

Abstract
Non-invasive enumeration of rare circulating cell populations in small animals is of great importance in many areas of biomedical research. In this work we describe a macroscopic fluorescence imaging system and automated computer vision algorithm that allows in vivo detection, enumeration and tracking of circulating fluorescently-labeled cells from multiple large blood vessels in the ear of a mouse. This imaging system uses a 660 nm laser and a high sensitivity electron-multiplied charge coupled device camera (EMCCD) to acquire fluorescence image sequences from relatively large (~5 × 5 mm2) imaging areas. The primary technical challenge was developing an automated method for identifying and tracking rare cell events in image sequences with substantial autofluorescence and noise content. To achieve this, we developed a two-step image analysis algorithm that first identified cell candidates in individual frames, and then merged cell candidates into tracks by dynamic analysis of image sequences. The second step was critical since it allowed rejection of >97% of false positive cell counts. Overall, our computer vision IVFC (CV-IVFC) approach allows single-cell detection sensitivity at estimated concentrations of 20 cells per mL of peripheral blood. In addition to simple enumeration, the technique recovers the cell’s trajectory, which in the future could be used to automatically identify, for example, in vivo homing and docking events.
Keywords: In vivo flow cytometry, automated, rare cell, computer vision
1. Introduction
There are many areas of pre-clinical biomedical research that require high-sensitivity detection and enumeration of rare circulating cell populations in the blood stream of small animals, including early stage cancer metastasis, immunology and novel stem cell therapies (1–5). Although extraction of peripheral blood (PB) samples and subsequent analysis with conventional flow cytometry is still the gold-standard method for cell enumeration, “in vivo flow cytometry” (IVFC) approaches are rapidly gaining acceptance since they allow continuous, non-invasive optical detection of circulating cells in situ. For example, fluorescence microscopy IVFC uses a laser line focused across a small arteriole in a mouse ear or retina. As fluorescently-labeled cells pass through this excitation beam, a transient fluorescence “spike” is detected with a photomultiplier tube (6,7). Since data can be obtained continuously, changes in cell populations that occur over minutes or hours can be measured. This is in contrast to more conventional techniques where PB samples can be drawn typically only once per day (8). In addition to fluorescence IVFC, two-photon IVFC (9,10), photoacoustic IVFC (11,12) and photothermal IVFC (13) designs have also been recently reported in the IVFC literature (14–17).
Despite these advances, one of the primary technical challenges associated with IVFC is the relatively small blood sampling volume. For most reported IVFC methods this on the order of 1 μL per minute, although Galanzha et. al. recently reported photoacoustic detection from a mouse aorta using a focused transducer where the flow rate is on the order of 1–2 mL per minute (18,19). Given that mice have approximately 2 mL of circulating blood, this limits the overall sensitivity of IVFC and in most cases means that very rare circulating cell populations (below about 103 cells per mL) are very difficult to detect. For experimental applications where circulating cell concentrations are sufficiently low (e.g. early-stage metastatic spread of cancer), mice must be euthanized and the entire PB analyzed, thereby eliminating the possibility of serial study of the same animal (20). As such, new higher-sensitivity IVFC designs that allow detection of very rare cell populations are needed.
One evident solution to the problem is simply to “zoom out” to a larger fluorescence imaging field-of-view (for example, to a larger region of the ear) so that more blood vessels and correspondingly larger blood volumes are optically sampled. In the context of rare-cell detection, the use of “macroscopic” fluorescence imaging with a wide field-of-view presents two significant technical challenges. First, this requires relatively high laser illumination intensity and high applied detector gain which results in detection of substantial non-specific tissue autofluorescence. Further, individual cells become small relative to the total image (1–5 pixels in dimension) and of comparable intensity to noise on autofluorescence. As we demonstrate, cells become difficult to distinguish from background autofluorescence and noise in a single image. Second, at low circulating cell concentrations (as we use in the experiments described herein) cells pass through the imaging field-of-view very infrequently, e.g. on the order of one cell per minute or less. As such, a method for automated detection and counting of cells to assist a human operator is highly desirable.
In this work we approached this problem by utilizing a simple a priori feature of circulating cells, i.e. that they are in motion. Circulating cells appear in multiple temporally-related frames of an image sequence. As we demonstrate, this simple property can be exploited to identify cells in noisy image sequences. To our knowledge, this macroscopic computer vision approach to rare cell fluorescence IVFC has never been studied previously. It is important note that the idea of computer vision “cell tracking” or “cell counting” is not novel (21–26). However, previously reported methods typically identify clearly defined objects with strong background contrast, for example, of cells in culture on a microscope slide. In the present case, our objective was to image circulating cells in situ with a widefield imager so that they appear as only a small cluster of pixels with comparable intensity to the noise on the autofluorescence background. Therefore, existing software packages for identifying or tracking cells (e.g. Imaris, Bitplane (27–29) or Volocity, Improvision (30–32)) in our experience are generally not suitable for tracking small moving cells in widefield fluorescence image sequences such as those presented here. This motivated us to develop a new computer vision algorithm as described in this work.
In this paper, we describe and validate our rare-cell ‘computer vision in vivo flow cytometry’ (CV-IVFC) method, first in flow phantom models and then in nude mice in vivo. As we demonstrate, this method allowed us to sample relatively large blood volumes and to detect circulating cells at very low concentrations. We typically imaged 3–4 large artery-vein pairs simultaneously along with the surrounding capillary bed of the ear. Based on reported blood flow rates of vasculature in the ear (6–19) we estimate that the instrument samples about 10–12 μL of peripheral blood minute. We demonstrate that we could detect injected concentrations of approximately 2.5 × 103 cells / mL Vybrant-DiD-labeled multiple myeloma (MM) cells. As we quantify in detail below, this algorithm enabled high detection sensitivity with a small false alarm rate (when compared to human operator), yielding an overall estimated system sensitivity of 20 circulating cells per mL. To our knowledge, CV-IVFC represents an entirely new, high-sensitivity but easily implementable approach to rare cell sensing and enumeration in pre-clinical small animal models.
