

Abstract
Based on numerous pharmacological studies that have revealed an interaction between cannabinoid and opioid systems at the molecular, neurochemical, and behavioral levels, a new series of hybrid molecules has been prepared by coupling the molecular features of two wellknown drugs, ie, rimonabant and fentanyl. The new compounds have been tested for their affinity and functionality regarding CB1 and CB2 cannabinoid and μ opioid receptors. In [35S]-GTPγS (guanosine 5′-O-[gamma-thio]triphosphate) binding assays from the post-mortem human frontal cortex, they proved to be CB1 cannabinoid antagonists and μ opioid antagonists. Interestingly, in vivo, the new compounds exhibited a significant dual antagonist action on the endocannabinoid and opioid systems.
Keywords: fentanyl, rimonabant, cannabinoid, opioid, behavioral assays
Introduction
Pharmacological data have revealed interactions between the cannabinoid and opioid systems at the molecular, neurochemical, and behavioral levels.1–7 Anatomical studies have shown that such pharmacological interactions are likely due to the close vicinity of CB1 and opioid receptors in the central nervous system structures involved in the control of nociception, motor activity, and brain reward. In this context, the development of a single molecule able to act on both the cannabinoid and opioid systems seems an interesting approach. Multivalent or multifunctional ligands constitute promising pharmacological tools and new targets for drug development. Moreover, targeting G protein-coupled receptors with hybrid molecules has been explored in different domains. The concept of hybridization has become of increasing interest for medicinal chemists. This strategy has resulted in promising leads that show significant therapeutic advantages.8,9 The main benefit compared with the administration of two drugs concerns the pharmacokinetic properties of a single molecule. In order to focus our efforts on an efficient designed multiple ligand approach, two well validated drugs were selected, ie, fentanyl and rimonabant (SR141716, Figure 1). Fentanyl is a synthetic μ opioid agonist widely used in clinical practice10 and rimonabant was the first selective CB1 cannabinoid receptor inverse agonist/antagonist to be approved for clinical use,11 even though its use was discontinued due to unwanted effects.12
Figure 1.

The designed multiple ligands are based on the CB1 cannabinoid receptor inverse agonist/antagonist rimonabant (SR141716) (Sanofi, Paris, France) and the μ opioid agonist fentanyl.13 X represents an n-alkyl chain from propyl to dodecyl, 1,3-phenyl, 1,3-benzyl, or biscyclohex-4-ylmethane.
Therefore, our aim was to explore the hybrid molecules represented in Figure 1 (4a–k) based on previous structural modification studies carried out on fentanyl and rimonabant. Fentanyl derivatives (Figure 2), in which a phenyl was replaced by an aliphatic chain bearing a guanidine moiety, have shown high affinity for the μ opioid receptor, with agonistic properties.13,14 Concerning the cannabinoid counterpart of the proposed hybrid molecules, good affinities for the CB1 receptor have been reported for diarylcarboxamides bearing alkyl chains (Figure 2).15,16
Figure 2.

Examples of fentanyl derivatives with selective μ opioid agonistic properties and rimonabant derivatives with CB1 receptor affinity.
Abbreviation: Ki, affinity constant.
Herein, we describe the synthesis, pharmacological profile, and behavioral effects in vivo of a series of hybrid molecules derived from fentanyl and rimonabant.
Materials and methods
General
All commercially available reagents were purchased from Sigma-Aldrich (Madrid, Spain) and used without further purification. Dry CH2Cl2 was obtained by distillation over calcium chloride. Flash column chromatography was performed using silica gel 60 (230–400 mesh) or on a medium pressure flash system with a prepacked silica gel cartridge (Biotage Flash 40 [SYMTA, Madrid, Spain], cartridges KP-Sil 40S [4 × 7 cm] or 4M [4 × 15 cm] with a particle size of 32–63 μm and 60 Å; FlashMaster Personal [SYMTA] with prepacked cartridges FlashPack of 2, 10, 20 or 50 g). Starting products 1 and 2 were prepared as previously described.17 N-[1-phenethyl-4-piperidyl]propanamide 3a–3k were prepared following a procedure previously described by our group.13,18
Chemistry
Compounds 4a–4k were prepared following the synthetic route depicted in Figure 3. Diaryl-1H-pyrazole-3-carboxylic acid 1 was previously prepared according to Krishnamurthy’s procedure.19 Next, pyrazole 1 was converted to the acid chloride 2 using the following procedure. A solution of the appropriate propanamide 3a–3k and triethylamine in dry CH2Cl2 was added dropwise to pyrazole-3-carboxylic acid chloride 2 dissolved in dry CH2Cl2 (5 mL). The reaction mixture was stirred at room temperature for 16 hours. The solvent was then removed under reduced pressure. The crude product was purified on silica gel by flash chromatography eluting with CH2Cl2/(MeOH/NH3) (100:0–100:2) to provide the corresponding pyrazole-3-carboxamides 4a–4k. The yields of the preparation and the structural assignments of 4a–4k are reported in the Supplementary material section (1H-NMR [nuclear magnetic resonance], 13C-NMR, MS [mass spectroscopy], and elemental analysis).
Figure 3.

Synthesis of the pyrazole carboxamide derivatives 4a–4k. Reagents and conditions: (i) SOCl2, toluene, Δ (reflex temperature); (ii) CH2Cl2, Et3N, room temperature.
Pharmacology
Post-mortem human brain CB1 and μ opioid binding assays
The ability of the new compounds to bind to the CB1 cannabinoid receptors and to the μ opioid receptor was evaluated in competitive displacement assays using, respectively, [3H]-CP55,940 (PerkinElmer, Boston, MA, USA) and [3H]-DAMGO ([DAla2, N-Me-Phe4, Gly5-ol]-enkephalin; American Radiolabeled Chemicals Inc, St Louis, MO, USA) as radioligands. Prefrontal cortex membranes from postmortem human brain were used as a source of receptors, given that CB1 receptors20 and μ opioid receptors are present in large amounts in this region. Neural membranes (P2 fractions) were prepared from the prefrontal cortex of human brains obtained at autopsy (Instituto Vasco de Medicina Legal, Bilbao, Spain) as previously reported by our research group.16 These samples belong to the Brain Collection of the University of the Basque Country registered with the National Biobank Register of the Spanish Health Department number (C 0000035). Specific [3H]-CP55,940 or [3H]-DAMGO binding was measured in 0.55 mL aliquots (50 mM Tris-HCl [Tris(hydroxymethyl) aminomethane hydrochloride], pH 7.5) of the neural membranes. The membranes were incubated with [3H]-CP55,940 (1 nM) for 60 minutes at 30°C or [3H]-DAMGO (2 nM) for 60 minutes at 25°C in the absence or presence of the competing compounds (10−12 M to 10−3 M, ten concentrations). Incubations were terminated by diluting the samples with 5 mL of ice-cold Tris-HCl incubation buffer (4°C). Membrane-bound radioligand was separated by vacuum filtration through Whatman GF/C glass fiber filters (GE Healthcare, Buckinghamshire, UK). The filters were then rinsed twice with 5 mL of incubation buffer and transferred to mini-vials containing 3 mL of OptiPhase HiSafe® II cocktail (PerkinElmer) and counted for radioactivity by liquid scintillation spectrometry. Specific binding was determined and plotted as a function of the compound concentration. Nonspecific binding was determined in the presence of WIN 55,212-2 (10−5 M) or naloxone (10−5 M), respectively. Analysis of competition experiments to obtain the inhibition constant (Ki) were performed by nonlinear regression using the GraphPad Prism program (GraphPad Software, Inc, San Diego, CA, USA). All experiments were analyzed assuming a one-site model of radioligand binding.
