

Abstract
The ability to detect single molecules in live bacterial cells enables us to probe biological events one molecule at a time and thereby gain knowledge of the activities of intracellular molecules that remain obscure in conventional ensemble-averaged measurements. Single-molecule fluorescence tracking and super-resolution imaging are thus providing a new window into bacterial cells and facilitating the elucidation of cellular processes at an unprecedented level of sensitivity, specificity and spatial resolution. In this Review, we consider what these technologies have taught us about the bacterial cytoskeleton, nucleoid organization and the dynamic processes of transcription and translation, and we also highlight the methodological improvements that are needed to address a number of experimental challenges in the field.
The advent of single-molecule detection 25 years ago1,2, which was followed soon after by single-molecule fluorescence imaging3 and the subsequent development of single fluorescent protein imaging at room temperature4, provided the means to investigate individually labelled objects without ensemble averaging. Beyond enabling fundamental investigations of the physics and chemistry of the emissive labels, one main advantage of this technology is its use in a wide range of biologically relevant tracking and imaging experiments. With the ability to express fluorescent fusion proteins in bacterial cells and to image and track single copies5, proteins have become the primary targets for in vivo labelling assays. Consequently, most single-molecule-based experiments in bacterial cells so far have focused on intracellular proteins.
In contrast to eukaryotic proteins, bacterial proteins are not confined to specific subcellular compartments by membrane-delimited organelles. Nonetheless, fluorescence imaging experiments have shown that certain bacterial proteins localize to specific subcellular locations at specific times6. Thus, in addition to proteins diffusing freely in the cytoplasm, bacteria have an intricate subcellular organization7. In this complex environment, single proteins mostly carry out their roles as individual entities that are embedded in their local surroundings, whereas other copies of the same protein occupy distinct locations and can be in different enzymatic or conformational states. Single-molecule studies can probe and exploit this heterogeneity by investigating one molecule at a time; for example, bound and unbound proteins can be distinguished from each other owing to differences in their diffusive properties. The ability to observe how single protein molecules behave inside bacterial cells has enabled us to ask where, when and how proteins act and interact, and how these events ultimately drive larger-scale cellular processes.
There are two primary experimental approaches for determining spatiotemporal information about bacterial proteins using the fluorescence of a single molecule. Both approaches are based on the same principle: the position of a single molecule — that is, its x and y (and in some cases z, see below) coordinates — can be extracted with high precision if its fluorescence emission profile on a wide-field detector does not significantly overlap with the emission profile from other molecules in the vicinity and if the molecule is not moving too quickly relative to the frame exposure time (BOX 1; FIG. 1a).
Box 1. Principles of super-resolution microscopy.
The terms super-resolution microscopy, subdiffraction microscopy, diffraction-unlimited microscopy and nanoscopy are often used synonymously. These techniques fall into two general classes. The first class, which includes methods that are based on deterministic detection, such as stimulated emission depletion (STED), reversible saturable optical fluorescence transitions (RESOLFT)113,114 and (saturated) structured illumination microscopy ((S)SIM)115,116, use a spatially patterned fluorescence excitation scheme and do not require single-molecule sensitivity. Therefore, we refer the reader to other reviews of these techniques applied to general cellular imaging (see REFS 117,118) and specifically to bacterial imaging (see REF 119).
In the second class, the diffraction limit is circumvented by applying an ‘active control’ method to limit the emitter concentration, so that the isolated fluorescence emission profiles that originate from individual molecules can be detected in each frame (FIG. 1a). By analysing the shape of these intensity profiles (either by centroid estimation or by fitting to a mathematical function), the positions of the molecules can be extracted with a precision that is mainly limited by the signal-to-background ratio of the measurement (that is, the number of photons that are detected above the background), as opposed to the optical diffraction limit. Approaches for actively controlling emitter concentration are diverse and include photoactivation120, photoswitching121 and photoinduced blinking4. In 2006, these methods were named (fluorescent) photoactivated localization microscopy (F)PALM10,11 and stochastic optical reconstruction microscopy (STORM)12. Since then, many different implementations of the underlying idea, as well as new approaches to limit emitter concentration, have emerged122–126. Furthermore, optical methods for achieving three-dimensional single-molecule imaging have been developed and are reviewed in detail in REF. 127.
