

Abstract
Blood–brain barrier (BBB) disruption is a pathological hallmark of severe traumatic brain injury (TBI) and is associated with neuroinflammatory events contributing to brain edema and cell death. The goal of this study was to elucidate the profile of BBB disruption after penetrating ballistic-like brain injury (PBBI) in conjunction with changes in neuroinflammatory markers. Brain uptake of biotin-dextran amine (BDA; 3 kDa) and horseradish peroxidase (HRP; 44 kDa) was evaluated in rats at 4 h, 24 h, 48 h, 72 h, and 7 days post-PBBI and compared with the histopathologic and molecular profiles for inflammatory markers. BDA and HRP both displayed a uniphasic profile of extravasation, greatest at 24 h post-injury and which remained evident out to 48 h for HRP and 7 days for BDA. This profile was most closely associated with markers for adhesion (mRNA for intercellular adhesion molecule-1) and infiltration of peripheral granulocytes (mRNA for matrix metalloproteinase-9 [MMP-9] and myeloperoxidase staining). Improvement of BBB dysfunction coincided with increased expression of markers implicated in tissue remodeling and repair. The results of this study reveal a uniphasic and gradient opening of the BBB after PBBI and suggest MMP-9 and resident inflammatory cell activation as candidates for future neurotherapeutic intervention after PBBI.
Key words: : blood–brain barrier dysfunction, inflammation, penetrating brain injury, PBBI, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability in the United States and represents one of the most prevalent types of injuries sustained by military personnel serving in Iraq and Afghanistan. Of more than 16 million military personnel involved in conflicts since 2001, 12–35% have sustained TBI. Recent reports indicate that the vast majority (more than 80%) of combat-related TBI result from blast explosion.1 Moreover, approximately 70% of severe blast-induced injuries are confounded by a penetrating injury to the brain.2 In contrast to non-penetrating TBI, few experimental studies have been conducted on penetrating TBI, which not only constitutes a major threat to our military,3 but is also the leading cause of TBI-related death in our civilian population.4–6
Penetrating ballistic-like brain injury (PBBI) in rats has been established as a military-relevant model of TBI and can be calibrated to simulate the leading pressure or shockwave to the brain from penetrating projectiles of low or high velocities.7 The 10% unilateral, frontal PBBI rodent model has been well characterized and reproduces acute neuropathological aspects of penetrating brain injury seen in humans, including lacerated brain damage, intracerebral hemorrhage, increased intracranial pressure (ICP), axonal degeneration, upregulation of pro-inflammatory cytokines, electrocortical disturbances, and neurofunctional impairment.7–14
The blood–brain barrier (BBB) is a highly specialized structure crucial for maintaining homoeostasis of the central nervous system (CNS) and limiting exposure to damaging cells and molecules. Breakdown of the BBB is a pathological hallmark of severe TBI and is associated with infiltration of peripheral fluid and leukocytes into the CNS leading to sequelae such as edema formation, neuroinflammation, and cell death.15–17 Status of the BBB is of critical importance in the treatment of CNS disorders because the majority of large- and small-molecule neurotherapeutic agents are unable to cross into the cerebrovasculature from the peripheral circulation.18 Elucidation of the dynamics of BBB dysfunction post-PBBI would provide important information to guide the selection of therapeutic agents and timing of treatment.
Methods
Subjects
Male Sprague-Dawley rats (245–325 g, Charles River Labs, Raleigh, VA) were used in the study. All procedures were approved by the Institutional Animal Care and Use Committee of Walter Reed Army Institute of Research. Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals, and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council (NRC) Publication, 1996 edition. Animals were housed individually under a 12 h light/dark cycle (lights on at 0600) in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
General surgical procedures
All surgical procedures were performed on anesthetized animals. Anesthesia was induced with isoflurane (2–5%) delivered in oxygen. Body temperature was maintained at 37±1°C throughout all surgical procedures by means of a homeothermic heating system (Harvard Apparatus, South Natick, MA). Indwelling intravenous (IV) cannulas were microsurgically placed into the right jugular vein of all animals for injection of horseradish peroxidase (HRP) or biotin-dextran amine (BDA) in accordance with the Thrivikraman protocol.19 After surgery, the animals were placed in a clean cage and kept warm by a circulating water-bath heating system (Gaymar Industries, Orchard Park, NY) until recovery from anesthesia. Food and water were provided ad libitum postoperatively.
Penetrating ballistic-like brain injury
Unilateral frontal PBBI was induced by stereotactic insertion of a custom probe through the right frontal cortex, and a rapid inflation/deflation (<40 msec) of an attached, elastic water-filled balloon to create a temporary cavity in the brain. The temporary cavity mimics that caused by the ballistic energy dispersion from a bullet or shrapnel.7,12 More specifically, the PBBI apparatus consisted of a computer-controlled hydraulic pressure generator (Mitre Corp., McLean VA), a PBBI probe (with balloon), and a stereotaxic frame equipped with a custom-designed probe holder. The probe was made of a 20 g stainless steel tube with fixed perforations along one end, which was sealed by a piece of elastic tubing (1 cm). The probe was secured on the probe holder attached to the stereotaxic frame angled at 50 degrees from the vertical axis and 25 degrees counterclockwise from the midline. The other end of the probe was connected to the hydraulic pressure generator, which, on activation by a computer, could inflate (and deflate) the elastic tubing on the probe to an elliptical-shaped balloon. The injury severity of the PBBI was determined by the size of the water-filled balloon under control of the hydraulic pressure. A balloon diameter calibrated to be 0.63 cm represents 10% of the total rat brain volume, and therefore defines the 10% PBBI.
During the PBBI surgery, the rat was secured in the stereotaxic frame under 2% isoflurane anesthesia. The scalp was incised along the midline and a craniectomy was performed (4 mm diameter, +4.5 mm anterioposterior (AP), +2 mm mediolateral (ML) from bregma) to expose the right frontal pole of the brain. The PBBI probe was manually advanced through the cranial window into the right frontal hemisphere to a distance of 1.2 cm from the surface of the cortex. Once it was in place, the hydraulic pressure generator was activated to inflate/deflate the water balloon. The probe was then retracted from the brain, the cranial opening sealed with sterile bone wax, and the incision closed with sterile wound clips followed by the administration of topical antibiotic. Sham controls received craniectomy without insertion of the PBBI probe.