CB2 binding evaluation
CB2 receptor affinity was evaluated from membrane fractions of human embryonic kidney (HEK293 EBNA) cells transfected with human CB2 receptors (RBXCB2M400UA; Analytical Sciences, Boston, MA, USA).21 The CB2 receptor membrane protein concentration was 5.20 pmol/mg or 6.20 pmol/mg and the protein concentration was 4.0 mg/mL or 3.6 mg/mL depending on the batch. The commercial membranes were diluted (approximately 1:20) with the binding buffer (50 mM Tris-HCl, 5 mM MgCl2 · H2O, 2.5 mM ethylene glycol tetraacetic acid, 1 mg/mL bovine serum albumin, pH 7.5). The final membrane protein concentration was 0.2 mg/mL of incubation volume. The radioligand used was [3H]-CP55,940 at a concentration of membrane KD (dissociation constant) ×0.8 nm, and the final volume was 600 mL. The 96-well plates and tubes necessary for the experiment were previously siliconized with Sigmacote (Sigma-Aldrich, St Louis, MO, USA). The membranes were resuspended in the corresponding buffer and were incubated with the radioligand and each compound (10−4–10−11 M) for 90 minutes at 30°C. Nonspecific binding was determined with 10 μM WIN 55,212-2, and 100% binding of the radioligand to the membrane was determined by its incubation with membrane without any compound. Filtration was performed by a Harvester® FilterMate (PerkinElmer) with Filtermat A GF/C filters pretreated with polyethylenimine 0.05%. After filtering, the filter was washed nine times with binding buffer, dried, and a melt-on scintillation sheet (MeltiLex™; PerkinElmer) was melted onto it. Radioactivity was then quantified by a liquid scintillation spectrophotometer (Wallac MicroBeta® TriLux; PerkinElmer). Competition binding data were analyzed using the GraphPad Prism program, and Ki values are expressed as the mean ± standard error of the mean (SEM) of at least three experiments performed in triplicate for each point.
Functionality binding assays
To determine the ability of the selected compounds to activate the CB1 and/or μ opioid receptors, [35S]-GTPγS (guanosine 5′-O-[gamma-thio]triphosphate) binding assays were performed in cortical membranes from post-mortem human brain. In this tissue, [35S]-GTPγS binds with high affinity to Gi/Go proteins.22 Thereby, agonists, inverse agonists, and antagonists can modulate this binding acting on a specific receptor, increasing (agonists) or decreasing (inverse agonists) the nucleotide binding or blocking the effect of an agonist (antagonists). The incubation buffer for measuring [35S]GTPγS binding to brain membranes contained 1 mM ethylene glycol tetraacetic acid, 3 mM MgCl2, 100 mM NaCl, 50 mM GDP (guanosine diphosphate), 50 mM Tris–HCl at pH 7.4, and 0.5 nM [35S]GTPγS (DuPont NEN, Brussels, Belgium) in a total volume of 500 μL. Protein aliquots were thawed and resuspended in the same buffer. The incubation was started by addition of the membrane suspension (40 μg of membrane proteins) to the previous mixture and was performed at 30°C for 120 minutes with shaking. In order to evaluate the influence of the compounds on [35S]GTPγS binding, ten concentrations (10−12–10−3 M) of the different compounds were added to the assay. Incubations were terminated by adding 3 mL of ice-cold resuspension buffer followed by rapid filtration through Whatman GF/C filters presoaked in the same buffer. The filters were rinsed twice with 3 mL of ice-cold resuspension buffer, transferred to vials containing 5 mL of OptiPhase HiSafe II cocktail, and the radioactivity trapped was determined by liquid scintillation spectrometry (Packard 2200CA; Packard Instrument Company, Meriden, CT, USA). The [35S]GTPγS bound was about 7%–14% of the total [35S]GTPγS added. Nonspecific binding of the radioligand was defined as the remaining [35S]GTPγS binding in the presence of 10 µM unlabeled GTPγS.
In vivo cannabinoid tetrad assays
Male imprinting control region mice weighing 25–30 g were used. Spontaneous behavior was always observed in the cage before treatment and/or performance of the different tests. Animals showing spontaneous behavioral modifications were discarded. To evaluate agonist effects, reference drugs and new compounds were administered 15 minutes (for the cannabinoid tetrad) and 30 minutes (for the opioid hot plate test) before starting the behavioral tests. When the compounds were tested as antagonists, they were administered 20 minutes before the reference agonists (WIN 55,212-2 or morphine). All drugs were given intraperitoneally. Separate groups of mice (n = 8–10 each) were given the following treatments: saline solution or vehicle (controls); WIN 55,212-2 1.5 mg/kg; 4d 10 mg/kg; 4e 5 mg/kg; rimonabant 1 mg/kg; rimonabant 1 mg/kg + WIN 55,212-2 1.5 mg/kg; 4d 2 mg/kg + WIN 55,212-2 1.5 mg/kg; 4d 4 mg/kg + WIN 55,212-2 1.5 mg/kg; 4d 8 mg/kg + WIN 55,212-2 1.5 mg/kg; and 4e 5 mg/kg + WIN 55,212-2 1.5 mg/kg. The tests were conducted consecutively at 5-minute intervals.
Hypothermia
Core mouse temperatures were measured using a lubricated thermometer inserted into the rectum to a constant depth of 1 cm. Temperature was evaluated twice in each animal, ie, before and after every treatment.
Locomotor activity
Spontaneous locomotor activity was evaluated using individual photocell activity chambers (Cibertec®, San Jose, Costa Rica). The mouse was placed in a chamber and, starting 10 minutes later, the number of interruptions of photocell beams was recorded over a 30-minute period. The mean number of crossings was compared with that obtained from a mouse control group that had received vehicle.
Nociception
The hot plate test was carried out using a hot plate at 55°C as the nociceptive stimulus. The latency time of licking of the front paw was taken as an index of nociception. The latency was measured before treatment (control latency) and after every treatment (latency after treatment). The cut-off time was 30 seconds and analgesia was quantified with the formula of the maximum possible effect (MPE), expressed as a percentage:
Catalepsy
Catalepsy was measured using a modified “ring test” as originally described by Pertwee.23 The mice were placed on a rubber-coated metal ring (6 cm in diameter) fixed horizontally at a height of 30 cm. CB1 cannabinoid agonists cause animals to become cataleptic, and the sum of all times during which the mice were immobile was registered for a 5-minute period and compared with the time registered in control animals. The criterion for immobility was the absence of all voluntary movement.
To confirm the duration of the effect of the new compounds, they were tested as antagonists on the hot plate test. Next, 4d (4 mg/kg, intraperitoneally) and 4e (5 mg/kg intraperitoneally) were administered 20 minutes before WIN 55,212-2 (1.5 mg/kg intraperitoneally) and the test was performed 1 hour after injection of this agonist.
In vivo opioid response on hot plate test
To assess the opioid activity of the new compounds 4d and 4e, the hot plate test was carried out as described above and separate groups of mice (n = 8–10 each) were treated with: saline solution or vehicle (controls); 4d 10 mg/kg; 4e 10 mg/kg; naloxone 1 mg/kg; rimonabant 1 mg/kg; morphine 10 mg/kg; 4d 10 mg/kg + morphine 10 mg/kg; 4e 10 mg/kg + morphine 10 mg/kg; naloxone 1 mg/kg + morphine 10 mg/kg; and rimonabant 1 mg/kg + morphine 10 mg/kg. The data are expressed as the mean ± SEM. For the in vitro assays, values for EC50 (half-maximal effective concentration) and 95% confidence limits of these values for agonists were calculated by nonlinear regression analysis using the equation for a sigmoid concentration-response curve (GraphPad Prism). For the effects of drugs on in vitro and in vivo tests, a one-way analysis of variance was used for statistical analysis of multiple comparisons within each group. When a significant difference was detected by one-way analysis of variance, the data were further analyzed using Newman–Keuls test. In each test, a P-value less than 0.05 was considered to indicate statistical significance. Drugs (WIN 55,212-2 mesylate and rimonabant) were obtained from Tocris (Biogen Científica SL, Madrid, Spain) and Sanofi, respectively. DAMGO, DL-dithiothreitol, GDP, GTP, GTPγS, fentanyl, and naloxone were purchased from Sigma-Aldrich. Morphine HCl was obtained from Alcaliber SA (Madrid, Spain). Morphine sulfate salt solution and naloxone hydrochloride dihydrate were obtained from Sigma-Aldrich (Madrid, Spain). All other chemicals were of the highest commercially available purity. Cannabinoids were dissolved in ethanol 1 mg:1 mL and subsequently in ethanol and Tween 80 (1:2), after which the ethanol was evaporated. Other drugs were dissolved in saline solution (0.9%).