2 Materials and Methods
2.1 Fluorescence Macroscope Design
A schematic and photograph of the video-rate fluorescence macroscope used in these experiments are shown in figure 1. The sample, either a tissue-mimicking flow phantom or a mouse ear (see below), was placed on an adjustable imaging platform and was transilluminated with the output of a 660 nm solid-state diode laser (DPSS-660; Crystalaser Inc., Reno, NV). The output beam was expanded to approximately 5 mm full-width at half maximum using a simple plano-convex lens pair (f = 50 mm and 200 mm; Edmund Optics, Barrington, NJ). A 660 nm “clean-up” filter (d660/20x Chroma Technology, Rockingham, VT) was also used in front of the laser to remove a small amount of out-of-band NIR output from the laser. The light intensity at the sample was 10 mW/cm2. At this intensity a small amount of tissue autofluorescence photobleaching (about 0.8% per minute on average) was observed during the in vivo experiments.
Figure 1.
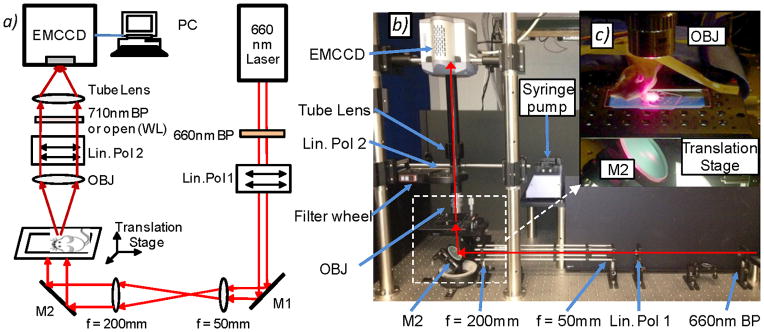
(a) Schematic and (b) photograph of the fluorescence macroscope used to acquire image sequences for this work (see text for details). Abbreviations: M – mirror, Lin Pol - linear polarizer, Obj – 2X objective. (c) Photograph of a mouse ear positioned on the imaging stage for in vivo experiments. As shown, a large region of the ear is illuminated and imaged.
We used a high-sensitivity, 14-bit electron multiplied charge coupled device (EMCCD) camera (iXonEM+855 Andor Technology, Belfast, Northern Ireland) fitted with a low-magnification objective with NA = 0.055 (2X Mitutoyo Plan Apo Infinity-Corrected Long WD Objective Edmund Optics, Barrington, NJ) and 200 mm 1X tube lens (Mitutoyo MT-1, Edmund), so that the imaging field of view was about 5 × 5 mm2. The depth of focus of this objective was 91 μm and we were able to resolve cells within 75 μm above or below this region (although cells were slightly blurry) so that the effective working depth of field was about 241 μm in tissue. This is well matched to the mouse ear, since the thickness is about 250–300 μm and most of the blood vessels are located within 100 μm of the ear surface (33,34). As such, essentially all cells traveling in blood vessels in the field of view were detectable. Fluorescence images were acquired with a 710 nm filter with 50 nm bandpass filter in place (et710/50m; Chroma) while the laser was illuminating the sample. The filter was mounted in a motorized 6-position filter wheel (FW102, Thorlabs Inc., Newton NJ). Crossed linear polarizers (25 mm diameter, Edmund) were also placed between the laser and the sample, and between the sample and the EMCCD to further reduce leakage of the laser light into the imager. In principle this polarizer pair was not required but was experimentally found to reduce laser leakage into the fluorescence channels, although this had the drawback of reducing the detected fluorescence by 50%. White light images of the sample were acquired by removing the fluorescence filter from the optics train (i.e. moving to an open position of the filter wheel) so that all wavelengths were detected by the camera and by back-illuminating the sample with an LED ring (Digi-Slave L-Ring 3200, Edmund). We chose to use a red laser and a near-infrared fluorophore for these experiments since tissue autofluorescence is markedly reduced compared to, for example, the blue-green wavelength region. Other lasers in the near-infrared range (such as a Titanium:Sapphire laser or diode-pumped solid state lasers) could similarly be used with near-infrared dyes such as Vybrant DiL, Alexafluor750 or Cyanine-7. In principle visible lasers and fluorophores could also be used (as is done routinely in microscopy IVFC (6,7)), but as we discuss this would result in an increase in tissue autofluorescence and potentially greater attenuation of light through the ~250–300 μm thick ear.
The exposure time for fluorescence images was typically 0.05 s, resulting in a frame rate of approximately 19 Hz (allowing time for data transfer to on-board memory). We also re-binned the 1024 × 1024 imaging array to 128 × 128 pixels in on-board camera hardware, which also increased the frame rate on the camera. Given our optics and camera configuration, a 10 μm diameter cell occupied an object only about 1–5 pixels in dimension in each image (rapidly moving cells were often recorded as a streak that were up to 5 pixels in length). Further, given the large imaging field of view relative to cell size, detection of fluorescently-labeled circulating cells required application of gain to the EMCCD camera. This could be configured with a personal computer running Andor software, and typically we operated this between 10–90 out of a maximum of 300 (arbitrary units). This resulted in substantial imaging noise (as opposed to background intensity) on the order of the detected cell intensity which necessitated the development of the imaging algorithms described here. Use of a higher magnification objective with a larger NA would have resulted in greater sensitivity and required less camera gain to resolve cells, however, this would have come at a cost of smaller imaging field of view and depth of focus, so that the overall cell detection sensitivity of the CV-IVFC instrument would have been reduced. During data collection we acquired 1000 frames per imaging sequence (52 s), but this could be repeated an arbitrary number of times with only about 0.5 s lag between image sequence acquisition to allow transfer of image sequences to the PC.
2.2 Computer vision algorithm
We developed a two-step algorithm to analyze image sequences and extract the tracks of moving cells from noisy widefield fluorescence images as shown schematically in figure 2. The overall strategy was as follows: in the first step we identified candidate cells in individual images in the sequence, and in the second step we connected cell candidates in multiple image frames into cell tracks. We first performed basic flat-field correction of the image (for the laser beam profile), followed by pixel-by-pixel background subtraction (Step-1A). This was done by taking the mean value mij of each image pixel pij in all N image frames in the sequence,
Figure 2.