Tracer administration
Experimental groups included PBBI+HRP and PBBI+BDA (n=5–6/group/time point) euthanized at 4 h, 24 h, 48 h, 72 h, and 7 days post-PBBI. Before euthanasia, animals received a single (IV) injection of either 5 mg HRP (44 kDa, Sigma-Aldrich, St. Louis, MO) or 3 mg BDA (3 kDa, Invitrogen, Carlsbad, CA) dissolved in 0.5 mL physiologic saline. The tracers were allowed to circulate for 30 minutes before perfusion with physiologic saline followed by 4% paraformaldehyde (∼3 min). Appropriate injured (PBBI alone, n=6) and non-injured (sham with either HRP or BDA, n=4/group) control groups were included at the 24 h post-injury time point only.
Histopathology
At the indicated time point, animals were fully anesthetized with ketamine/xylazine (70 and 6 mg/kg, intramuscular [IM], respectively) and transcardially perfused with physiologic saline followed by ice-cold 4% paraformaldehyde. Brains were extracted, immersed in 4% paraformaldehyde for 24 h, and then transferred to 0.1 M phosphate buffer containing 20% sucrose (pH 7.4, 4°C). All brain tissue was sent to FD Neurotechnologies (Ellicott City, MD) for histopathological and immunohistochemical processing. After immersion in 0.1 M phosphate buffer (PB) containing 20% sucrose for 72 h at 4°C, brains were snap frozen and stored at −75°C. Serial sections (40 mm thick) were cut coronally through the cerebrum from +4.0 to −7.0 mm (AP) from bregma. Six sets of serial sections (22 sections per set per rat) were collected at 1 mm intervals. The first set was mounted on Superfrost plus microscopic slides and were stained with hematoxylin and eosin (H&E) for gross morphological assessment of injury. The remaining sets were stored as free floating sections.
Immunohistochemistry
Brain sections from animals injected with BDA were first treated with hydrogen peroxidase to block the endogenous peroxidase activity. After thorough rinsing (3×10 min) in 0.01 M phosphate-buffered saline (PBS; pH 7.4), sections were processed according to the previously established avidin-biotin complex method20 using the Vectastin elite ABC kit (Vector Lab., Burlingame, CA). Sections were incubated in PBS containing 0.3% Triton X-100 (Sigma, St. Louis, MO) and avidin-biotinylated HRP complex for 1 h. Sections were then incubated for 10 min in 0.05 M Tris buffer (pH 7.2) containing 0.03% 3′,3′-diaminobenzidine (Sigma) and 0.0075% H2O2. All steps were performed at room temperature, and each step was followed by washes in PBS. After thorough rinses in distilled water, all sections were mounted on slides, dehydrated in ethanol, cleared in xylene, and coverslipped in Permount® (Fisher Scientific, Fair Lawn, NJ).
Brain sections from animals injected with HRP were rinsed (3×10 min) in 0.01 M PBS (pH 7.4) then incubated free floating in PBS containing 1% normal horse serum (Vector Lab., Burlingame, CA), 0.3% Triton X-100 (Sigma, St. Louis, MO), and a mouse monoclonal HRP (2H11) antibody (1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA) for 3 days at 4°C. Subsequently, the immunoreaction product was visualized according to the same avidin-biotin complex method using Vectastin elite ABC kit detailed above with one alteration. Before incubation with avidin-biotin complex, sections were incubated in PBS containing biotinylated horse anti-mouse IgG, Triton-X, and normal horse serum for 1 h.
Additional sets of free floating sections were processed for glial fibrillary acidic protein (GFAP) for detection of activated astrocytes, rat MHC class I (OX-18) for detection of activated microglia, and myeloperoxidase (MPO) for detection of neutrophils,21 respectively. After inactivating the endogenous peroxidase activity with hydrogen peroxidase, sections were incubated separately with avidin and biotin solutions (Vector Lab, Burlingame, CA) for blocking nonspecific binding of biotin, biotin-binding protein, and lectins. Sections were then incubated free floating in 0.01 M PBS (pH 7.4) containing 1% normal blocking serum, 0.3% Triton X-100, and one of the following specific antibodies: rabbit anti-GFAP (1:8,000; DAKO, Carpinteria, CA), mouse anti-OX-18 (1: 6,000; AbD Serotec, Raleigh, NC), and rabbit anti-MPO (1:8,000; DAKO) for 1 h. Subsequently, the immunoreaction product was visualized according to the avidin-biotin complex method with the Vectastin elite ABC kit detailed above for BDA processing. Critically, all tissue sections were processed simultaneously in batches based on each respective antibody to ensure consistent and uniform staining results.
Immunohistochemical analysis
For unbiased evaluation of histopathology, digital images of immunostained sections were captured using a BX61 microscope (Olympus, PA) at uniform criteria for sensitivity and exposure time. For HRP and BDA, digital images (2×) were captured for four consecutive sections spaced 200 μm apart from +2.26 mm to −3.80 mm to bregma. Digital images of MPO, GFAP, and OX-18 immunostaining were taken at 4×magnification to evaluate positive staining in regions of interest (i.e., the injured hemisphere and perilesional regions). Additional, high magnification (20×for GFAP; 40×for OX-18) images were captured from these sections to quantify reactive astrocytes (GFAP) and activated microglia (OX-18). Threshold quantification of GFAP-positive staining on 20×pictures was assessed in the thalamus and hippocampus (CA1 and CA3 regions). Threshold quantification of OX-18-positive staining on 40×pictures was evaluated in the parietal cortex, thalamus, and hippocampus (CA1 and CA3 regions).
Computerized thresholding analysis was performed using ImageJ software (NIH, Version 1.44) as it has been used similarly in previous studies22–24 to measure the extent of positive staining in areas of different size/shape. In this study, the area (mm2) of specific regions of interest may vary across animals because of animal variability, especially in the area along the injury tract.12 Thus, we chose to apply the thresholding “area” technique to provide more accurate and objective assessments of the extent of protein expression for better comparison across different protein markers of interest. In part, this was based on our previous work demonstrating that threshold analysis provides a sensitive and reliable measure of neuroinflammation in a closed-head concussive model of mild TBI.22
The threshold values (i.e., grey level index) were set to consistently detect maximal positive staining of extravasated tracer or immunohistochemical targets with minimal artifacts. To ensure objective quantification, the same threshold value was applied to all brain sections for each respective protein marker. Threshold measurements on low magnification (i.e., 2×and 4×) images were made on injured and non-injured hemispheres (demarcated by tissue borders) whereas threshold measurements at high magnification (20×and 40×) were made using the entire image. Positive staining was quantified as number of pixels detected above the selected threshold for a given area. All pixel measurements were automatically converted into area measurements (mm2) according to the scale (pixels per mm) for each image. Area of positive staining was averaged across all analyzed sections for a given animal.
