [DOI] [PubMed] [Google Scholar]
- 33. Harwerth RS, Wheat JL, Rangaswamy NV. Age-related losses of retinal ganglion cells and axons. Invest Ophthalmol Vis Sci. 2008; 49: 4437–4443 [DOI] [PubMed] [Google Scholar]
- 34. Samuel MA, Zhang Y, Meister M, Sanes JR. Age-related alterations in neurons of the mouse retina. J Neurosci. 2011; 31: 6033–16044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Higashide T, Kawaguchi I, Ohkubo S, Takeda H, Sugiyama K. In vivo imaging and counting of rat retinal ganglion cells using a scanning laser ophthalmoscope. Invest Ophthalmol Vis Sci. 2006; 47: 2943–2950 [DOI] [PubMed] [Google Scholar]
- 36. Henrich-Noack P, Voigt N, Prilloff S, Fedorov A, Sabel BA. Transcorneal electrical stimulation alters morphology and survival of retinal ganglion cells after optic nerve damage. Neurosci Lett. 2013; 543: 1–6 [DOI] [PubMed] [Google Scholar]
- 37. Agarwal R, Gupta SK, Agarwal P, Saxena R, Agarwal SS. Current concepts in the pathophysiology of glaucoma. Indian J Ophthalmol. 2009; 57: 257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuehn MH, Fingert JH, Kwon YH. Retinal ganglion cell death in glaucoma: mechanisms and neuroprotective strategies. Ophthalmol Clin North Amer. 2005; 18: 383–395 [DOI] [PubMed] [Google Scholar]
- 39. Conejero-Goldberg C, Davies P, Ulloa L. Alpha7 nicotinic acetylcholine receptor: a link between inflammation and neurodegeneration. Neurosci Biobehav Rev. 2008; 32: 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Famiglietti EV Jr. ‘Starburst' amacrine cells and cholinergic neurons: mirror-symmetric on and off amacrine cells of rabbit retina. Brain Res. 1983; 261: 138–144 [DOI] [PubMed] [Google Scholar]
- 41. Masland RH. Amacrine cells. Trends Neurosci. 1988; 11: 405–410 [DOI] [PubMed] [Google Scholar]
- 42. Perry VH, Walker M. Amacrine cells, displaced amacrine cells and interplexiform cells in the retina of the rat. Proc R Soc Lond B Biol Sci. 1980; 208: 415–431 [DOI] [PubMed] [Google Scholar]
- 43. Kielczewski JL, Pease ME, Quigley HA. The effect of experimental glaucoma and optic nerve transection on amacrine cells in the rat retina. Invest Ophthalmol Vis Sci. 2005; 46: 3188–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Damji KF, Behki R, Wang L. Canadian perspectives in glaucoma management: Setting target intraocular pressure range. Canadian J Ophthalmol. 2003; 38: 189–197 [DOI] [PubMed] [Google Scholar]