When imaging live cells, it is possible to monitor the change of a structure over time128–130 and/or to use distinguishable features in the measured single-molecule signals to alter the selection of detected molecules for inclusion in the final reconstructed images. As illustrated in part a of the figure, molecules that have different diffusive properties produce very different intensity profiles that also depend on the camera acquisition time. The simulated images show the effect of Brownian motion on the intensity profile of three molecules with different diffusion coefficients (D) that are representative of cytoplasmic, membrane-attached and essentially fixed molecules. The three rows in part b of the figure show three different experimental strategies for varying the effective frame exposure time. The image of a rapidly diffusing molecule is spread out to undetectable levels with a 200 ms image acquisition time and continuous illumination. This approach is often used in live cells to selectively image a quasi-static structure in the presence of rapidly diffusing molecules37,93. By contrast, a rapidly diffusing molecule produces a clear diffraction-limited image when a short exposure time of 20 ms and a stroboscopic illumination time of 2 ms is used, which makes this the method of choice for tracking the fast motion of free proteins in the cytoplasm131. For very slowly diffusing (or fixed) proteins, all three exposure time approaches are suitable.
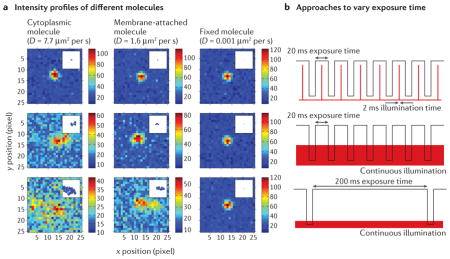
Figure 1. Principles of single-molecule tracking and imaging.

a | The position of a single molecule is determined by fitting the measured fluorescence intensity profile to a mathematical function. The intensity profile is the diffraction-limited image of a point source of light, and its width is determined by the point spread function (PSF) of the microscope. The measured profile is well approximated by a Gaussian bell-shaped curve, which makes it possible to estimate the position of the point source, indicated by the arrow. The uncertainty in the position parameter (standard deviations of 10–40 nm are typical for a few thousand detected photons) is much smaller than the width of the diffraction-limited PSF (200–300 nm). b | In single-molecule tracking, the same labelled protein moving dynamically through the cell is repeatedly localized in subsequent frames when a fast frame rate is used. This provides snapshots of the spatial trajectory of the protein over time, which enables the track of the protein to be reconstructed (right). If a slow frame rate is used, the fluorescence emission of the molecule is blurred by diffraction and becomes part of the background autofluorescence (left). c | In super-resolution imaging, the morphology of an extended structure is determined by measuring the positions of different copies of labelled molecules in the structure. If all labels emit at once, the structure is blurred by diffraction (left). The ring-like structure at mid-cell is sampled by stochastically determining the positions of different labelled monomeric subunits in sequential frames. Using the list of localization measurements, a reconstructed image is then computationally assembled during post-acquisition processing (right).
The first approach, which is known as single-molecule tracking (FIG. 1b), involves the expression5 or activation8,9 of a labelled protein construct — most commonly a fluorescent fusion protein (BOX 2) — at a very low concentration to produce only one or two concurrently emitting molecules per cell. By repeatedly detecting and then localizing the precise position of the same molecule over time, its spatial trajectory (which is estimated on the basis of sequential position measurements) can be determined5. Analysis of the resulting single-molecule tracks then provides information on the mode of motion of the labelled protein, which may be diffusive, motor-directed, confined or a mixture of these. As intracellular crowding and protein-binding kinetics influence the trajectory of a single protein, these measurements enable us to ‘watch’ the motion of the protein as it carries out its function in vivo.
Box 2. The pros and cons of fluorescent protein labels.
Almost all single-molecule tracking and super-resolution studies in bacteria so far have used fluorescent protein fusion constructs, owing to the many advantages of this labelling approach. Bacterial strains can be constructed to express fluorescent proteins fused to almost any protein of interest inside the cell132. Endogenous expression of fluorescent proteins eliminates the challenge of delivering fluorescent molecules across the cell membrane and cell wall, and provides a level of labelling specificity and efficiency that is unmatched by other methods. Several fluorescent proteins are available for bacterial single-molecule super-resolution experiments, but they need to be paired with the appropriate active control mechanism. Photoactivatable fluorescent proteins, such as photoactivatable GFP (PAGFP) and PAmCherry1, can be converted from a non-fluorescent dark state to a fluorescent state by a pulse of short-wavelength light133,134; photoswitchable fluorescent proteins, such as Dendra2 and mEos2 (mEosFP variant), change their fluorescent activation and emission wavelength upon illumination with short-wavelength light135–137; and GFP-variants, such as enhanced yellow fluorescent protein (eYFP), can be induced to blink by high-intensity illumination138 and can also be restored from a dark state to an emissive state by short-wavelength light38. The different spectral characteristics of different fluorescent proteins have also made it possible to distinguish two or more different labels using multicolour, single-molecule super-resolution microscopy64,71,139.