RNA isolation
A 3 mm (100 μg) coronal section starting at the frontal pole of the brain was divided into ipsilateral and contralateral hemisections and the ipsilateral hemisection was used for RNA isolation. Purified total RNA was isolated using the RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA). Briefly, tissue samples were homogenized using a cell disruptor in QIAzol Lysis Reagent. Chloroform was added, and the homogenate was separated into aqueous and organic phases by centrifugation at 13,000 rpm for 15 min.
The upper, aqueous layer was collected and 70% ethanol added to provide appropriate binding conditions. The sample was applied to an RNeasy spin column, where the total RNA bound to the column's membrane while the phenol and other contaminants were washed away. The RNA was then incubated in RNase-free DNase (Qiagen, Valencia, CA) for 15 min at 37°C. After DNase treatment, the membranes were washed with RW1 and RPE buffers and spun at 10,000 rpm. Lastly, the membranes were eluted with RNase-free water. Sample concentrations were measured and then samples were stored at −80°C.
cDNA synthesis for quantitative real-time polymerase chain reaction (PCR)
AffinityScript QPCR cDNA synthesis kit (Agilent technologies, Santa Clara, CA) was used for synthesis of cDNA. 20 μL reactions contained 3 μg of total RNA, 0.3 μg of oligo(dt) primers, 10 μL of Mastermix, and 1 μL of AffinityScript RT/RNase enzyme. The reaction was incubated at 25°C for 5 min, 42°C for 45 min, 95°C for 5 min, and 4°C for 5 min. cDNA was diluted 1:10 in RNase/DNase free water and then stored at −20°C until further use.
Quantitative real-time PCR
Relative amounts of mRNA were measured by QRT-PCR using Taqman assays and primer/probes (Applied Biosystems) for icam-1 (species R norvegicus, Rn00564227_m1), vcam-1 (species R norvegicus, Rn00563627_m1), sel e (species R norvegicus, Rn00594072_m1), mmp-9 (species R norvegicus, Rn01423075_g1), mmp-3 (species R norvegicus, Rn00591740_m1), and mmp-2 (species R norvegicus, Rn01538170_m1). Each sample was tested in triplicate. ΔCt levels for β-actin were used as an endogenous control to normalize ΔCt levels for each sample, and relative quantities were calculated using the formula RQ=2-ΔΔCt. Analysis of real-time amplification data was done; relative quantities were calculated using 7500 software V.2.0.1 (Applied Biosystems). Sham data were collected at the 24 h time point and used for comparison to all experimental PCR groups.
Statistical analysis
All statistical analysis of immunohistochemical quantification was performed with IBM SPSS Statistics version 20.0 using the Student t test or one-way analysis of variance (ANOVA) (using time as a factor) followed by Bonferroni post hoc analysis when appropriate. The one-way ANOVA was used to analyze significant trends over time, whereas the Student t test was used to identify between-group trends (*p<0.05; PBBI vs. 24 h sham) at specific post-injury time points. Statistical analysis of relative mRNA levels was performed with Graphpad Prism version 5.0 using one-way ANOVA followed by Dunnett and Bonferroni post hoc tests when appropriate. All data are presented as mean±standard error of the mean (SEM).
Results
Histology
As previously described,7,12 unilateral 10% frontal PBBI produced a consistent pattern of severe brain damage along the injury track. At 24 h post-PBBI, H&E-stained brain sections revealed a hemorrhagic core lesion that permeated the frontal cortex and progressed through the dorsolateral striatum toward the lateral amygdala. The lesion included laceration damage surrounded by a region of pale H&E staining representing the lack of parenchymal neuronal cells and tissues (Fig. 1).
FIG. 1.
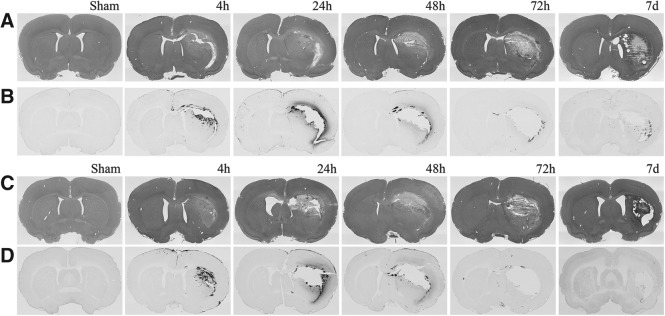
Photomicrographs of hematoxylin and eosin (H&E) staining and tracer coronal sections at 0.26 mm to bregma. H&E (A, C) and tracer specific (B, D) immunostains of tissue sections from animals injected with biotin-dextran amine (A, B) and horseradish peroxidase (C, D) shown at 2×magnification across all time points.
BBB disruption
Digitized slides for animals from PBBI+BDA, PBBI+HRP, and BDA and HRP sham control groups were evaluated microscopically to quantify the degree of tracer extravasation into parenchyma. Positive staining for both tracers appeared brown to dark-brown and could be visualized diffusely within the perilesional parenchyma (Fig. 1). The one-way ANOVA showed that both BDA and HRP displayed a uniphasic distribution of tracer extravasation that was evident by 4 h post-injury, peaked at 24 h, and was significantly reduced by 48 h post-injury, albeit not to sham levels. HRP extravasation levels remained significant out to 48 h (3.30±0.86 mm2) post-PBBI but were no longer evident at 72 h post-injury (Fig. 2). In contrast, BDA extravasation levels remained significantly elevated out to 7 days (0.80±0.18 mm2) post-PBBI despite a significant decrease at 72 h compared with levels at 24 h (Fig. 2). In addition, overall measurements of the mean area of extravasation demonstrated the extent of positive staining was greater for BDA (13.01±4.18 mm2) than HRP (6.83±3.55 mm2) at 24 h post-PBBI (p<0.05).
FIG. 2.

Quantification of tracer extravasation. Mean area of BDA (A) and HRP (B) extravasation from 4 h to 7 days post-PBBI. Data presented as means±standard error of the mean. Grey dashed line represents level of tracer extravasation in 24 h sham rats. *p<0.05 compared with sham rats by Student t test. †p<0.05 compared with the 24 h experimental group by one-way analysis of variance.