Alcohol intake assays
The operant ethanol self-administration was examined following a protocol essentially based on a previously published alcohol relapse model in Wistar rats.24
Administration, distribution, metabolism, excretion (ADME) properties in silico
ADME properties were calculated using QikProp version 3.5 integrated in Maestro (Schrödinger, LLC, New York, NY, USA). The global minimum energy conformer used as input was generated using the program Spartan ’08 (Wave Function, Inc, Irvine, CA, USA). Ab initio energy minimizations were performed at the Hartree-Fock 6-31G* level.
Results and discussion
Receptor affinities and structure-activity relationship
Concerning the CB1 cannabinoid binding, Table 1 displays the Ki values obtained for 4a–4k and for AM251 (Tocris, Bristol, UK) and rimonabant as reference CB1 ligands. Some of the new compounds (4a, 4b, 4d–4f, 4k) showed low to medium affinity (Ki = 0.19–3.99 μM), while others (4c, 4g–4j) did not show any affinity for the CB1 receptor in this assay (Ki >10 μM). The ligands showing the highest affinity were compounds 4b (Ki = 0.57 μM), and 4e (Ki = 0.70 μM), containing a butyl and a heptyl chain linker, respectively. Now, if we refer to our recent published binding data on bivalent cannabinoid ligands25 (Figure 2), it is interesting to note that the bivalent molecule with the heptyl linker also showed the best CB1 affinity. Actually, the alkyl chain length does not correlate with CB1 receptor affinity. However, the values obtained for 4g–4j indicate that aromatic spacers and longer alkyl chains lead to a loss of CB1 cannabinoid receptor affinity. Interestingly, replacing the linear alkyl chain of nine methylene units (4g; Ki >10 μM) with a biscyclohexylene (4k; Ki =2.06 μM) as a conformation constrainer, resulted in increased affinity for the CB1 receptor.
Table 1.
Binding affinity of compounds 4a–4k for CB1 cannabinoid and μ opioid receptors
| Compound | X | [3H]-CP55,940 CB1 Ki (μM)a | [3H]-DAMGO μ Ki (μM)a |
|---|---|---|---|
| AM251 | 0.005±0.002 | ND | |
| Rimonabant | 0.004±0.002 | 0.20±0.12 | |
| Fentanyl | ND | 0.003±0.001 | |
| 4a | –(CH2)3– | 0.19±0.07 | 3.81±0.39 |
| 4b | –(CH2)4– | 0.57±0.20 | 1.23±0.43 |
| 4c | –(CH2)5– | >10 | 0.17±0.10 |
| 4d | –(CH2)6– | 2.29±1.86 | 0.30±0.06 |
| 4e | –(CH2)7– | 0.70±0.57 | 6.54±0.95 |
| 4f | –(CH2)8– | 3.99±1.37 | 1.24±0.79 |
| 4g | –(CH2)9– | >10 | 0.11±0.06 |
| 4h | –(CH2)12– | >10 | 6.90±1.58 |
| 4i |
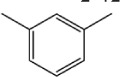
|
>10 | 1.25±0.67 |
| 4j |
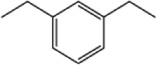
|
>10 | 1.02±0.25 |
| 4k |
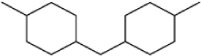
|
2.06±0.60 | 0.66±0.37 |
Notes:
Values are expressed as the mean ± SEM of at least three experiments. Rimonabant (Sanofi, Paris, France); Fentanyl.13
Abbreviations: ND, not determined; SEM, standard error of the mean; DAMGO, [D-Ala2,N-MePhe4,Gly-ol]-enkephalin.
The μ opioid binding component is reflected in Table 1. Excluding 4a, 4e, and 4h, which showed a Ki higher than 3.8 μM, the Ki values of 4b–d, 4f–g, and 4i–k, that ranged from 205 nM to 1.3 μM, indicate moderate affinity for the μ opioid receptor. Compounds 4g, 4c, and 4d showed the best Ki values (108, 166, and 295 nM, respectively).
These CB1 and opioid competitive displacement assays were performed in the post-mortem human brain, where CB1 and opioid receptors are present in large amounts and where the presence of dimers and heteromers has been proposed. The fact that the new compounds with affinity for both receptors have the shortest linkers suggests that they bind to CB1 and to opioid monomers. Spacers ranging from 10 to 20 atoms are necessary for the molecule to bridge heteromer receptors. It is unlikely that these hybrid molecules target a CB1 opioid heteromer. However, they bind individually to the respective receptors. Regarding CB2 receptor binding assays, compounds 4a–4j did not show any significant affinity (>40 μM), thus indicating CB1 selectivity. CB2 binding assay for 4k could not be carried out for solubility reasons. The new compounds have a CB1 activity profile.
Functionality
Although these new compounds showed modest affinity, evaluation of their functionality has yielded interesting results. We selected 4b and 4e for their CB1 cannabinoid affinity and 4d for its μ opioid affinity along with a micromolar CB1 affinity constant value. [35S]-GTPγS binding assays have been performed in cortical membranes from the post-mortem human brain to determine the ability of the selected compounds to activate the CB1 and/or μ opioid receptors.
We first evaluated the direct effect of compounds 4b, 4d, and 4e. Compound 4b produced an inhibition of basal [35S]-GTPγS binding (EC50 25±4 μM; Figure 4), demonstrating its inverse agonist effect. This effect is similar to that induced by the well known CB1 inverse agonist/antagonist rimonabant, used as reference (EC50 19±4 μM; Figure 4).26 Compounds 4d, 4e, and the opioid-selective antagonist naloxone did not induce any significant change in basal [35S]-GTPγS binding values, indicating a lack of agonist or inverse agonist properties (Figure 4).
Figure 4.

Concentration-response curves for stimulation of [35S]-GTPγS binding by 4b, 4d, 4e, rimonabant, and naloxone to cortical membranes in the post-mortem human brain. The data are expressed as the mean (± SEM) values for 4–6 experiments performed in different brain samples.
Notes: Rimonabant (Sanofi, Paris, France); naloxone (Sigma-Aldrich, Madrid, Spain).
Abbreviations: SR, rimonabant; Nx, naloxone; SEM, standard error of the mean; GTPγS, guanosine 5′-O-[gamma-thio]triphosphate.
The next step was to characterize their antagonist effect on cannabinoid receptors. The maximum agonist effects (Emax) of WIN 55,212-2 (Emax 184%±4%; EC50 1.1±0.22 μM) were reverted by each of the new compounds (4b, 4d, and 4e), as well as by rimonabant, at a single concentration (10−5 M, Figure 5A). They produced a rightward displacement of the WIN 55,212-2 dose-response curve (EC50 for rimonabant, 21±3 μM; EC50 for 4b, 33±8 μM; EC50 for 4d, 21±2 μM; EC50 for 4e, 16±2 μM). These data show that 4b, 4d, and 4e behave as antagonists of the CB1 cannabinoid receptor with potency similar to that of rimonabant. Conversely, the opioid antagonist naloxone (10−5 M) did not change the WIN 55,212-2 stimulation (EC50 1.3±0.17 μM; Emax 187%±4%).
Figure 5.