Although fluorescent molecules are seldom directly harmful to cellular processes, their fusion to target proteins can perturb the native function of the target protein. Numerous proteins function as dimers, multimers and polymers, or they function in close proximity to physical barriers, such as membranes. The binding interfaces of proteins are often highly specific for their interaction partners, and care must be taken to introduce the bulky fluorescent protein (for example, the 28 kDa GFP has 238 amino acid residues) at a location that does not sterically interfere with these binding interactions. For these reasons, monomeric target proteins that freely diffuse in the cytoplasm are generally more likely to continue to function correctly as fluorescent fusion proteins. In addition, some fluorescent proteins artificially induce clustering under certain conditions, which may alter the native localization pattern of the fusion partners140.
To confirm the validity of any fluorescent imaging studies, the functionality of the labelled construct should be independently verified. The most robust method to test this involves replacing the gene that encodes the wild-type protein with the gene that encodes the fusion protein, and then evaluating the mutant cells for any phenotypic changes compared to wild-type cells. Genomic replacements have the additional advantage of ensuring that the fusion protein is expressed at native levels, which can vary considerably for cell cycle-regulated proteins, for example. If the fluorescent fusion protein does produce an altered phenotype, in some instances it might still be possible to use it as a label by expressing it in addition to the wild-type protein (the degree of functional perturbation then becomes concentration-dependent). The increased spatial resolution that is gained using single-molecule techniques (BOX 1) ideally requires full functionality of labelled protein constructs, as even slight alterations in protein localization patterns are now becoming detectable.
The second approach is known as single-molecule super-resolution imaging10–12 (FIG. 1c). As opposed to tracking of the same molecule, this approach determines the positions of different copies of labelled molecules. If these labelled copies are incorporated into a larger structure, such as a polymeric protein filament, then their positions randomly sample this structure and thereby provide information about its overall shape and position in the cell. A point-by-point reconstruction can then be assembled by combining the localized positions of all detected molecules in a computational post-processing step. Importantly, because all molecules are localized with a precision of tens of nanometres, this approach circumvents the diffraction limit that otherwise limits image resolution to 200–300 nm in conventional fluorescence microscopy. Such a large improvement in resolution (a factor of five or more, which results in resolutions that are typically in the 20–40 nm range), means that structures that were previously unresolvable are now clearly visible, which enables us to address one long-standing question in bacterial cell biology: what are the locations and (three-dimensional (3D)) architectures of bacterial protein superstructures at a length scale of 200–300 nm?
In this Review, we describe a selection of recent single-molecule tracking and super-resolution imaging studies in bacteria that best illustrate the novel insights that have been gained. Thus far, Caulobacter crescentus and Escherichia coli are the two model systems that have been most studied by single-molecule methods, although applications in Bacillus subtilis are beginning to emerge. We consider the many diverse aspects of bacterial cell biology that have been examined, including structural (cytoskeletal) proteins, nucleoid organization, chromosome segregation and partitioning, and transcription and translation13. Owing to space constraints, not all topics are covered, such as recent studies14,15 determining the stoichiometry and component exchange of the replisome, which are reviewed in REF. 16. We conclude with a discussion of the present limitations that future work in the field needs to address and the potential for future discoveries.
Structural proteins: the bacterial cytoskeleton
The bacterial cytoskeleton consists of polymeric protein filaments that provide the cell with structural scaffolds to coordinate cellular processes over spatial distances that are larger than the size of individual protein monomers. These processes include cell wall synthesis17, cell division18 and chromosome segregation19. The first bacterial cytoskeletal filaments to be identified were the cell division protein FtsZ20 (a homologue of tubulin) and the rod shape-maintaining protein MreB21,22 (a homologue of actin). Additional cytoskeletal proteins that lack eukaryotic homologues continue to be discovered and have been reviewed in detail in REFS 23, 24.
Filament-forming cytoskeletal proteins have been frequent subjects of super-resolution experiments; as they have a presumed quasi-static and directional nature, filaments are readily inferred, even from sparsely labelled samples. However, these filamentous structures can be challenging to label with bulky fluorescent proteins, as polymeric packing can be perturbed by the addition of a bulky fluorescent label at or near the binding interface (BOX 2). In this section, we review the biological insight that has been gained using single-molecule methods for MreB, FtsZ and the polar organizing protein PopZ (FIG. 2).
Figure 2. Cytoskeletal and structural proteins imaged by single-molecule methods.