Immunohistochemistry
Microscopically, MPO was visualized as a dark-brown cytoplasmic stain. Cells staining positively for MPO were observed to be small granulocytes (predominantly neutrophils and to a lesser extent monocytes). After PBBI, immunoreactivity for MPO was significantly elevated from 4–72 h with peak elevations evident from 24 h (0.41±0.08 mm2) to 48 h (0.38±0.05 mm2) post-PBBI (p<0.05; Fig. 3). Examined sections confirmed perilesional infiltration of granulocytes into the parenchyma evident by 4 h post-injury that was significantly increased from 24–48 h (p<0.05 compared with 4 h and 72 h post-injury) and was less apparent at 72 h (Fig. 3).
FIG. 3.

Myeloperoxidase (MPO) immunohistochemical quantification and photomicrographs. (A) Mean area of MPO-positive staining from 4 h to 7 days post-PBBI. (B) Representative sections (4×and 20×magnification) in the perilesional region at 24 h and 72 h post-injury. Data presented as means±standard error of the mean. Grey dashed line represents level of positive staining in 24 h sham rats. *p<0.05 compared with sham rats by Student t test. †p<0.05 compared with the 24 h and 48 h experimental groups by one-way analysis of variance. Scale bar=50 μm.
High magnification microscopic examination of GFAP-positive astrocytes showed an activated morphology of increased dentritic branching with thicker processes and strongly staining stroma (Fig. 4). Similar to MPO, a significant increase in GFAP-positive staining in the injured hemisphere (including perilesional regions) was evident by 4 h, reached maximal levels 24–48 h after injury, and declined significantly at 72 h. Unlike MPO, however, GFAP reactivity remained significantly elevated out to 7 days post-PBBI (Fig. 4). Notably, although GFAP reactivity in perilesional and subcortical (thalamic) regions was significantly elevated (compared with sham) across all post-injury time points, ANOVA results showed that GFAP expression was significantly higher at 48 h (1.28±0.14 mm2) and 7 days (1.11±0.10 mm2) compared with 72 h (0.62±0.05 mm2) post-injury (p<0.05) suggesting a biphasic pattern of GFAP reactivity in the thalamic region (Fig. 4). Measurements of GFAP staining in the CA1 and CA3 regions of the hippocampus were not significant at any time point compared with sham controls.
FIG. 4.

Glial fibrillary acidic protein (GFAP) immunohistochemical quantification and photomicrographs. Mean area of GFAP-positive staining in the injured hemisphere (A), perilesional region (B), and subcortical region (C) from 4 h to 7 days post-PBBI. (D) Representative sections (20×) of the subcortical and perilesional immunostains at 48 h, 72 h, and 7 days. The image in the lower right corner depicts the measured regions. Data presented as means±standard error of the mean. Grey dashed line represents level of positive staining in 24 h sham rats. *p<0.05 compared with sham rats by Student t test. (A) †p<0.05 compared with the 48 h experimental group by one-way analysis of variance (ANOVA). (C) †p<0.05 compared with the 48 h and 7 day experimental groups by one-way ANOVA. Scale bar=50 μm.
Morphologic examination of sections confirmed that in regions staining intensely for OX-18, the majority of cells reactive to the OX-18 stain were activated microglia. Staining cells observed in the cortical, perilesional, and subcortical (thalamic) regions of the injured hemisphere exhibited the typical bushy, amoeboid morphology at 7 days (Fig. 5). ANOVA revealed a biphasic increase in OX-18 expression detected primarily in the perilesional regions of the injured hemisphere. Notably, a significant increase in OX-18 expression was not detected until 24 h post-PBBI, and maximum upregulation was detected at 48 h (2.67±0.26 mm2) and 7 days (3.25±0.68 mm2) post-injury. In contrast, a significant increase in subcortical expression of OX-18 was detected only at 7 days post-PBBI (p<0.05; Fig. 5). No significant increases in OX-18 expression were detected in hippocampal or cortical regions of interest.
FIG. 5.
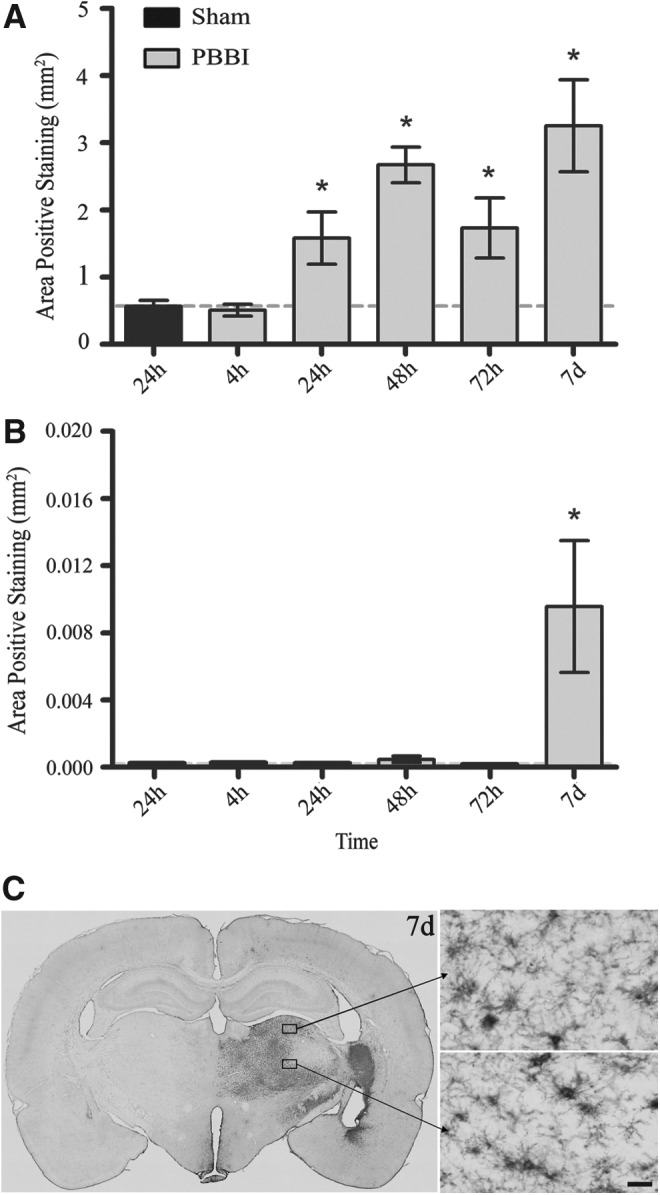
OX-18 Immunohistochemical quantification and photomicrographs. Mean area of OX-18-positive staining in the injured hemisphere (A) and subcortical region (B) from 4 h to 7 days post-PBBI. Representative sections (4×and 40×) of subcortical immunostains (C) at 7 days post-PBBI. Data presented as means±standard error of the mean. Grey dashed line represents level of positive staining in 24 h sham rats. *p<0.05 compared with sham rats by Student t test. Scale bar=30 μm.