Concentration-response curves for stimulation of [35S]-GTPγS binding by WIN 55,212-2 (Tocris/Biogen Científica SL, Madrid, Spain) (A) or fentanyl (B) in the absence (○) or presence of a single concentration (10−5 M) of 4b, 4e, 4d, naloxone, or rimonabant. The experiments were performed using cortical membranes in the post-mortem human brain. The data are expressed as the mean ± SEM values from 4–8 experiments carried out in different brain samples.
Abbreviations: SR, rimonabant; Nx, naloxone; WIN, WIN 55,212-2; Fen, fentanyl; SEM, standard error of the mean; GTPγS, guanosine 5′-O-[gamma-thio] triphosphate.
Finally, compounds 4b, 4d, and 4e have been evaluated for their antagonism at the μ opioid receptor. The agonist effects of the opioid fentanyl (Emax 146%±2%; EC50 0.28±0.04 μM) were blocked by 4b, 4d, 4e, and naloxone at a single concentration (10−5 M, Figure 5B). All of these caused a rightward displacement of the fentanyl dose-response (EC50=456±60 μM for naloxone; EC50=24±5 μM for 4b; EC50=33±2 μM for 4d; EC50=3±1 μM for 4e). These data confirm that 4b, 4d, and 4e behave as opioid antagonists.
With the above functionality studies, we clearly demonstrate that 4b, 4d, and 4e have dual functionality, ie, CB1 receptor antagonism with potency similar to that of rimonabant and μ opioid receptor antagonism with potency superior to that of naloxone. Since previous studies suggested therapeutic interest in coadministration of a cannabinoid antagonist and an opioid antagonist,27–30 we considered that it would be interesting to follow on with in vivo studies.
Behavioral properties
The in vivo cannabinoid properties of 4d and 4e were characterized on the basis of their behavioral effects in mice. Cannabinoid agonists have been shown to induce a drop in body temperature, catalepsy on an elevated ring, acute analgesia on a hot plate, and a decrease of spontaneous activity in an open field.31 Compound 4d, at the three tested doses (intraperitoneal administration; Figure 6) antagonized all the effects produced by the cannabinoid agonist WIN 55,212-2 (1.5 mg/kg), the difference being statistically significant for rectal temperature, catalepsy, and analgesia. Three (catalepsy, analgesia, and reduction of the spontaneous activity) of the four effects induced by WIN 55,212-2 (1.5 mg/kg) on mice were significantly prevented by 4e (5 mg/kg, intraperitoneal administration; Figure 7). These results support the antagonistic CB1 cannabinoid properties of 4d and 4e. It is unlikely that this antagonism could be due to loss of CB1 receptors as a consequence of a downregulation process since it is normally the result of chronic rather than acute administration, as in this case.
Figure 6.

Effects of 4d on the cannabinoid tetrad. Bars show modifications induced by treatment with WIN 55,212-2 1.5 mg/kg, 4d 10 mg/kg, rimonabant 1 mg/kg, and WIN 55,212-2 1.5 mg/kg after treatment with rimonabant 1 mg/kg or 4d 2, 4, or 8 mg/kg. °C represents body temperature; Seconds of immobility represents catalepsy on an elevated ring; Cross number represents the number of interruptions of photocell beams.
Notes: Values show the mean ± SEM (n=10). #P<0.05; ##P<0.01; and ###P<0.001 indicate a statistically significant difference versus vehicle; *P<0.05; **P<0.01, and ***P<0.001 indicate a statistically significant difference versus WIN (one-way analysis of variance, Newman–Keuls post hoc test). WIN55,212-2 (Tocris/Biogen Científica SL, Madrid, Spain).
Abbreviations: W, WIN 55,212-2; SR, rimonabant; SEM, standard error of the mean; MPE, maximum possible effect; C, control; V, vehicle.
Figure 7.

Effects of 4e on the cannabinoid tetrad. Bars show modifications induced by treatment with WIN 55,212-2 1.5 mg/kg, 4e 5 mg/kg, rimonabant 1 mg/kg, and WIN 55,212-2 1.5 mg/kg after treatment with rimonabant 1 mg/kg or 4e 5 mg/kg. °C represents body temperature; Seconds of immobility represents catalepsy on an elevated ring; Cross number represents the number of interruptions of photocell beams.
Notes: Values show the mean ± SEM (n=10). #P<0.05, ##P<0.01, and ###P<0.001 indicate a statistically significant difference versus vehicle; *P<0.05, **P<0.01, and ***P<0.001 indicate a statistically significant difference versus WIN (one-way analysis of variance, Newman–Keuls post hoc test). Rimonabant (Sanofi, Paris, France).
Abbreviations: W, WIN 55,212-2; SR, rimonabant; SEM, standard error of the mean; MPE, maximum possible effect.
To assess the duration of effect of 4d and 4e, the hot plate test was carried out after intraperitoneal injection of WIN 55,212-2 (1.5 mg/kg). Both compounds (4d at 4 mg/kg and 4e at 5 mg/kg, administered intraperitoneally) completely and significantly (P<0.001, one-way analysis of variance) reversed the effect of the cannabinoid agonist WIN 55,212-2 (percent of MPE for 4d =3.5±2.5 and percent MPE for 4e =5.7±4.7; values shown as the mean ± SEM [n=6]).
The in vivo opioid properties of 4d and 4e were evaluated using a hot plate test at 55°C as the nociceptive stimulus (Figure 8). Treatment with the μ opioid agonist morphine induced antinociception that was reversed by the opioid antagonist naloxone but not by the cannabinoid antagonist rimonabant. The data described above show that intraperitoneal administration of 4d (10 mg/kg) or 4e (10 mg/kg) did not induce any analgesic effect. However, when 4d (10 mg/kg, intraperitoneally) or 4e (10 mg/kg, intraperitoneally) were administered 20 minutes before injection of morphine (10 mg/kg), the antinociceptive effect of morphine was partially antagonized. This effect reached statistical significance in the case of 4e. From these findings, it can be concluded that 4d and 4e exhibit a μ opioid antagonist profile.
Figure 8.

Antinociception: effects of 4d and 4e on the hot plate test. Bars show percent MPE (mean ± SEM, n≤10) induced by morphine 10 mg/kg, 4d 10 mg/kg, 4e 10 mg/kg, naloxone 1 mg/kg, and rimonabant 1 mg/kg administered 20 minutes after treatment with 4d 10 mg/kg, 4e 10 mg/kg, naloxone 1 mg/kg, or rimonabant 1 mg/kg. Statistically significant versus control value; ###P<0.001. Significant difference versus morphine; **P<0.01 (one-way analysis of variance, Newman–Keuls post hoc test).
Notes: Morphine (Sigma-Aldrich, Madrid, Spain); naloxone (Tocris/Biogen Científica SL, Madrid, Spain); rimonabant (Sanofi, Paris, France).
Abbreviations: MF, morphine; Nx, naloxone; SEM, standard error of the mean; SR, rimonabant; MPE, maximum possible effect.
An extensive literature search suggests that the endocannabinoid and μ opioid systems are involved in the development of alcohol dependence. In rodent models, rimonabant was found to decrease alcohol consumption.24 Treatment with the nonselective opioid antagonist naltrexone was effective in modulating aspects of alcohol-seeking behavior.32 Due to the μ opioid and CB1 cannabinoid dual antagonist activity on behavioral evaluations of 4e, the effects of this compound on operant ethanol self-administration were examined following a protocol essentially based on the alcohol relapse model in Wistar rats. For this purpose, the alcohol deprivation effect model previously reported by our group was used.33 The animals did not show significant changes in number of alcohol responses compared with the vehicle group during treatment with 4e at doses of 0.5 mg/kg, 2.0 mg/kg, and 8.0 mg/kg (data not shown).