Flow chart of the two-step automated computer vision analysis of fluorescence image sequences as well as example images obtained after each step. The algorithm identified cell candidates (Step 1) from image sequences, and then connected them dynamically into cell tracks (Step 2). The position of the cell is indicated with a red arrow and dotted circle. See text for details.
| (1) |
In Step-1B we converted the 14-bit image sequence into a binary image sequence by subtracting this mean value (pixel-by-pixel) and comparing the resulting value to a threshold value τ as follows,
| (2) |
Therefore, in general there was no condition that a cell be a specific size or shape. Rather at least one pixel must have exceeded the threshold for a cell to be identified as a candidate. As we discuss, selection of the particular threshold τ for each sequence was very important since it altered the performance of the algorithm with respect to overall sensitivity and false alarm rate (see section 2.5 and sections 3.3 below for greater detail). We observed substantial inter-experimental variability in overall image intensity (~50%) and therefore determined that a fixed threshold across all experiments was not suitable. Instead, we set the threshold as a function of percentile of all pixel intensities (ranging from 0 to 16383) in the image sequence; as we demonstrate, threshold percentiles in the range of 99.93 to 99.99 were empirically determined to work well in mice in vivo. We further note that the raw image intensity data sets directly from the EMCCD were processed by the algorithm and no prior image manipulation filters (such as edge, sharpen, contrast etc.) were employed since these can often exacerbate existing noise or introduce additional artifacts in the image sequence.
Following this operation, the resulting binary image sequence contained the set of circulating cell candidates which in practice greatly outnumbered the actual number of cells. Specifically, many post-threshold cell candidates were due to image noise (as we show, for in vivo data this was about 97% of candidates). Therefore, in Step 2 cells were distinguished from background noise through dynamic analysis of image sequences to identify cell “tracks”. First, (Step 2A) cell candidates were connected into tracks in consecutive frames. For each cell candidate, a search §k was performed on pixels pij in the next image frame inside a radius equal to two times the dimension (e.g. diameter) of the candidate cell Φk from the center of the candidate cell Pij:
| (3) |
Typically this diameter was about 1–3 pixels, but could be slightly longer for fast moving cells where ~5 pixel long “streaks” were observed. This search was performed for all candidates in the image sequence. When multiple cell candidates were observed inside this radius, the closest candidate was selected to merge to the track. Because cells were relatively dim and the intensity could intermittently drop below the threshold, gaps in a given cell’s track (of up to ~10 frames) were often observed after Step 2A. As such, in Step 2B these were connected together by merging individual tracks that occurred relatively close together in space and time as follows: when the final position of one track (identified in Step 2A) occurred within a radius of 15 pixels and within 15 image frames from the first position of the next identified track in the image sequence, the tracks were merged. We then applied a second condition for merging cell tracks in Step 2C that combined the start and end points of consecutive identified tracks SmT(start) and SnT-t(end) observed at times T and T-t, respectively,
| (4) |
where M was the condition for merging the tracks. The search area was determined by analyzing the final position and velocity v of the previous track (SmT-t),
| (5) |
where τ was the inverse of the frame rate (in this case 0.052 s) and by extrapolating the final position assuming a speed in the range of 0.5v to 2v as follows,
| (6) |
where t was the elapsed time since the end of the previous track. The three search regions §m1, §m2 and §m3 centered on Pm1, Pm2 and Pm3 with a radius of 0.5vt, vt and 2vt, respectively; in practice, this produced a cone-shaped search area. Therefore Step 2B and Step 2C differed in the search region for merging as follows: Step 2B used a circular search radius whereas Step 2C used a cone oriented in the direction of cell movement. At the end of Step 2, remaining cell candidates that did not connect to any track (i.e. were observed in only single frames) were discarded from the analysis. As we show, this was the case for about 97% of cell candidates that were identified in Step 1. As such Step 2 was critical in the rejection of false positive counts. Last, for visualization, cell tracks were overlaid onto the white light image of the sample (mouse ear vasculature). A total cell count in an image sequence was obtained by simply counting the total number of identified cell tracks detected in an image sequence.
2.3 Phantom Measurements
We first tested our imager and algorithm in an optical flow phantom model which was intended to mimic a blood vessel in a mouse ear. Disk shaped phantoms (figure 3a) approximately 2 mm thick by 20 mm in diameter were made from polyester resin (Casting Craft, Fields Landing, CA) with Titanium Oxide (TiO2; Signa-Aldrich Inc., St. Louis, MO) and India ink added at 50 parts per million to yield optical properties similar to biological tissue at near-infrared wavelengths. Specifically, based on previously published characterization of similar phantoms (35) the final optical properties at 700 nm were estimated to be as follows: reduced scattering coefficient μ′s = 15 cm−1 and absorption coefficient μa = 0.1 cm−1. Strands of microbore Tygon tubing (250 μm internal diameter, TGY-010C, Small Parts, Inc, Seattle, WA) were embedded in the phantom in either an arc or in a straight line (N = 2 each) before hardening to mimic a blood vessel. The position of the clear tubing is indicated with an overlaid dotted line in figure 2a. These were attached to a syringe mounted on a precision microsyringe pump (70–2209, Harvard Apparatus, Holliston, MA), so that solutions of fluorescent micropsheres suspended in PBS could be passed through the phantom at controlled linear flow rates between 0.5 and 10 mm/s, therefore approximately matching the blood flow rates in large ear blood vessels reported in the literature (6,12). We used 6 μm diameter fluorescent microspheres with absorption maxima at 645 nm, and emission maxima near 695 nm (Peakflow Claret, P-24670, Invitrogen, Calsbad, CA), to match commonly used Cyanine5.5 and Alexafluor-680 organic fluorophores. Microspheres were suspended at a concentration of 3,000 spheres / mL. For these experiments, the EMCCD gain was set to 10 (arbitrary units; out of a maximum of 300). Image sequences were acquired for a total of 5 minutes for each phantom.
Figure 3.

(a) Photograph of an ear-mimicking phantom with curved length of embedded Tygon tubing. The position of the clear tube is indicated with the dotted black line. (b–f) Raw fluorescence image sequence (separated by 0.25 s) showing a fluorescent microsphere (red arrows and dotted circles) flowing in the phantom. The stationary white points were autofluorescent inclusions in the flow tube, simulating stationary tissue autofluorescence observed in vivo. (g–k) the same image sequence is shown after application of Step 1 of the algorithm. (l–p) The CV-IVFC algorithm successfully connected the sphere path in a trajectory over the arc. This trajectory was over-laid on the white light image of the phantom (green line).