Inflammatory gene expression profiles
QRT-PCR was used to track the time course of alterations in mRNA for selected cellular adhesion molecules (CAM) and matrix metalloproteinases (MMP) post-PBBI. The relative quantities of mRNA for icam-1 and vcam-1 were significantly upregulated within 4 h of injury. Greatest increases in CAM mRNA expression relative to 24 h sham were as follows: icam-1 (8.02±0.89 fold at 4 h; Fig. 6), and vcam-1 (1.56±0.09 fold at 4 h; Fig. 6). icam-1 and vcam-1 mRNA levels returned to sham by 24 h post-injury. Uniquely, vcam-1 mRNA levels followed an inverse parabolic curve after the significant decrease between 4 h and 24 h, reaching a minimum of 0.29±0.02 fold at 48 h, then undergoing a second significant rise to 1.36±0.17 fold mRNA (p<0.05) at 7 days (Fig. 6). Relative quantities of mRNA for sel e were not significantly changed at any time point compared with those measured in sham rats (not shown).
FIG. 6.

Relative mRNA levels for adhesion molecule genes. Quantities of relative mRNA for icam-1 (A) and vcam-1 (B) from 4 h to 7 days post-PBBI. Data presented as means±standard error of the mean. Grey dashed line represents level of relative mRNA in sham rats at 24 h post-PBBI. *p<0.05 compared with sham rats by one-way analysis of variance (ANOVA). †p<0.05 compared with the 4 h and 7 d experimental groups by one-way ANOVA.
Relative levels of mmp-9 mRNA were most increased at 24 h (4.12±1.32 fold) and returned to approximate sham levels at 72 h post-injury (Fig. 7). Alternatively, relative quantity of mmp-2 mRNA showed a delayed 3.72±0.47 fold (p<0.05) increase at only 7 days post-PBBI (Fig. 7). Relative quantities of mmp-3 were not significantly changed at any time point compared with those measured in sham rats (not shown).
FIG. 7.

Relative mRNA levels for matrix metalloproteinase genes. Quantities of relative mRNA for mmp-9 (A), mmp-2 (B) from 4 h to 7 days post-PBBI. Data presented as means±standard error of the mean. Grey dashed line represents level of relative mRNA in sham rats at 24 h post-PBBI. *p<0.05 compared with sham rats by one-way analysis of variance.
Discussion
In this study, we defined the temporal profile of BBB dynamics after a PBBI injury using more sensitive markers of BBB integrity and compared the time-course of PBBI-induced BBB dysfunction with measured changes in markers of neuroinflammation. Using BDA (3 kDa) and HRP (44 kDa) extravasation, the present study defined a profile of BBB disruption that was uniphasic and gradient after PBBI. This uniphasic profile corresponded closely to markers for adhesion (icam-1 mRNA) and infiltration (MPO staining and mmp-9 mRNA) of peripheral granulocytes, and improvement of BBB dysfunction coincided with increasing markers implicated in tissue remodeling and repair (OX-18 staining and mmp-2 mRNA).
BBB disruption time course
Loss of BBB integrity has long been considered a pathological hallmark of severe TBI, linked to neuroinflammation and to development of later neuropathological disorders.1,25,26 Using Evan's blue (EB; albumin-bound, ∼66 kDa) extravasation, we previously reported evidence of a biphasic pattern of BBB disruption after PBBI consistent with what others have reported in preclinical models of TBI or ischemia.27–30 In that study, we detected two peak phases of EB extravasation evident at 4 h post-PBBI and then again at 48–72 h post-injury.26 PBBI, however, produces significant mechanical damage to the microvasculature that may result in residual trapping of albumin-bound EB within the lumen of damaged blood vessels perhaps partly caused by interaction of albumin and albumin aggregates with both damaged vascular endothelium and thrombus. This trapping may lead to overestimates of BBB permeability using EB extravasation methods.31,32 One goal of the present study was to more clearly delineate the BBB permeability profile after PBBI by using molecular tracers, such as BDA and HRP, which have been reported as providing measures of BBB disruption that may be less confounded by damaged or clotted cerebral vasculature.31,33
Results of the current study revealed a uniphasic and gradient profile of BBB disruption after PBBI. Extravasation of both BDA and HRP tracers was initially detected at 4 h post-PBBI, reached maximal extravasation at 24 h post-injury, and remained evident out to either 48 h for HRP or 7 days for BDA. Differences between the extent and duration of BDA and HRP extravasation support the theory that the BBB permeability is not an all-or-none event but rather occurs in an incremental (or gradient) manner after PBBI. These findings are supported by previous studies in other TBI models that have shown that the BBB remains permeable to smaller molecular weight tracers (i.e., BDA) at post-injury times when barrier permeability to larger molecules (i.e., HRP) has resolved.31,33 Continued diffusion of BDA at later time points (>72 h) indicates persistent underlying BBB dysfunction despite functional improvement sufficient to prohibit passage of HRP into the CNS. The presence of sustained BBB permeability to compounds of smaller molecular weights may be clinically significant with regard to the therapeutic window for small (≤3 KDA) molecular weight compounds after severe penetrating TBI.
Critically, significant adverse effects were detected after administration of HRP, including edema and mild cyanosis evident in the distal limbs (i.e., the hind and forepaws) resulting in distress to the animals. In addition, conservative interpretation of HRP extravasation is needed because of evidence supporting facilitated and passive transfer of the peroxidase across membranes that might lead to overestimations of BBB permeability.34,35 Based on these observations, it is strongly recommended that the use of HRP be limited to anesthetized animals, or avoided completely.
BBB and neuroinflammatory markers
Overall, the primary objective of this study was to compare the time course of PBBI-induced BBB permeability with measured changes in markers of neuroinflammation: activation of resident inflammatory cells, upregulation of pro-inflammatory genes, and markers of peripheral inflammatory cell infiltration. From the current results, it is clear that the regulation of these markers is dynamically altered in the first week after PBBI. The neuroinflammatory processes occurring after PBBI are implicated in the progression of TBI via secondary mediators such as reactive oxygen species, nitric oxide, pyrogenic and pro-apoptotic cytokines, and matrix metalloproteinases.36–41 For the purpose of this discussion, the post-PBBI BBB permeability/neuroinflammatory response is broken down into the following three phases: Phase 1 (0–4 h), Phase 2 (24–72 h), and Phase 3 (>72 h).