ADME parameters in silico
A computational approach is considered to be an appropriate tool in early-stage drug discovery. Therefore, in silico ADME predictions can provide significant insights into drug-like properties.34 Calculation of ADME properties of 4e was performed on the conformer of global minimum energy using QikProp version 3.5 integrated in Maestro with a set of 34 physicochemical descriptors. Based on our predicted data, Lipinski35 and Jorgensen36 pharmacokinetic rules are followed. With regard to blood–brain barrier penetration (logBB −0.8; −3.0/1.2 for 95% of drugs), compound 4e was predicted to penetrate the brain. This prediction is consistent with its high lipophilicity (logP[octanol/water] = 9.9), and with the results obtained in the in vivo assays. As expected, compound 4e is poorly water-soluble (logS −12.6; −6.5/0.5 for 95% of drugs). However, the predicted human oral absorption is good (100%). Finally, in silico studies indicate unfavorable hERG (human Ether-à-go-go Related Gene) K+ channel blockade, with a logIC50 (inhibitory concentration 50) of −9.671 (range 95% of drugs; logIC50<−5). This in silico approach suggests that 4e has an acceptable ADME and cardiotoxicity profile.
Conclusion
Using the CB1 cannabinoid receptor inverse agonist/antagonist rimonabant (SR141716) as a scaffold, a series of pyrazole carboxamides containing part of the opioid fentanyl structure were prepared (4a–4k). The structural variations were focused on the linker separating the two pharmacophores, ie, aliphatic chains, cycloalkanes, and aromatic rings. Competitive binding assays and [35S]-GTPγS functionality tests were carried out in post-mortem human prefrontal cortex membrane preparations because receptors for both CB1 and μ opioids are expressed in this tissue, with the CB1 receptor being expressed in higher proportions.20 Selected compounds (4b, 4d, and 4e) displayed opioid and cannabinoid antagonism in these assays. Although the new compounds have modest affinity for both CB1 and μ receptors, an interesting finding is that 4b, 4d, and 4e behaved as CB1 cannabinoid receptor antagonists with a potency similar to that of rimonabant.
Subsequently, the in vivo cannabinoid antagonism of 4d and 4e, based on behavioral effects in mice, support the [35S]-GTPγS bioassay results. With regard to their opioid properties in vivo, the hot plate test confirmed that 4d and 4e had a µ opioid antagonist profile. In an alcohol relapse model in Wistar rats, treatment with 4e was not significantly effective in modulating relapse-like behavior.
Although these new compounds were designed as analogs of the opioid agonist fentanyl, the fact that they behave as opioid antagonists can be accounted for by subtle alteration of ligand structures that may lead to differences in affinity or intrinsic activity. However, these alterations occur more commonly with a morphine-like structure than with a fentanyl structure. While this paper was being prepared, Le Naour et al36 published on hybrid compounds based on rimonabant and morphine molecules. Unlike our case, the reported structural modification of morphine did not modify its opioid agonist nature.
The in silico ADME predictions for compound 4e suggest that it is likely to penetrate the blood–brain barrier, and this is consistent with the in vivo data.
In conclusion, this is the first description, to our knowledge, of hybrid compounds with cannabinoid and opioid antagonist properties in vitro and/or in vivo. It has been suggested in the literature that coadministration of cannabinoid antagonists and μ opioid antagonists could offer therapeutic advantages.27–29 Opioid-cannabinoid interactions in the regulation of appetite are of particular interest. Very recently, Wright and Rodgers30 reported a study showing that the pruritic effect of rimonabant can be attenuated by the opioid receptor antagonist naloxone. In this context, the promising dual activity of the cannabinoid/opioid compounds presented here offers an attractive starting point for future therapeutic applications.
Supplementary material
Synthesis procedures and characterization of new compounds
General
Melting points were determined with a Reichert Jung Thermovar apparatus (Reichert Optische Werke, Vienna, Austria). Mass spectra (MS) were recorded using electrospray positive mode. Elemental analysis was performed on a Heraeus CHN-O rapid analyzer (Foss Heraeus GmbH, Hanau, Germany). Analyses indicated by symbols of the elements or functions were within ±0.4% of theoretical values. Analytical high-performance liquid chromatography (HPLC) was run on a Waters 6000 with a Delta Pak C 18.5 μm (Waters, Cerdanyola del Vallès, Spain), 300 Å, 3.9×150 mm column, using CH3CN/H2O 95/5 (0.05% trifluoroacetic acid [TFA]) as eluent; flow rate 1 mL per minute; 254 nm. Nuclear magnetic resonance (NMR, 1H, 13C) spectra were recorded on Varian 300 (Agilent, Barcelona, Spain) and 400 unity spectrometers. All chemical shifts are reported in ppm. s: singlet; t: triplet; m: multiplet; brt: broad triplet; dp: double quadruplet; brm: broad multiplet; p: pentuplet; q: quadruplet; brd: broad doublet; brs: broad signal.
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(N-(1-phenethylpiperidin-4yl)propionamido)propyl)-1H-pyrazole-3-carboxamide (4a)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (triethylamine) (26 μL, 0.19 mmol) and N-(3-aminopropyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3a) (82 mg, 0.11 mmol), affording 4a as a yellow oil (44%): 1H NMR (399.93 MHz, CDCl3) δ7.43–7.04 (m, 12H), 4.46 (m, 1H), 3.43 (m, 2H), 3.37 (t, 2H, J=7.1 Hz), 3.28 (m, 2H), 3.1 (m, 2H), 2.77 (m, 2H), 2.59 (m, 2H), 2.35 (s, 3H), 2.32 (m, 2H), 2.05 (m, 2H), 1.84 (m, 2H), 1.69 (m, 2H), 1.10 (t, 3H, J=7.3 Hz); 13C NMR (99.98 MHz, CDCl3) δ 173.7, 162.8, 145.2, 142.8, 140.1, 136.0, 135.7, 134.9, 132.9, 130.8, 130.6, 130.2, 128.9, 128.8, 128.6, 128.4, 127.8, 126.1, 117.5, 60.3, 55.3, 53.2, 51.5, 41.1, 39.4, 36.9, 33.8, 30.1, 26.9, 26.8, 9.7, 9.6, 9.4; MS (ES+) (electrospray) m/z (%): 680 (100) [M+H]+; HPLC 99% purity; anal. (elemental analysis) C36H40Cl3N5O2.H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-(N-(1-phenethylpiperidin-4-yl)propionamido)butyl)-1H-pyrazole-3-carboxamide (4b)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(4-aminobutyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3b) (63 mg, 0.19 