2.4 In Vivo Measurements
We performed in vivo testing of our system and algorithm using six nude (nu/nu) mice injected intravenously with Vybrant-DiD-labeled Multiple Myeloma (MM) cells. All mice were handled in accordance with Northeastern University’s Division of Laboratory Animal Medicine rules on animal treatment and care. MM cells were grown in culture and suspended in RPMI with 0.1% Bovine Serum Albumin (BSA) at a concentration of 1 × 106 cells / mL. Cells were labeled with 1 μmol/L of Vybrant-DiD and incubated for 30 minutes at 37°C and then spun down and re-suspended at a final concentration of 5 × 104 cells / mL prior to injection. Mice were anesthetized with a combination of Ketamine (100 mg/kg) and Xylazine (5 mg/kg). 100 μL of the cell suspension were injected intravenously via the tail vein, so that the injected cell population was 5 × 103 cells, or approximately 2.5 × 103 cells per mL of PB (assuming about 2 mL of mouse blood). Mice were then placed on the translation stage and positioned so that the ear was flat on a microscope slide in the imager field of view. A glass cover-slip was also added (with a drop of water) to keep the ear flat during imaging. For in vivo experiments we increased the EMCCD gain to 90 (arbitrary units; out of a maximum of 300) due to the relative dimness of the labeled MM cells compared to the fluorescent microspheres. As above, image exposure times were set to 0.05 s, yielding a frame rate of about 19 Hz. Images were acquired for approximately 30 minutes for each of the six mice.
2.5 Performance Metrics
We used two standard metrics to assess the performance of our imaging system and cell detection algorithm, specifically, sensitivity = TP / (TP + FN) and false alarm rate (FAR) = FP / minute. Here TP is “true positive” cell count, FN are the “false negative” counts (in this case, cells that were undetected by the algorithm) and FP are the “false positive” counts. As noted in section 2.2, we tested a range of histogram threshold percentiles from 99.93 to 99.99, and quantified each performance metric at each level. To determine the “true” cell counts, a human operator manually counted the circulating cells appearing in the image sequences and compared them with those identified by the algorithm. Therefore, these metrics do not reflect circulating cells that may have been missed if they were not sufficiently bright to be visible on image sequences. However, the number of detected cells was generally in good agreement given the low concentration of injected cells and peripheral blood volume in the imager field-of-view.
Results
3.1 Optical Flow Phantom Testing
We first tested our CV-IVFC approach in tissue-mimicking optical flow phantoms (fig 3a) with fluorescent microspheres. We tested a range of flow speeds (0.5 to 10 mm/s) as well as straight and curved flow channel geometric configurations. An example set of fluorescence images acquired during the experiment are shown in figs 3b–f, along with the corresponding mean background subtracted and thresholded image sequence (after Step 1) in figs 3g–k. In this case, a phantom with a curved flow channel and linear flow speed of 5.1 mm/s is shown. By inspection of figs 3b–f, fluorescence contrast was excellent in the optical flow phantom model and this allowed us to verify that the algorithm could correctly identify and track small (~1–3 pixels diameter) fluorescent targets (indicated by red arrows and dotted circles), even when significant directional changes were observed. The extracted microsphere track (green curve) was overlaid on the white light images of the phantom as shown in figs. 3l–p. It is important to note that the algorithm successfully distinguished moving fluorescent targets from stationary bright pixels or pixel groups. For example, two stationary bright pixels can be clearly observed in the raw fluorescence images in figures 3b–f, but these points were rejected (primarily) during Step 1 of the CV-IVFC algorithm (fig 3g–k). Residual stationary pixels that were not rejected in Step 1 (for example due to photon counting noise as in figs. 3g,h) were rejected in Step 2, since they did not form trajectories in subsequent image frames (see section 2.2). In general, microspheres were successfully tracked at speeds up to 10 mm / s, which exceeds the expected flow speeds in blood vessels in the mouse ear in vivo of 0.5 to 5 mm / s from literature values (6,12). Since fluorescence contrast was extremely high in the flow phantom models, it was not necessary to adjust the threshold value in Step 1 (as opposed to the in vivo experiments where this was necessary; see below) so that we used a single threshold value of 99.96% of maximum. The overall system performance over all four phantoms and flow speeds tested was sensitivity = 0.993 and the false alarm rate = 0.074 / minute.
3.2 Testing in Mice In Vivo
We next tested the CV-IVFC in nude mice injected with very low concentrations (injected concentrations of ~2,500 cells / mL peripheral blood) of fluorescently-labeled multiple myeloma (MM) cells. An example fluorescence image sequence acquired in vivo is shown in figure 4a–e. By inspection, the noise and background autofluorescence (for example, from sebaceous glands on the ear (28)) observed was significantly higher than in phantoms, and made discrimination of cells in individual frames (red arrows with dotted circles) more difficult. As is evident in figs 4f–j, a much larger number of “cell candidates” (i.e. pixels that exceed the threshold after background subtraction) were identified after Step 1, which greatly outnumbered the true numbers of cells. However, the vast majority of these cell candidates did not form cell tracks as defined in Step 2 and were therefore rejected by our algorithm (please see section 3.3 below). The trajectories of the subset of cell candidates that did form tracks (in Step 2) were stored and counted by the CV-IVFC algorithm. Example tracks of two circulating cells are shown in fig 4k–o, overlaid on white light images of the mouse ear vasculature. In this case (and in about 95% of cells observed), these cell tracks appeared to correlate to the large blood vessels indicated by dark regions in the white light image. This specific example also demonstrates the ability of the algorithm to distinguish between multiple circulating cell tracks in the same image sequence (indicated by green and yellow tracks). Likewise, figure 5 shows an example case where a single cell changed speed and direction rapidly as it moved from one blood vessel to another (in figs 5c,h,m). Other example cell detection events are shown in the movie files included online (“cell_detection1.avi” to “cell_detection4.avi”). It is possible that a given volume of peripheral blood may pass twice through the imager field of view so that this could potentially result in over-counting of cells. In the future, this effect could be corrected by considering the direction of movement (distal or proximal) and adjusting the cell count accordingly.
Figure 4.

(a–e) An example fluorescence image sequence obtained from the ear of a mouse, where each image is separated by 0.4 s. Two fluorescently-labeled MM cells (red arrows and dotted circles) passed through the field of view, with the second cell appearing in (c) and the first disappearing in (e). By inspection, these were difficult to discriminate from autofluorescence in a single image. (f–j) The corresponding image sequence is shown after the background subtraction and thresholding operation performed in Step 1. The relatively large number of “false alarm” cell candidates in the sequence are evident, but these were rejected by dynamic analysis of the image sequence in Step 2 (see text section 2.2. for details). (k-0) The extracted tracks of the two cell candidates are shown (here indicated by green and yellow curves), overlaid on the white light image of the mouse ear vasculature.