During Phase 1 (0–4 h post-PBBI), significant increases in BBB permeability occurred in conjunction with an upregulation in astrocytic GFAP expression as well as icam-1 and vcam-1 mRNA levels. Although global mRNA changes in these inflammatory markers do not allow for cell-specific analysis, they are highly indicative of recruitment of peripheral immune cells. Previous research using both pre-clinical and clinical models of TBI has demonstrated that the acute post-injury phase is characterized by increased expression of pro-inflammatory cytokines10,14,42 that have been implicated in triggering the upregulation of CAMs.43 CAMs play a pivotal role in the migration of peripheral leukocytes into the CNS, and the interactions between ICAM-1, VCAM-1, and their ligands facilitate adhesion and transmigration of peripheral inflammatory cells.25,44–46 The PBBI-induced release of plasma and cellular components likely triggers rapid activation of astrocytes and endothelial cells, resulting in astrocytic cytokine production and upregulation of CAM genes in endothelial cells.14,46 These alterations would facilitate acute migration of peripheral inflammatory cells as well as contribute to a secondary phase of increased vascular permeability and continued leukocyte infiltration.
In support of this, the current results demonstrated a progressive activation of resident inflammatory cells and peripheral leukocyte infiltration (MPO-positive granulocytic infiltration, mmp-9 mRNA) in conjunction with a rapid decrease of icam-1 and vcam-1 mRNA production from 24–72 h post-PBBI (Phase 2). Astrocytes and microglia reached peak activation levels during this period, while icam-1 and vcam-1 mRNA levels were sharply decreased. This was consistent with the results of our previous PBBI studies demonstrating glial activation during this period.14,47 Novel to this study is the finding of a direct correlation between the peak phase of both BBB permeability and infiltrating granulocytes, and corresponding increase in mmp-9 mRNA. These results, however, should be interpreted conservatively, because further work is needed to confirm/identify corresponding patterns of protein expression using immunohistochemistry. That notwithstanding, the mRNA results in the present study provide critically important information regarding which proteins and time points should be targeted for further examination.
The current results suggest that release of MMP-9 into the injured brain tissue is an important factor in sustaining increased BBB permeability and recruiting peripheral inflammatory cells in the second phase of the PBBI-BBB disruption time course. MMP are zinc-dependent proteinases that are collectively capable of degrading all components of the extracellular matrix and basement membrane. MMP-2, -3, and -9 have been linked to severe TBI and the associated BBB dysfunction in both preclinical and clinical studies.48–50 Of note, MMP-9 (gelatinase B) plays an important role in the degradation of basement membrane components, including collagen type IV and type V, fibronectin, and elastin.50 Past work using fluid percussion injury models has established that MMP-9 is acutely upregulated after TBI and is sensitive both to injury severity and to manipulations in core brain temperature.40,41
Studies of ischemic brain injury have implied that MMP-9 is predominantly leukocyte-derived, its release into tissues likely resulting from rapid degranulation of neutrophils.37,38 In the current study, concurrent increases in mmp-9 mRNA and MPO-positive granulocytes detected in the injured hemisphere post-PBBI provide further support for a granulocytic origin for early MMP-9. The actions of MMP-9 on the extracellular matrix and basement membrane have been implicated in both the acute disruption of the BBB and in migration of early inflammatory cells.37,50 While further work is ongoing to identify the localization of MMP-9 activation in brain tissue, the progression of increased mRNA for CAMs, increasing BBB permeability, and infiltration of peripheral leukocytes supports a strong association between the degree of BBB disruption and the relocation of peripheral immune cells to the site of CNS injury.
Phase 3 (>72 h post-PBBI) is characterized by measures that may be indicative of a “wound healing” response in the injured brain including increased microglial activation, sustained astrocytic activation, and an abrupt increase in mmp-2 mRNA. Notably, the time frame during which this wound healing response emerges (i.e., 3–7 days post-injury) appears to coincide with the resolution of post-injury edema and EB extravasation as well as acute spontaneous recovery of motor function evaluated previously using neurological and balance beam assessments within 3 days post-injury in the same PBBI model.9,28
In the current study, both mmp-2 mRNA and glial activation increased sharply in the injured brain tissue during the same time frame that BBB permeability to tracer molecules appears to be resolving. Microglia have long been considered the resident macrophages of the CNS. They serve several functions necessary for neuronal survival after injury including clearing toxic debris, producing anti-inflammatory mediators, and facilitating repair.36 While a subset of activated microglia was detected at 7 days post-PBBI in the subcortical (thalamic) region, the majority were perilesional and well-placed to support tissue repair near the site of injury.
The relationship between astrocytes and microglial activation appears to be both spatial and temporal. While the activation of astrocytes and microglia were detected in similar brain regions, the temporal profiles of activation of these cell types differed. Astrocytic activation was detected more intensely in the injured hemisphere and, to a lesser degree, in the subcortical regions throughout the post-injury period. In contrast, activation of microglia appeared to evolve more slowly in both the injured hemisphere and areas distal to the site of injury. These differential patterns of protein expression were similar to what was previously reported in this PBBI model,7,14 indicating that both cell types are exquisitely sensitive to brain injury, but they may play different roles in the development of neuroinflammation after PBBI.
Recent studies have also begun to suggest that MMP-2 plays an important role in the reparative aspects of injury response and tissue remodeling in the CNS. At>5 days post-injury, MMP-2 (gelatinase A) has been localized to both reactive astrocytes and macrophages surrounding the resolving injury site.38,49 Similarly increased expression of MMP-2 has been associated with augmented axonal outgrowth in mammalian spinal cord injury and localized to astrocytes surrounding the proximal nerve stump and lesion after rat optic nerve crush injury.51 Correspondingly, genetic upregulation of MMP-2 was evident solely in the latest period of the current study beginning between 3–7 days after PBBI. The background of continued astrocytic activation, delayed increases in perilesional microglial activation, and the upregulation of mmp-2 mRNA suggests that upregulation of MMP-2 may contribute to post-PBBI CNS repair mechanisms. Further work is warranted to identify the regional and/or cellular profile of MMP-2 protein activation in the injured brain.