mmol), affording 4b as a yellowish solid (99%): melting point 80°C–82°C; 1H NMR (399.93 MHz, CDCl3) δ 7.42–7.05 (m, 12H), 4.51 (m, 1H), 3.43 (m, 2H), 3.23 (m, 2H), 2.89 (m, 2H), 2.78 (m, 2H), 2.61 (m, 2H), 2.37 (s, 3H), 2.32 (m, 2H), 2.10 (m, 2H), 1.92 (m, 2H), 1.64 (m, 2H), 1.12 (t, 3H, J=73 Hz); 13C NMR (99.98 MHz, CDCl3) δ 173.9, 162.7, 145.0, 143.0, 139.9, 135.9, 135.8, 134.9, 132.9, 130.7, 130.5, 130.2, 129.1, 128.8, 128.8, 128.4, 126.1, 117.6, 60.2, 55.1, 53.1, 43.12, 38.2, 33.6, 30.6, 28.7, 27.3, 26.8, 9.7, 9.4; MS (ES+) m/z (%): 696 (100) [M+H]+; HPLC 99% purity; anal. C37H42Cl3N5O2.1.5H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(5-(N-(1-phenethyl piperidin-4-yl)propionamido)pentyl)-1H-pyrazole-3-carboxamide (4c)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (49 mg, 0.12 mmol), TEA (21 μL, 0.15 mmol) and N-(5-aminopentyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3c) (52 mg, 0.15 mmol), affording 4c as a yellow solid (73%): 1H NMR (399.93 MHz, CDCl3) (mixture of rotamers) δ 7.40–6.92 (m, 12H), 5.80 (m, 0.4H), 4.55 (m, 0.3H), 3.83 (m, 0.6H), 3.70 (m, 0.7H), 3.44 (m, 2H), 3.10 (m, 2H), 2.74 (m, 2H), 2.56 (m, 2H), 2.35 (s, 3H), 2.30 (m, 2H), 2.07 (m, 2H), 1.80 (m, 2H), 1.65–1.30 (m, 8H), 1.12 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 173.8, 173.0, 162.7, 144.9, 138.8, 136.0, 134.9, 133.0, 130.8, 130.6, 130.5, 130.3, 128.9, 128.6, 128.4, 127.9, 126.1, 117.7, 60.4, 55.3, 53.3, 53.0, 43.3, 42.6, 38.7, 33.6, 30.7, 31.3, 29.5, 29.2, 9.8, 9.6, 9.4; MS (ES+) m/z (%): 710 (100) [M+H]+; HPLC 99% purity; anal. C38H44Cl3N5O2 (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(6-(N-(1-phenethy lpiperidin-4-yl)propionamido)hexyl)-1H-pyrazole-3-carboxamide (4d)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(6-aminohexyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3d) (68 mg, 0.19 mmol), affording 4d as a white solid (54%): melting point 69°C–71°C; 1H NMR (399.93 MHz, CDCl3) (mixture of rotamers) δ 7.43–6.92 (m, 12H), 4.46 (m, 0.6H), 3.55 (m, 0.4H), 3.41 (m, 2H), 3.20–3.04 (m, 4H), 2.79 (m, 2H), 2.59 (m, 2H), 2.37 (s, 3H), 2.32 (m, 2H), 2.06 (brt, 2H, J=11.7 Hz), 1.86 (dq, 2H, J=3.4 and 12.2 Hz), 1.68 (brm, 2H), 1.60, 1.54, 1.39 and 1.31 (m, 8H), 1.14 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 173.7, 162.7, 145.0, 143.1, 140.1, 136.0, 135.8, 134.9, 133.0, 130.8, 130.6, 130.5, 130.3, 128.9, 128.6, 128.4, 127.9, 126.1, 117.7, 60.4, 54.5, 53.2, 51.4, 43.4, 42.0, 38.8, 33.9, 30.9, 31.6, 30.0, 29.8, 26.9, 26.7, 26.6, 9.8, 9.7, 9.4; MS (ES+) m/z (%): 724 (100) [M+H]+; HPLC 99% purity; anal. C39H46Cl3N5O2 (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(7-(N-(1-phenethylpiperidin-4yl)propionamido)heptyl)-1H-pyrazole-3-carboxamide (4e)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(7-aminoheptyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3e) (41 mg, 0.11 mmol), affording 4e as an orange oil (55%): 1H NMR (399.93 MHz, CDCl3) (mixture of rotamers) δ7.41–6.91 (m, 12H), 4.45 (m, 0.7H), 3.53 (m, 0.3H), 3.39 (p, 2H, J=7.0 Hz), 3.17 (t, 2H, J=8.0 Hz), 3.10 (m, 2H), 2.79 (m, 2H), 2.60 (m, 2H), 2.36 (s, 3H), 2.30 (m, 2H), 2.05 (brt, 2H, J=11.7 Hz), 1.84 (q, 2H, J=11.3 Hz), 1.67 (brd 2H, J=10.7 Hz), 1.58, 1.51, 1.35, 1.26 (m, 10H), 1.30 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 173.7, 172.9, 162.6, 145.0, 143.0, 140.1, 135.9, 135.8, 134.8, 132.9, 130.8, 130.5, 130.3, 117.6, 60.4, 55.2, 53.1, 51.2, 43.4, 38.9, 38.8, 33.8, 29.6, 27.0, 26.9, 9.8, 9.6, 9.4, 42.1, 31.6, 30.8, 28.9, 27.1; MS (ES+) m/z (%): 738 (100); [M+H]+; HPLC 99% purity; anal. C40H48Cl3N5O2.0.5H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(8-(N-(1-phenethylpiperidin-4-yl)propionamido)octyl)-1H-pyrazole-3-carboxamide (4f)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(8-aminooctyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3f) (40 mg, 0.10 mmol), affording 4f as an orange solid (87%): melting point 59°C–60°C; 1H NMR (399.93 MHz, CDCl3) (mixture of rotamers) δ 7.40–6.93 (m, 12H), 4.45 (m, 0.8H), 3.45 (m, 0.2H), 3.38 (m, 2H), 3.12 (m, 2H), 3.04 (m, 2H), 2.79 (m, 2H), 2.61 (m, 2H), 2.35 (s, 3H), 2.31 (m, 2H), 2.02 (m, 2H), 1.87 (m, 2H), 1.68 (m, 2H), 1.56–1.28 (m, 12H), 1.13 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 173.8, 172.9, 162.6, 145.1, 144.8, 140.5, 136.0, 134.9, 133.0, 130.8, 130.5, 130.3, 128.9, 128.6, 128.4, 127.9, 126.1, 117.7, 60.4, 53.3, 53.2, 43.5, 39.0, 33.9, 31.7, 30.9, 29.3, 29.2, 27.2, 29.7, 26.9, 26.7, 9.8, 9.6, 9.4; MS (ES+) m/z (%): 750 (100) [M+H]+; HPLC 98% purity; anal. C41H50Cl3N5O2.H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(9-(N-(1-phenethylpiperidin-4-yl)propionamido)nonyl)-1H-pyrazole-3-carboxamide (4g)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(9-aminononyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3g) (30 mg, 0.07 mmol) affording 4g as a yellow oil (85%): 1H NMR (399.93 MHz, CDCl3) (mixture of rotamers) δ 7.73–6.92 (m, 12H), 4.45 (m, 0.7H), 3.53 (m, 0.3H), 3.39, 3.10 (m, 2H), 3.07 (m, 2H), 2.80 (m, 2H), 2.57 (m, 2H), 2.35 (s, 3H), 2.30 (m, 2H), 2.08 (m, 2H), 1.84 (q, 2H, J=12.2 Hz), 1.67 (m, 2H), 1.58, 1.49 and 1.26 (m, 14H), 1.13 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 173.7, 172.9, 162.6, 145.1, 143.1, 135.9, 134.8, 132.9, 130.8, 130.6, 130.5, 130.3, 128.9, 128.6, 128.4, 127.8, 127.2, 60.4, 55.3, 53.2, 51.4, 43.5, 39.0, 33.8, 31.6, 30.9, 30.0, 29.5, 29.2, 27.3, 27.2, 26.7, 26.9, 9.8, 9.6, 9.4; MS (ES+) m/z (%): 764 (100) [M+H]+; HPLC 98% purity; anal. C42H52Cl3N5O2.1.25H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(12-(N-(1-phenethylpiperidin-4-yl)propionamido)dodecyl)-1H-pyrazole-3-carboxamide (4h)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(12-aminododecyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3h) (78 mg, 0.19 mmol), affording 4h as an orange oil (99%): 1H NMR (399.93 MHz, CDCl3) δ 7.40–6.91 (m, 12H), 4.41 (m, 1H), 3.36 (q, 2H, J=7.5 Hz), 3.14–2.96 (m, 4H), 2.78 (m, 2H), 2.58 (m, 2H), 2.30 (m, 3H), 2.23 (m, 2H), 1.97 (m, 2H), 1.80 (m, 2H), 1.62 (m, 2H), 1.55–1.19 (m, 20H), 1.08 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 174.2, 173.3, 163.0, 145.5, 143.4, 136.4, 136.3, 135.3, 133.4, 131.2, 130.9, 130.7, 129.3, 129.1, 128.9, 128.3, 127.8, 126.5, 118.1, 60.9, 55.7, 53.8, 53.7, 53.5, 44.1, 42.7, 39.5, 34.26, 31.4, 30.2, 29.9, 29.8, 29.7, 27.8, 27.7, 27.4, 27.2; MS (ES+) m/z (%): 808 (100) [M+H]+; HPLC 