Figure 5.

(a–e) Example fluorescence image sequence of a single MM cell (red arrows and dotted circles) that was observed to rapidly change direction in two blood vessels in the mouse ear. Images shown were separated by 0.4 s. (f–j) Corresponding images sequence after thresholding operation performed in Step 1, and (k-0) the full track of the MM cell that was correctly recovered and over-laid on the white light image of the mouse ear.
Cells that were moving rapidly were often recorded as “streaks” rather than single points (for example, as observed in “cell_dection3.mov”, about 1.5 s into the video). In general this had the effect of reducing the recorded intensity, i.e. since the intensity was divided over the pixels in the streak. However, as long as the intensity exceeded the threshold (Step 1B) and the object appeared in multiple frames of an image sequence (Step 2B and 2C) it was recorded as a cell track. Moreover, the ear was easily immobilized (by securing the mouse and using a drop of water on the ear) so that image sequences were generally free of breathing artifacts. It is also worth re-iterating that the injected cell concentrations used here were extremely low and were generally below the operating range of other reported optical IVFC techniques. Therefore, occurrences of circulating cells passing through the imager field of view were very rare compared to the total length of video sequences (specifically about 0.5 per minute on average) thereby underscoring the importance of the automated detection algorithm.
While it is difficult to exactly quantify the number of cells in circulation during these experiments (as opposed to the injected concentration), we estimate this concentration as follows: we injected on average 5 × 103 labeled MM cells suspended in 100 μL of media, which were diluted in the ~2 mL blood volume for a total injected concentration of approximately 2.5 × 103 cells per mL of peripheral blood. From previous analysis of extracted PB samples, it is estimated that about approximately 10–20% of injected cells were retained in circulation 5 minutes after injection, with the balance either trapped at the site of the tail vein injection, or trapped rapidly in the lungs and in the spleen after injection of the initial bolus. Moreover, MM cells are known to home to the bone marrow continuously during circulation, so that we estimate that overall there was 250–500 cells / mL in circulation during these experiments. At this concentration, we observed 0.5 cells per minute over all experiments performed. Assuming that the lower limit of sensitivity for our system would occur when only 1 cell observed in a 60 minute period, this is about 17 cells / mL. Conservatively then, we estimate that the practical lower limit of detection sensitivity of our approach is about 20 cells / mL. To better quantify the true circulating cell population in the future, we could extract peripheral blood samples and analyze them using conventional flow cytometry. However, the low concentrations of circulating cells used here would necessitate euthanizing the mouse and analyzing peripheral blood volumes on the order of 1 mL. Moreover, measurement of cell populations at multiple time points in the 30 minute period following injection would be required because MM cells are known to clear rapidly from circulation (6,7).
3.3 Performance Metrics
In figs 4 and 5, the threshold value τ used in Step 1 (eqn 2) was arbitrarily selected to be the 99.96th percentile of all measured pixels in the image sequence. However, the overall performance of the algorithm could be controlled by adjusting this threshold. The concept is shown in figure 6a, where the distribution of background (autofluorescence) noise is shown for a typical mouse (blue curve), along with the distribution of fluorescence intensities of circulating cells (red curve). Here, all pixel values for all image frames of full 30 minute acquisition for a single mouse are represented. Data is presented following mean background subtraction (i.e. Step 1A), so that that mean value for the background is zero but the standard deviation was 980 counts. Likewise the mean and standard deviation of the cell intensities were 7304 counts and 1955 counts, respectively. The dotted vertical line indicates the value of the 99.96th percentile (i.e. the threshold) which in this case was 4200 counts. Although only a small overlap between the blue and red curves is evident by inspection, it should be emphasized that the number of points in the blue distribution outnumbers the number of points in the red distribution by a factor of 104; as such, this small overlap results in many potential false positive cell candidates that are removed in Step 2 of the algorithm. We therefore quantified the performance of our CV-IVFC method over all 6 in vivo data sets according to sensitivity and FAR as a function of the selected threshold. As shown in fig. 6b, use of a lower threshold (i.e. 99.93rd percentile) resulted in a sensitivity of better than 0.9, but resulted in a relatively high FAR of 1.5 per minute (fig. 6c). Increasing this threshold (e.g. to 99.99th percentile) reduced to false alarm rate to only 0.04 per minute on average, but likewise this resulted in a reduction of sensitivity to 0.65. Therefore, it is possible for the end user to adjust this threshold to trade-off sensitivity and FAR depending on the specific cell-counting application. This is illustrated explicitly in the operating curve shown in fig 6d. As we have noted (and by inspection of the image sequences) the use of simple background subtraction and thresholding (in Step 1) was not sufficient to identify circulating cells in the image sequences. This is shown in fig. 6e, where the FAR realized after Step 1 (threshold step) and Step 2 (merging step) as described in detail in fig. 2 and section 2.2 above shown. As indicated, the dynamic merging analysis performed in Step 2 reduced that FAR by at least 97% for all threshold values used. As such, the second step was critical for the CV-IVFC approach, as noted primarily due to substantial tissue autofluorescence and EMCCD imager noise. Moreover, we note that less brightly labeled cells (e.g. with a fluorophore with smaller extinction coefficient or quantum yield) would result in a shift of the dotted curve to the left, so that more overlap between the distributions and a greater potential increase in the false alarm rate would be observed. Similarly, an increase in autofluorescence noise (e.g. if a visible fluorescent dye were used) would result in increased overlap. In this case, the threshold in Step 1b could be increased, or the radius of merging (Steps 2b,c) decreased to reduce the FAR, but this would most likely come at a reduction in sensitivity. Likewise, a microscope objective with greater magnification could be used to reduce the autofluorescence noise, but this would come at a cost of a smaller imaging area and less overall sensitivity.
Figure 6.