Conclusion
The current results indicate that there is a strong temporal and spatial association between the degree of BBB disruption and the ability of circulating inflammatory cells to quickly relocate to the site of CNS injury. Although these findings significantly enhance the understanding of BBB dysregulation after penetrating brain injury, further studies of relevant structures (i.e., basement membrane components and junctional proteins) are warranted. In addition, examination of cell-specific changes rather than global changes in inflammatory markers would provide greater resolution in identifying specific targets for potential therapies.
Acknowledgments
Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official, or as reflecting true views of the Department of the Army or Department of Defense.
All procedures described in this article were approved by the Institutional Animal Care and Use Committee of Walter Reed Army Institute of Research. Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals (NRC). The animals were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Masel B.E., Bell R.S., Brossart S., Grill R.J., Hayes R.L., Levin H.S., Rasband M.N., Ritzel D.V., Wade C.E., and DeWitt D.S. (2012). Galveston Brain Injury Conference 2010: clinical and experimental aspects of blast injury. J. Neurotrauma 29, 2143–2171 [DOI] [PubMed] [Google Scholar]
- 2.Bell R.S., Vo A.H., Neal C.J., Tigno J., Roberts R., Mossop C., Dunne J.R., and Armonda R.A. (2009). Military traumatic brain and spinal column injury: a 5-year study of the impact blast and other military grade weaponry on the central nervous system. J. Trauma 66, Suppl 4, S104–S111 [DOI] [PubMed] [Google Scholar]
- 3.Sapsford W. (2003). Penetrating brain injury in military conflict: does it merit more research? J. R. Army Med. Corps 149, 5–14 [DOI] [PubMed] [Google Scholar]
- 4.Martins R.S., Siqueira M.G., Santos M.T., Zanon-Collange N., and Moraes O.J. (2003). Prognostic factors and treatment of penetrating gunshot wounds to the head. Surg. Neurol. 60, 98–104 [DOI] [PubMed] [Google Scholar]
- 5.Shaffrey M.E., Polin R.S., Phillips C.D., Germanson T., Shaffrey C.I., and Jane J.A. (1992). Classification of civilian craniocerebral gunshot wounds: a multivariate analysis predictive of mortality. J. Neurotrauma 9, Suppl 1, S279–S285 [PubMed] [Google Scholar]
- 6.Thurman D.J., Alverson C., Dunn K.A., Guerrero J. N. and Sniezek J.E. (1999). Traumatic brain injury in the United States: A public health perspective. J. Head Trauma Rehabil. 14, 602–615 [DOI] [PubMed] [Google Scholar]
- 7.Williams A.J., Hartings J.A., Lu X.C., Rolli M.L. and Tortella F.C. (2006). Penetrating ballistic-like brain injury in the rat: differential time courses of hemorrhage, cell death, inflammation, and remote degeneration. J. Neurotrauma 23, 1828–1846 [DOI] [PubMed] [Google Scholar]
- 8.Lu X.C., Hartings J.A., Si Y., Balbir A., Cao Y., and Tortella F.C. (2011). Electrocortical pathology in a rat model of penetrating ballistic-like brain injury. J. Neurotrauma 28, 71–83 [DOI] [PubMed] [Google Scholar]
- 9.Shear D.A., Lu X.C., Bombard M.C., Pedersen R., Chen Z., Davis A., and Tortella F.C. (2010). Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. J. Neurotrauma 27, 1911–1923 [DOI] [PubMed] [Google Scholar]
- 10.Wei H.H., Lu X.C., Shear D.A., Waghray A., Yao C., Tortella F.C., and Dave J.R. (2009). NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating ballistic-like brain injury in rats. J. Neuroinflammation 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei G., Lu X.C., Yang X., and Tortella F.C. (2010). Intracranial pressure following penetrating ballistic-like brain injury in rats. J. Neurotrauma 27, 1635–1641 [DOI] [PubMed] [Google Scholar]
- 12.Williams A.J., Hartings J.A., Lu X.C., Rolli M.L., Dave J.R., and Tortella F.C. (2005). Characterization of a new rat model of penetrating ballistic brain injury. J. Neurotrauma 22, 313–331 [DOI] [PubMed] [Google Scholar]
- 13.Williams A.J., Ling G.S., and Tortella F.C. (2006). Severity level and injury track determine outcome following a penetrating ballistic-like brain injury in the rat. Neurosci. Lett. 408, 183–,188 [DOI] [PubMed] [Google Scholar]
- 14.Williams A.J., Wei H.H., Dave J.R., and Tortella F.C. (2007). Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J. Neuroinflammation 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adelson P.D., Whalen M.J., Kochanek P.M., Robichaud P., and Carlos T.M. (1998). Blood brain barrier permeability and acute inflammation in two models of traumatic brain injury in the immature rat: a preliminary report. Acta neurochir. Suppl. 71, 104–106 [DOI] [PubMed] [Google Scholar]
- 16.Marmarou A. (1994). Traumatic brain edema: an overview. Acta Neurochir. Suppl. (Wein) 60, 421–424 [DOI] [PubMed] [Google Scholar]
- 17.Marmarou A., Portella G., Barzo P., Signoretti S., Fatouros P., Beaumont A., Jiang T., and Bullock R. (2000). Distinguishing between cellular and vasogenic edema in head injured patients with focal lesions using magnetic resonance imaging. Acta Neurochir. Suppl. 76, 349–351 [DOI] [PubMed] [Google Scholar]
- 18.Pardridge W.M. (2002). Targeting neurotherapeutic agents through the blood-brain barrier. Arch. Neurol. 59, 35–40 [DOI] [PubMed] [Google Scholar]
- 19.Thrivikraman K.V., Huot R.L., and Plotsky P.M. (2002). Jugular vein catheterization for repeated blood sampling in the unrestrained conscious rat. Brain Res. Brain Res. Protoc. 10, 84–94 [DOI] [PubMed] [Google Scholar]
- 20.Hsu S.M., Raine L., and Fanger H. (1981). Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 29, 577–580 [DOI] [PubMed] [Google Scholar]
- 21.Biagas K.V., Uhl M.W., Schiding J.K., Nemoto E.M., and Kochanek P.M. (1992). Assessment of posttraumatic polymorphonuclear leukocyte accumulation in rat brain using tissue myeloperoxidase assay and vinblastine treatment. J. Neurotrauma 9, 363–371 [DOI] [PubMed] [Google Scholar]