98% purity; anal. C45H58Cl3N5O2.H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(N-(1-phenethylpiperidin-4-yl)propionamido)phenyl)-1H-pyrazole-3-carboxamide (4i)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (15 μL, 0.11 mmol) and N-(3-aminophenyl)-N-(1-phenethyl-piperidin-4-yl)propionamide (3i) (35 mg, 0.09 mmol), affording 4i as a yellow solid (48%): melting point 105°C–108°C; 1H NMR (399.93 MHz, CDCl3) δ 7.53–6.73 (m, 16H), 4.58 (m, 1H), 2.91 (brs, 2H, J=10.5 Hz), 2.61 (m, 2H), 2.43 (m, 2H), 2.33 (s, 3H), 2.07 (brt, 2H, J=11.5 Hz), 1.91 (m, 2H), 1.75 (brt, 2H, J=14.0 Hz), 1.39 (m, 2H), 0.94 (t, 3H, J=7.5 Hz); 13C NMR (99.98 MHz, CDCl3) δ 173.4, 160.5, 144.5, 143.6, 139.5, 138.9, 136.2, 135.7, 135.1, 133.0, 132.9, 130.8, 130.5, 130.4, 129.6, 129.0, 128.6, 128.3, 127.9, 126.8, 126.0, 125.8, 121.4, 119.2, 118.3, 60.4, 53.1, 52.2, 33.8, 30.6, 28.5, 9.6, 9.5; MS (ES+) m/z (%): 716 (100) [M+H]+; HPLC 99% purity; anal. C39H38Cl3N5O2.3H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(3-(N-(1-phenethylpiperidin-4yl)propionamido)benzyl)-1H-pyrazole-3-carboxamide (4j)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (60 mg, 0.15 mmol), TEA (26 μL, 0.19 mmol) and N-(3-(aminomethyl)benzyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3j) (60 mg, 0.15 mmol), affording 4j as a yellow solid (60%): melting point 83°C–85°C; 1H NMR (399.93 MHz, CDCl3) (mixture of rotamers) δ 7.40–7.06 (m, 16H), 4.60 (m, 4H), 4.51 (m, 1H), 3.67 (m, 1H), 3.00 (m, 2H), 2.74 (m, 2H), 2.55 (m, 2H), 2.48 (q, 0.5H, J=7.3 Hz), 2.40 (s, 3H), 2.23 (q, 1.5H, J=7.3 Hz), 2.11 (m, 2H), 1.79 (m, 2H), 1.65 (m, 2H), 1.20 (t, 1H, J=7.3 Hz), 1.07 (t, 2H, J=7.3 Hz); 13C NMR (99.98 MHz, CDCl3) δ 174.7, 173.9, 162.6, 144.7, 143.1, 140.1, 139.1, 138.3, 136.0, 135.9, 134.9, 132.9, 130.8, 130.5, 130.3, 129.0, 128.9, 128.6, 128.4, 127.9, 127.2, 126.5, 126.1, 124.8, 124.5, 117.9, 60.3, 53.0, 51.5, 50.7, 46.3; MS (ES+) m/z (%): 742 (100) [M+H]+; HPLC 99% purity; anal. C41H42N5O2.0.5H2O (C, H, N, O).
5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(4-[(4-N-(1-phenethylpiperidin-4-yl)propionamido)cyclohexyl]methyl)cyclo-hexyl-1H-pyrazole-3-carboxamide (4k)
5-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (2) (49 mg, 0.12 mmol), TEA (21 μL, 0.15 mmol) and N-(4-((4-aminocyclohexyl)methyl) cyclohexyl)-N-(1-phenethylpiperidin-4-yl)propionamide (3k) (70 mg, 0.15 mmol), affording 4k as a yellow solid (73%): 1H NMR (399.93 MHz, CDCl3) δ 7.36–6.69 (m, 12H), 4.12 (m, 1H), 3.84 (m,1H), 3.47 (m, 1H), 3.05 (m, 2H), 2.57 (m, 2H), 2.53 (m, 2H), 2.31 (s, 3H), 2.27 (m, 2H), 2.02 (m, 2H), 1.86 (m, 2H), 1.63 (m, 2H), 1.99–1.31 (m, 20H), 1.19 (m, 3H); 13C NMR (99.98 MHz, CDCl3) δ 172.8, 161.9, 145.2, 142.9, 140.0, 136.0, 135.9, 134.8, 132.9, 130.8, 130.6, 130.3, 128.8, 128.6, 128.4, 127.8, 126.1, 117.7, 60.5, 56.0, 53.3, 48.5, 45.5, 37.5, 34.4, 33.1, 32.1, 32.0, 29.6, 29.3, 33.8, 28.6, 24.5, 9.6, 9.4; MS (ES+) m/z (%): 816 (100) [M+H]+; HPLC 99% purity; anal. C46H56Cl3N5O2.2H2O (C, H, N, O).
Acknowledgments
This work was supported by grants from the Spanish Ministry of Economy and Competitivity (SAF2012-40075, SAF2009-12422, SAF2010-20521, SAF2011-26818), Red de Trastornos Adictivos (RETICS RD06/001), the Madrid Government (CANNAB-CM, S2010/BMD-2308), the University of the Basque Country (UFI 11/35), the Basque Government (IT-199-07, SAIOTEK S-PE10UN14), and the Instituto de Salud Carlos III, Centro de Investigación Biomédica en Red de Salud Mental, CIBERSAM. AME is the recipient of a predoctoral fellowship from the Basque Government. PM is the recipient of a fellowship (JAE-Pre-2010-01119) from Junta para la Ampliación de Estudios, cofinanced by the European Social Fund. The authors thank Laura Hernández-Folgado for her help in preparation of the manuscript.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Manzanares J, Corchero J, Romero J, Fernandez-Ruiz JJ, Ramos JA, Fuentes JA. Pharmacological and biochemical interactions between opioids and cannabinoids. Trends Pharmacol Sci. 1999;20(7):287–294. doi: 10.1016/s0165-6147(99)01339-5. [DOI] [PubMed] [Google Scholar]
- 2.Vigano D, Rubino T, Parolaro D. Molecular and cellular basis of cannabinoid and opioid interactions. Pharmacol Biochem Behav. 2005;81(2):360–368. doi: 10.1016/j.pbb.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 3.Abrams DI, Couey P, Shade SB, Kelly ME, Benowitz NL. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90(6):844–851. doi: 10.1038/clpt.2011.188. [DOI] [PubMed] [Google Scholar]
- 4.Maldonado R, Berrendero F. Endogenous cannabinoid and opioid systems and their role in nicotine addiction. Curr Drug Targets. 2010;11(4):440–449. doi: 10.2174/138945010790980358. [DOI] [PubMed] [Google Scholar]
- 5.Christie MJ. Opioid and cannabinoid receptors: friends with benefits or just close friends? Br J Pharmacol. 2006;148(4):385–386. doi: 10.1038/sj.bjp.0706756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trang T, Sutak M, Jhamandas K. Involvement of cannabinoid (CB1)-receptors in the development and maintenance of opioid tolerance. Neuroscience. 2007;146(3):1275–1288. doi: 10.1016/j.neuroscience.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 7.Scavone JL, Sterling RC, Van Bockstaele EJ. Cannabinoid and opioid interactions: Implications for opiate dependence and withdrawal. Neuroscience. 2013;248:637–654. doi: 10.1016/j.neuroscience.2013.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morphy R, Rankovic Z. Designing multiple ligands – medicinal chemistry strategies and challenges. Curr Pharm Des. 2009;15(6):587–600. doi: 10.2174/138161209787315594. [DOI] [PubMed] [Google Scholar]
- 9.Shonberg J, Scammells PJ, Capuano B. Design strategies for bivalent ligands targeting GPCRs. Chem Med Chem. 2011;6(6):963–974. doi: 10.1002/cmdc.201100101. [DOI] [PubMed] [Google Scholar]
- 10.Davis MP. Fentanyl for breakthrough pain: a systematic review. Expert Rev Neurother. 2011;11(8):1197–1216. doi: 10.1586/ern.11.63. [DOI] [PubMed] [Google Scholar]
- 11.Jagerovic N, Fernandez-Fernandez C, Goya P. CB1 cannabinoid antagonists: structure-activity relationships and potential therapeutic applications. Curr Top Med Chem. 2008;8(3):205–230. doi: 10.2174/156802608783498050. [DOI] [PubMed] [Google Scholar]
- 12.Christopoulou FD, Kiortsis DN. An overview of the metabolic effects of rimonabant in randomized controlled trials: potential for other cannabinoid 1 receptor blockers in obesity. J Clin Pharm Ther. 2011;36(1):10–18. doi: 10.1111/j.1365-2710.2010.01164.x. [DOI] [PubMed] [Google Scholar]