Performance metrics of the computer vision algorithm as a function of the threshold level (as a function of maximum pixel intensity – see text for details) over all 6 mice studied. (a) Example distributions of background signal (blue curve) and cell fluorescence signal (red curve) following mean background subtraction. Data shown is for all pixels in a 30 minute image sequence from a single mouse. The dotted vertical line indicates a threshold of 99.96 percentile, which in this case was 4200 counts. b) Sensitivity of the CV-IVFC method in vivo as a function of threshold level. (c) The corresponding false alarm rate (FAR) as a function of threshold, and (d) the sensitivity and false alarm rate operating curve. (e) The overall average false alarm rate (FAR) as a function of threshold, obtained over the six mice studied after Step 1 and Step 2 of the detection algorithm. As shown, the dynamic analysis in Step 2 resulted in rejection of >97% of false alarms over all thresholds tested.
Finally, we note that in addition to detection and counting, our computer vision IVFC approach allows us to automatically extract cell behavior information. For example, as shown in the histogram in figure 7, we extracted the speed of circulating cells from the detected cell tracks. These data were generated from a total of 85 tracked cells, and the mean speed from each cell is displayed (i.e. since cells frequently changed speed during the track). A wide range of speeds were observed, and the mean speed was about 1.2 mm / s over the experiments performed which is in good agreement with literature values. We also observed events such as rapid changes in flow speed (supplemental video “cell_detection3.avi”); in this case, the cell rapidly changed speeds from 7.4 mm / s to 1.5 mm / s, presumably as it entered a smaller region of the blood vessel. Generally the range of cells speeds observed were consistent with previously reported literature values in the mouse ear. For example Novak et. al. (6) reported flow speeds of 1–3 mm / s, and Zharov et. al. (12) reported flow speeds of 0.5–4 mm / s. It is possible that faster moving cells were recorded less frequently than slower moving cells due to the streak effect described above. However, observed tracks consistently coincided with the location of larger blood vessels with larger blood flow rates so that we do not believe this was a major effect. MM cells were also observed to stop at a site in the tissue (e.g. supplemental video “cell_detection4.avi”), which we interpret to have been a possible docking event at the blood vessel wall. In the future, we could modify our algorithm to automatically identify and characterize homing and extravasation events for example, for immune cells in response to inflammatory injury (37). As such, this approach can yield information that cannot be obtained with existing IVFC techniques that simply count cell events.
Figure 7.
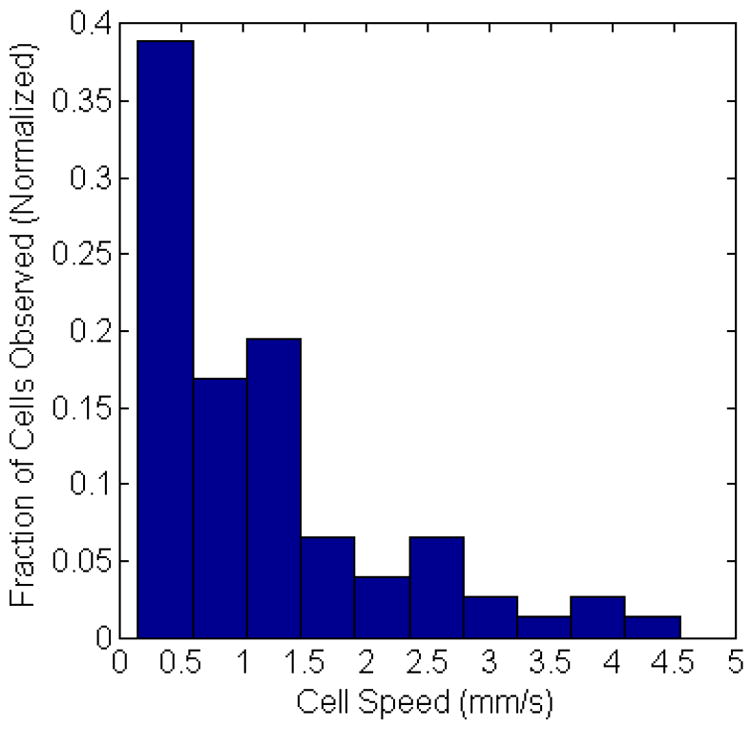
Incidence of cell speed range observed from all tracked cells in vivo, normalized to the total number of cell observations. The average cell speed was about 1.1 mm/s.
Discussion and Conclusions
In summary, we have developed a new computer vision IVFC method for detection and enumeration of very rare circulating cells. The CV-IVFC algorithm utilized a fairly straightforward “detect-and-connect” methodology that nonetheless resulted in a powerful IVFC instrument. The main advantage of the CV-IVFC approach is the high detection sensitivity, owing to the relatively large imaging area (5 × 5 mm2) and correspondingly large blood sampling volume. In the experiments shown here, 3–4 large artery-vein pairs in the ear and the surrounding capillary bed were typically simultaneously sampled. To our knowledge, this has never been implemented previously. In future work, it could also allow us to automatically characterize cell behavior in vivo, for example, in studying the adhesion, rolling and extravasation of immune cells in response to insult. Moreover, although we have chosen to focus on rare circulating cells in the present work in principle the algorithm would also work with higher circulating cell concentrations. As shown in figure 4, multiple cells could be tracked simultaneously in an image sequence, so that operation with several orders of magnitude greater cell concentrations is feasible.
While in principle the automated computer vision algorithm is “optional” (since a human operator could manually count cells in a fluorescence video sequence) in the case of the rare circulating cell populations shown here the frequency of cell detection events was 0.5 per minute or less on average, thereby making this extremely tedious in practice. Another advantage of our computer vision IVFC approach is that false positives identified by the algorithm can easily be rejected by a human operator, simply by reviewing the video sequence and checking the identified cells (again, this is not possible with many existing IVFC systems). The use of the dynamic analysis (Step 2) rejected > 97% of false positives, making this a much less time consuming activity.