- 22.Chen Z., Leung L.Y., Mountney A., Liao Z., Yang W., Lu X.C., Dave J., Deng-Bryant Y., Wei G., Schmid K., Shear D.A., and Tortella F.C. (2012). A novel animal model of closed-head concussive-induced mild traumatic brain injury: development, implementation, and characterization. J. Neurotrauma 29, 268–280 [DOI] [PubMed] [Google Scholar]
- 23.Ruifrok A.C. (1997). Quantification of immunohistochemical staining by color translation and automated thresholding. Anal. Quant. Cytol. Histol. 19, 107–113 [PubMed] [Google Scholar]
- 24.Zhao J., Pati S., Redell J.B., Zhang M., Moore A.N., and Dash P.K. (2012). Caffeic Acid phenethyl ester protects blood-brain barrier integrity and reduces contusion volume in rodent models of traumatic brain injury. J. Neurotrauma 29, 1209–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlos T.M., Clark R.S., Franicola-Higgins D., Schiding J.K., and Kochanek P.M. (1997). Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J. Leukoc. Biol. 61, 279–285 [DOI] [PubMed] [Google Scholar]
- 26.Tomkins O., Feintuch A., Benifla M., Cohen A., Friedman A., and Shelef I. (2011). Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc. Psychiatry Neurol. 2011, 765923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baskaya M.K., Rao A.M., Dogan A., Donaldson D., and Dempsey R.J. (1997). The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett. 226, 33–36 [DOI] [PubMed] [Google Scholar]
- 28.Shear D.A., Lu X.C., Pedersen R., Wei G., Chen Z., Davis A., Yao C., Dave J., and Tortella F.C. (2011). Severity profile of penetrating ballistic-like brain injury on neurofunctional outcome, blood-brain barrier permeability, and brain edema formation. J. Neurotrauma 28, 2185–2195 [DOI] [PubMed] [Google Scholar]
- 29.Zhao J., Moore A.N., Redell J.B., and Dash P.K. (2007). Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 27, 10240–10248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belayev L., Busto R., Zhao W., and Ginsberg M.D. (1996). Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 739, 88–96 [DOI] [PubMed] [Google Scholar]
- 31.Habgood M.D., Bye N., Dziegielewska K.M., Ek C.J., Lane M.A., Potter A., Morganti-Kossmann C., and Saunders N.R. (2007). Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 25, 231–238 [DOI] [PubMed] [Google Scholar]
- 32.Webber M.M. (1977). Labeled albumin aggregates for detection of clots. Semin. Nucl. Med. 7, 253–261 [DOI] [PubMed] [Google Scholar]
- 33.Lotocki G., de Rivero Vaccari J.P., Perez E.R., Sanchez-Molano J., Furones-Alonso O., Bramlett H.M., and Dietrich W.D. (2009). Alterations in blood-brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post-traumatic hypothermia. J. Neurotrauma 26, 1123–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietrich W.D., Alonso O., and Halley M. (1994). Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J. Neurotrauma 11, 289–301 [DOI] [PubMed] [Google Scholar]
- 35.Povlishock J.T., Becker D.P., Sullivan H.G., and Miller J.D. (1978). Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. 153, 223–239 [DOI] [PubMed] [Google Scholar]
- 36.Block M.L., Zecca L., and Hong J.S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69 [DOI] [PubMed] [Google Scholar]
- 37.Gidday J.M., Gasche Y.G., Copin J.C., Shah A.R., Perez R.S., Shapiro S.D., Chan P.H., and Park T.S. (2005). Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. Heart Circ. Physiol. 289, H558–H568 [DOI] [PubMed] [Google Scholar]
- 38.Romanic A.M., White R.F., Arleth A.J., Ohlstein E.H., and Barone F.C. (1998). Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 39.Shojo H., Kaneko Y., Mabuchi T., Kibayashi K., Adachi N., and Borlongan C.V. (2010). Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience 171, 1273–1282 [DOI] [PubMed] [Google Scholar]
- 40.Jia F., Pan Y.H., Mao Q., Liang Y.M., and Jiang J.Y. (2010). Matrix metalloproteinase-9 expression and protein levels after fluid percussion injury in rats: the effect of injury severity and brain temperature. J. Neurotrauma 27, 1059–1068 [DOI] [PubMed] [Google Scholar]
- 41.Truettner J.S., Suzuki T., and Dietrich W.D. (2005). The effect of therapeutic hypothermia on the expression of inflammatory response genes following moderate traumatic brain injury in the rat. Brain Res. Mol. Brain Res. 138, 124–134 [DOI] [PubMed] [Google Scholar]
- 42.Lenzlinger P.M., Morganti-Kossmann M.C., Laurer H.L., and McIntosh T.K. (2001). The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol. 24, 169–181 [DOI] [PubMed] [Google Scholar]
- 43.Carlos T.M., and Harlan J.M. (1994). Leukocyte-endothelial adhesion molecules. Blood 84, 2068–2101 [PubMed] [Google Scholar]
- 44.Arvin B., Neville L.F., Barone F.C., and Feuerstein G.Z. (1996). The role of inflammation and cytokines in brain injury. Neurosci. Biobehav. Rev. 20, 445– 52 [DOI] [PubMed] [Google Scholar]
- 45.Smith C.W. (1993). Endothelial adhesion molecules and their role in inflammation. Can. J. Physiol. Pharmacol. 71, 76–87 [DOI] [PubMed] [Google Scholar]
- 46.Zimmerman G.A., Prescott S.M., and McIntyre T.M. (1992). Endothelial cell interactions with granulocytes: tethering and signaling molecules. Immunol. Today 13, 93–100 [DOI] [PubMed] [Google Scholar]
- 47.Lu X.C., Chen R.W., Yao C., Wei H., Yang X., Liao Z., Dave J.R., and Tortella F.C. (2009). NNZ-2566, a glypromate analog, improves functional recovery and attenuates apoptosis and inflammation in a rat model of penetrating ballistic-type brain injury. J. Neurotrauma 26, 141–154 [DOI] [PubMed] [Google Scholar]
- 48.Grossetete M., Phelps J., Arko L., Yonas H. and Rosenberg G.A. (2009). Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery 65, 702–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenberg G.A. (2002). Matrix metalloproteinases in neuroinflammation. Glia 39, 279–291 [DOI] [PubMed] [Google Scholar]
- 50.Yong V.W., Power C., Forsyth P., and Edwards D.R. (2001). Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verslegers M., Lemmens K., Van Hove I., and Moons L. (2013). Matrix metalloproteinase-2 and -9 as promising benefactors in development, plasticity and repair of the nervous system. Prog. Neurobiol. 105, 60–78 [DOI] [PubMed] [Google Scholar]