- 13.Montero A, Goya P, Jagerovic N, et al. Guanidinium and aminoimidazolinium derivatives of N-(4-piperidyl)propanamides as potential ligands for mu opioid and I2-imidazoline receptors: synthesis and pharmacological screening. Bioorg Med Chem. 2002;10(4):1009–1018. doi: 10.1016/s0968-0896(01)00356-x. [DOI] [PubMed] [Google Scholar]
- 14.Dardonville C, Fernandez-Fernandez C, Gibbons SL, et al. Synthesis and pharmacological studies of new hybrid derivatives of fentanyl active at the mu-opioid receptor and I-2-imidazoline binding sites. Bioorg Med Chem. 2006;14(19):6570–6580. doi: 10.1016/j.bmc.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Thomas BF, Francisco MEY, Seltzman HH, et al. Synthesis of long-chain amide analogs of the cannabinoid CB1 receptor antagonist N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-py razole-3-carboxamide (SR141716) with unique binding selectivities and pharmacological activities. Bioorg Med Chem. 2005;13(18):5463–5474. doi: 10.1016/j.bmc.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Fernandez C, Decara J, Bermúdez-Silva FJ, et al. Description of a bivalent cannabinoid ligand with hypophagic properties. Arch Pharm. 2013;346(3):171–179. doi: 10.1002/ardp.201200392. [DOI] [PubMed] [Google Scholar]
- 17.Sasmal PK, Reddy DS, Talwar R, et al. Novel pyrazole-3-carboxamide derivatives as cannabinoid-1 (CB1) antagonists: journey from non-polar to polar amides. Bioorg Med Chem Lett. 2011;21(1):562–568. doi: 10.1016/j.bmcl.2010.10.055. [DOI] [PubMed] [Google Scholar]
- 18.Dardonville C, Jagerovic N, Callado LF, Meana JJ. Fentanyl derivatives bearing aliphatic alkaneguanidinium moieties: a new series of hybrid molecules with significant binding affinity for mu-opioid receptors and I2-imidazoline binding sites. Bioorg Med Chem Lett. 2004;14(2):491–493. doi: 10.1016/j.bmcl.2003.10.048. [DOI] [PubMed] [Google Scholar]
- 19.Krishnamurthy M, Li W, Moore BM., 2nd Synthesis, biological evaluation, and structural studies on N1 and C5 substituted cycloalkyl analogues of the pyrazole class of CB1 and CB2 ligands. Bioorg Med Chem. 2004;12(2):393–404. doi: 10.1016/j.bmc.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 20.De Jesus ML, Salles J, Meana JJ, Callado LF. Characterization of CB1 cannabinoid receptor immunoreactivity in postmortem human brain homogenates. Neuroscience. 2006;140(2):635–643. doi: 10.1016/j.neuroscience.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Cumella J, Hernandez-Folgado L, Giron R, et al. Chromenopyrazoles: non-psychoactive and selective CB1 cannabinoid agonists with peripheral antinociceptive properties. ChemMedChem. 2012;7(3):452–463. doi: 10.1002/cmdc.201100568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez-Maeso J, Rodriguez-Puertas R, Gabilondo AM, Meana JJ. Characterization of receptor-mediated [35S]GTPgammaS binding to cortical membranes from postmortem human brain. Eur J Pharmacol. 2000;390(1–2):25–36. doi: 10.1016/s0014-2999(99)00827-4. [DOI] [PubMed] [Google Scholar]
- 23.Pertwee RG. The ring test: a quantitative method for assessing the ‘cataleptic’ effect of cannabis in mice. Br J Pharmacol. 1972;46(4):753–763. doi: 10.1111/j.1476-5381.1972.tb06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serra S, Carai MA, Brunetti G, et al. The cannabinoid receptor antagonist SR 141716 prevents acquisition of drinking behavior in alcohol-preferring rats. Eur J Pharmacol. 2001;430(2–3):369–371. doi: 10.1016/s0014-2999(01)01379-6. [DOI] [PubMed] [Google Scholar]
- 25.Erdozain AM, Diez-Alarcia R, Meana JJ, Callado LF. The inverse agonist effect of rimonabant on G protein activation is not mediated by the cannabinoid CB1 receptor: evidence from postmortem human brain. Biochem Pharmacol. 2012;83(2):260–268. doi: 10.1016/j.bcp.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Rowland NE, Mukherjee M, Robertson K. Effects of the cannabinoid receptor antagonist SR 141716, alone and in combination with dexfenfluramine or naloxone, on food intake in rats. Psychopharmacology. 2001;159(1):111–116. doi: 10.1007/s002130100910. [DOI] [PubMed] [Google Scholar]
- 27.Robledo P, Berrendero F, Ozaita A, Maldonado R. Advances in the field of cannabinoid-opioid cross-talk. Addict Biol. 2008;13(2):213–224. doi: 10.1111/j.1369-1600.2008.00107.x. [DOI] [PubMed] [Google Scholar]
- 28.Tallett AJ, Blundell JE, Rodgers RJ. Effects of acute low-dose combined treatment with naloxone and AM 251 on food intake, feeding behaviour and weight gain in rats. Pharmacol Biochem Behav. 2009;91(3):358–366. doi: 10.1016/j.pbb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Wright FL, Rodgers RJ. Low dose naloxone attenuates the pruritic but not anorectic response to rimonabant in male rats. Psychopharmacology. 2013;226(2):415–431. doi: 10.1007/s00213-012-2916-5. [DOI] [PubMed] [Google Scholar]
- 30.Pertwee RG, Stevenson LA, Elrick DB, Mechoulam R, Corbett AD. Inhibitory effects of certain enantiomeric cannabinoids in the mouse vas deferens and the myenteric plexus preparation of guinea-pig small intestine. Br J Pharmacol. 1992;105(4):980–984. doi: 10.1111/j.1476-5381.1992.tb09088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heyser CJ, Moc K, Koob GF. Effects of naltrexone alone and in combination with acamprosate on the alcohol deprivation effect in rats. Neuropsychopharmacology. 2003;28(8):1463–1471. doi: 10.1038/sj.npp.1300175. [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Moreno JA, Gonzalez-Cuevas G, Rodriguez de Fonseca F, Navarro M. Long-lasting increase of alcohol relapse by the cannabinoid receptor agonist WIN 55,212-2 during alcohol deprivation. J Neurosci. 2004;24(38):8245–8252. doi: 10.1523/JNEUROSCI.2179-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowen JP, Guner OF. A perspective on quantum mechanics calculations in ADMET predictions. Curr Top Med Chem. 2013;13(11):1257–1272. doi: 10.2174/15680266113139990032. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54(3):355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 36.Le Naour M, Akgun E, Yekkirala A, et al. Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB) receptors are potent analgesics devoid of tolerance. J Med Chem. 2013;56(13):5505–5513. doi: 10.1021/jm4005219. [DOI] [PMC free article] [PubMed] [Google Scholar]