Although we have chosen to use a cell membrane-labeling fluorophores (Vybrant-DiD) for these proof of concept experiments, in principle the CV-IVFC system could be used for a wide range of biological models using, for example, constitutively expressed red fluorescent proteins (RFPs), although to date we have not explicitly tested this. RFPs would most likely yield less-brightly labeled cells than the Vybrant-DiD dye used here. Comparison of the extinction coefficients quoted in the product literature, for example, for Turbo-FP650 (Evrogen Inc.) and Vybrant DiD (Life Technologies Inc.) suggests that RFP-MM labeled cells would be on the order of 3 times less brightly-labeled than the cells used here (although a literature search failed to reveal the relative quantum yields and expected local fluorophore concentrations of labeled cells which also contribute to brightness). The fluorescence distributions presented in fig. 6a imply that cells 3 times less bright would be detectable, but with a greater false alarm rate (or decreased sensitivity). Again, this could be accommodated by adjustment of the merging algorithm in Step 2 or use of a higher powered objective with smaller field of view but better contrast. Likewise, receptor targeted near-infrared fluorescent nano-particles (29,30) in principle could be used with the instrument. Moreover, we have chosen to use a near-infrared dye here but in principle the CV-IVFC approach could be used for virtually any visible or near-infrared fluorophore, simply by changing the laser and filter combinations of the instrument. Again, we have not yet explicitly tested this and we anticipate that the associated increase in background autofluorescence noise may result in increased false alarm rates, which would also necessitate optimization of the instrument and cell tracking algorithm. Optimization of the collection optics, for example, through alternative selection of filters or removal of the second linear polarizer may also improve collection of fluorescence light and further improve sensitivity of the instrument in future versions.
Finally, we note that the false alarm rate could be reduced simply by restricting the area under consideration to regions corresponding to large artery-vein pairs (i.e. the dark regions) on white light images. We chose not to do this here since we observed a small fraction of cells (about 5%) moving between these larger vessels, presumably in the capillary bed in the ear. As such this improvement in FAR would have come at a penalty of about 5% in sensitivity, which was deemed to be more important for the CV-IVFC approach. Moreover, MM cells were intravenously injected immediately prior to imaging in the case and therefore were generally circulating in larger blood vessels. In the case of, for example, a metastatic cancer model this fraction could be significantly higher and have even greater effect on the overall instrument sensitivity.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R21HL098750; NHLBI) and the Massachusetts Life Sciences Center to M. Niedre. The assistance of Mr. Dwayne Vickers in these experiments is greatly appreciated.
Footnotes
Conflicts of Interest:
The authors declare no conflicts of interest.
References
- 1.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–91. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 2.Liu MC, Warren RD, Cohen P, Wilkinson M, Occaviano YL, Rao SB, Eng-Wong J, Seillier-Moiseiwitsch F, Noone A, Isaacs C. Circulating Tumor Cells: A Useful Predictor of Treatment Efficacy in Metastatic Breast Cancer. J Clin Oncol. 2009;27:5153–5159. doi: 10.1200/JCO.2008.20.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Economos CM, Vessella RL. Circulating tumor cells as a marker of response: implications for determining treatment efficacy and evaluating new agents. Curr Opin Urol. 2012;22:190–196. doi: 10.1097/MOU.0b013e3283519b58. [DOI] [PubMed] [Google Scholar]
- 4.Mocellin S, Hoon D, Ambrosi A, Nitti D, Rossi CR. The prognostic value of circulating tumor cells in patients with melanoma: a systematic review and meta-analysis. Clin Cancer Res. 2006;12:4605–13. doi: 10.1158/1078-0432.CCR-06-0823. [DOI] [PubMed] [Google Scholar]
- 5.Allard JM, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW, Terstappen LWMM. Tumor Cells Ciculate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients With Nonmalignant Diseases. Clin Cancer Res. 2004;10:6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 6.Novak J, Georgakoudi I, Wei X, Prossin A, Lin CP. In vivo flow cytometer for real-time detection and quantification of circulating cells. Opt Lett. 2004;29:77–9. doi: 10.1364/ol.29.000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alt C, Veilleux I, Lee H, Pitsillides CM, Cote D, Lin CP. Retinal flow cytometer. Opt Lett. 2007;32:3450–2. doi: 10.1364/ol.32.003450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoff J. Methods of Blood Collection in the Mouse. Lab Animal. 2000:47–53. [Google Scholar]
- 9.Tkaczyk ERZC, Ye JY, Myc A, Thomas T, Cao Z, Duran-Struuck R, Luker KE, Luker GD, Norris TB, Baker JR. In Vivo Monitoring of Multiple Circulating Cell Populations Using Two-photon Flow Cytometry. Opt Commun. 2008;281:888–894. doi: 10.1016/j.optcom.2007.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong CF, Tkaczyk ER, Thomas T, Ye JY, Myc A, Bielinska AU, Cao Z, Majoros I, Keszler B, Baker JR, et al. Quantitative two-photon flow cytometry--in vitro and in vivo. J Biomed Opt. 2008;13:034008. doi: 10.1117/1.2931077. [DOI] [PubMed] [Google Scholar]
- 11.Zharov VP, Galanzha EI, Shashkov EV, Kim JW, Khlebtsov NG, Tuchin VV. Photoacoustic flow cytometry: principle and application for real-time detection of circulating single nanoparticles, pathogens, and contrast dyes in vivo. J Biomed Opt. 2007;12:051503. doi: 10.1117/1.2793746. [DOI] [PubMed] [Google Scholar]
- 12.Zharov VP, Galanzha EI, Shashkov EV. In vivo photoacoustic flow cytometry for monitoring of circulating single cancer cells and contrast agents. Opt Lett. 2006;31:3623–3625. doi: 10.1364/ol.31.003623. [DOI] [PubMed] [Google Scholar]
- 13.Zharov VP, Galanzha EI, Tuchin VV. Photothermal image flow cytometry in vivo. Opt Lett. 2005;30:628–30. doi: 10.1364/ol.30.000628. [DOI] [PubMed] [Google Scholar]
- 14.Tuchin VV, Tarnok A, Zharov VP. Towards in vivo flow cytometry. J Biophoton. 2009;2:457–547. doi: 10.1002/jbio.200910546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tuchin VV. Advanced Optical Cytometry: Methods and Disease Diagnoses. Front Matter. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2011. pp. I–XXXVII. [Google Scholar]
- 16.Tuchin VV, Tarnok A, Zharov VP, editors. Special Issue: In Vivo Flow Cytometry. Cytometry A. 2011;79A:737–883. doi: 10.1002/cyto.a.21143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuchin VV, Tarnok A, Zharov VP. In Vivo Flow Cytometry: A Horizon of Opportunities. Cytometry A. 2011;79A:737–745. doi: 10.1002/cyto.a.21143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galanzha EISE, Spring P, Suen JY, Zharov VP. In Vivo, Non-invasive, Label- Free Detection and Eradication of Circulating Metastatic Melanoma Cells using Two-Color Photoacoustic Flow Cytometry with a Diode Laser. Cancer Res. 2009;69:7926–7934. doi: 10.1158/0008-5472.CAN-08-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galanzha EIZV. Photoacoustic flow cytometry. Methods. 2012;57:280–96. doi: 10.1016/j.ymeth.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azab AK, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, Maiso P, Sun JD, Hart CP, Roccaro AM, Sacco A, Ngo HT, Lin CP, Kung AL, Carrasco RD, Vanderkerken K, Ghobrial IM. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119:5782–94. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forero MG, Hidalgo A. DeadEasy Neurons: Automatic Counting of HB9 Neuronal Nuclei in Drosophila. Cytometry. 2012;77A:371–378. doi: 10.1002/cyto.a.20877. [DOI] [PubMed] [Google Scholar]
- 22.Bauer KD, Diel IJ, Hawes D, Decker WJ, Priddy C, Bossy B, Ludmann S, Yamamoto K, Masih AS, Espinoza FP, Harrington DS. Reliable and Sensitive Analysis of Occult Bone Marrow Metastases Using Automated Cellular Imaging. Clin Cancer Res. 2000;6:3552–3559. [PubMed] [Google Scholar]
- 23.Lamprecht MR, Carpenter AE. CellProfiler™: free, versatile software for automated biological image analysis. BioTechniques. 2007;42:71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- 24.Wang C-W. Fast automatic quantitative cell replication with fluorescent live cell imaging. BMC Bioinformatics. 2012;13:1–15. doi: 10.1186/1471-2105-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraeft S-K, Gravelin L, Hu G-H, Ferland LH, Richardson P, Elias A, Chen LB. Detection and Analysis of Cancer Cells in Blood and Bone Marrow Using a Rare Event Imaging System. Clin Cancer Res. 2000;6:434–442. [PubMed] [Google Scholar]
- 26.Lowes LE, Keeney M, Allan AL. Image Cytometry Analysis of Circulating Tumor Cells. Methods Cell Biol. 2011;102:261–290. doi: 10.1016/B978-0-12-374912-3.00010-9. [DOI] [PubMed] [Google Scholar]
- 27.Waldmeier L, Diepenbruck M, Christofori G. Py2T Murine Breast Cancer Cells, a Versatile Model of TGFb-Induced EMT In Vitro and In Vivo. PLoS One. 2012;7:e48651. doi: 10.1371/journal.pone.0048651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barral PP, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, Batista FD. CD169+ macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol. 2010;11:303–312. doi: 10.1038/ni.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Worbs TRM, Bölter J, von Andrian UH, Förster R. CCR7 ligands stimulate the intranodal motility of T lymphocytes in vivo. JEM. 2007;204:489–495. doi: 10.1084/jem.20061706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdulreda MH, Molano RD, Lopez-Cabezas M, Molina J, Tan Y, Ron Echeverria OA, Zahr-Akrawi E, Rodriguez-Diaz R, Edlund PK, Leibiger I, Bayer AL, Perez V, Ricordi C, Caicedo A, Pileggi A, Berggren P-O. High-resolution, noninvasive longitudinal live imaging of immune responses. Proc Natl Acad Sci USA. 2011;108:12863–8. doi: 10.1073/pnas.1105002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srinivas S, Clements M, Smith JC, Beddington RSP. Active cell migration drives the unilateral movements of the anterior visceral endoderm. Development. 2004;131:1157–1164. doi: 10.1242/dev.01005. [DOI] [PubMed] [Google Scholar]
- 32.Cseresnyés MBaZ. Cell rearrangements, cell divisions and cell death in a migrating epithelial sheet in the abdomen of Drosophila. Development. 2009;136:2403–2411. doi: 10.1242/dev.035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao Q, Cai W, Li ZB, Chen K, He L, Li HC, Hui M, Chen X. PET imaging of acute and chronic inflammation in living mice. Eur J Nucl Med Mol Imaging. 2007;34:1832–42. doi: 10.1007/s00259-007-0451-0. [DOI] [PubMed] [Google Scholar]
- 34.Knosp CA, Carroll HP, Elliott J, Saunders SP, Nel HJ, Amu S, Pratt JC, Spence S, Doran E, Cooke N, Jackson R, Swift J, Fitzgerald DC, Heaney LG, Fallon PG, Kissenpfennig A, Johnston JA. SOCS2 regulates T helper type 2 differentiation and the generation of type 2 allergic responses. J Exp Med. 2011;208:1523–31. doi: 10.1084/jem.20101167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baeten J, Niedre M, Dunham J, Ntziachristos V. Development of fluorescent materials for Diffuse Fluorescence Tomography standards and phantoms. Opt Express. 2007;15:8681–94. doi: 10.1364/oe.15.008681. [DOI] [PubMed] [Google Scholar]
- 36.Dintzis PMTaS. Comparative Anatomy and Histology: A Mouse and Human Atlas. Waltham, MA: Elsevier; 2011. p. 464. [Google Scholar]
- 37.Saxena A, Thompson B, McCarthy JR, Iwamoto Y, Lin CP, Jaffer FA. High-Resolution Optical Mapping of Inflammatory Macrophages Following Endovascular Arterial Injury. Mol Imaging Biol. 2012 doi: 10.1007/s11307-012-0599-2. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutton EJ, Henning TD, Pichler BJ, Bremer C, Daldrup-Link HE. Cell tracking with optical imaging. Eur Radiol. 2008;18:2021–32. doi: 10.1007/s00330-008-0984-z. [DOI] [PubMed] [Google Scholar]
- 39.Filonov GS, Piatkevich K, Ting LM, Zhang J, Kim K, Verkhusha VV. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol. 2011;29:757–61. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giepmans BNG, Ellisman MH, Tsien RY. The Fluorescent Toolbox for Assessing Protein Location and Function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 41.Pitsillides CM, Spencer JA, Zhi L, Wu MX, Lin CP. Cell labeling approaches for fluorescence-based in vivo flow cytometry. Cytometry A. 2011;79A:758–765. doi: 10.1002/cyto.a.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo S, Zhang E, Su Y, Cheng T, Shi C. A review of NIR dyes in cancer targeting and imaging. Biomaterials. 2011;32:7127–38. doi: 10.1016/j.biomaterials.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 43.Tung CH. Fluorescent Peptide Probes for In Vivo Diagnostic Imaging. Biopolymers. 2004;76:391–403. doi: 10.1002/bip.20139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.