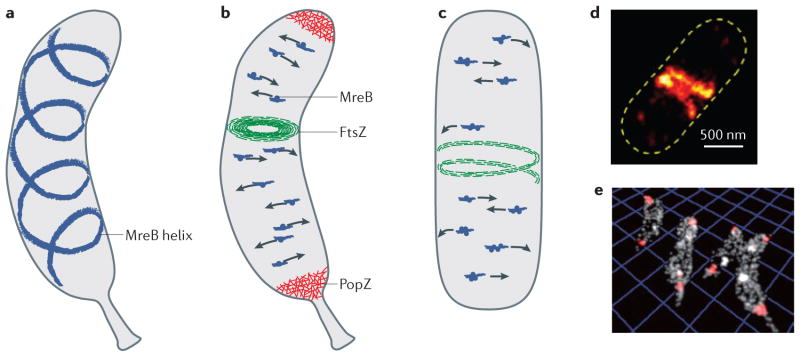
a | Model for the diffraction-limited and super-resolution images of enhanced yellow fluorescent protein (eYFP)–MreB in Caulobacter crescentus, which suggests that the protein adopts a quasi-helical structure that spans the length of the cell21,25,27,28,38. However, the presence of this helix was later shown to be an artefact that originated from the fluorescent protein label42,43. b | Model of the localization and motion patterns of MreB, FtsZ and PopZ in C. crescentus. Single eYFP–MreB molecules move across the cells in directions that are perpendicular to the long axis of the cell, at a speed of approximately 6 nm per second, which suggests that they move in a circumferential manner37. The MreB molecules (dark blue dots) are hypothesized to be incorporated into short actin-like filaments (blue streaks) that may provide a scaffold for the peptidoglycan synthetic complex17,46,17,37,46. FtsZ molecules assemble into short filaments50 that organize into a narrow ring structure. The three-dimensional (3D) super-resolution reconstructions show that this FtsZ bundle extends further into the cell interior, such that a cytoplasmic opening of only ~150–300-nm in diameter is present57. c | Model of cytoskeletal proteins imaged by single-molecule methods in Escherichia coli and Bacillus subtilis. MreB patches, which consist of multiple labelled MreB molecules that emit at the same time (group of dark blue dots), move together across the cell (at a speed of 6.7 nm per second in E. coli and 20–60 nm per second in B. subtilis) in directions that are perpendicular to the long axis of the cell, similar to the single-molecule tracks that are observed in C. crescentus. The speed of MreB movement depends on the organism and the environmental conditions39–41. d | In unconstricted E. coli cells, FtsZ filaments assemble into a compressed helical structure before collapsing into a constriction ring at mid-cell51. e | The polar PopZ molecules (red) form a 3D polymeric network that fills the polar spaces of C. crescentus cells64. Analysis of the single-molecule localization data revealed the shapes of PopZ nanodomains as well as the fact that these nanodomains have a uniform volume density, which suggests that this structural protein has a conserved architectural property (as shown in part b). The image in part d is reproduced from REF. 51. The image in part e is reproduced, with permission, from REF. 64 © (2013) American Chemical Society Publications.
MreB
Rod-shaped bacteria maintain their shape by tightly regulating the insertion of new peptidoglycan units into their rigid cell wall; however, the mechanism by which an organized insertion pattern is achieved remains unclear. Rod-shaped cells develop abnormal lemon-shaped morphologies if they are depleted of MreB25,26 or if they are exposed to the MreB polymerization inhibitor A22 for long periods of time (for example, 10 hours)27–30. These observations have led to the suggestion that MreB is involved in directing the insertion of new peptidoglycan units into the cell wall and thereby maintaining the rod shape21,22,31–33.
Traditional epifluorescence microscopy in rod-shaped bacteria has shown that fluorescent MreB fusion proteins have a cell-spanning helical ultrastructure21,25,27,28 (FIG. 2a). Many other fluorescent fusion proteins (for example, the peptidoglycan synthetic enzyme penicillin- binding protein 2 (REFS 25,28,34,35)) also localize in a helical arrangement. Thus, a widely accepted model was proposed, which posited that the MreB helix directs the pattern of rod-shaped cell wall growth by recruiting essential components of the cell wall synthetic machinery31,36. However, knowledge of the mechanistic details of MreB function has advanced considerably in recent years, owing to results that were obtained using a combination of single-molecule methods, single-particle tracking (see centroid estimation) and cryo-electron tomography.
The first single-molecule tracking study of MreB used amino-terminal enhanced yellow fluorescent protein (eYFP)–MreB fusion proteins that were expressed at extremely low levels from a xylose-inducible promoter in C. crescentus37. The low concentration of eYFP–MreB made it possible to track the motion of single molecules along the cell membrane. Analysis of the tracks showed that eYFP–MreB is present in two populations: a fast-diffusing population of monomers and a second population of polymerized MreB that exhibited slow and directed motion. The slow-moving molecules travelled at 6 nm per second, and the tracks had an average length of 332 nm and were orientated in a roughly perpendicular arrangement relative to the long axis of the cell. These data suggest that MreB monomers might treadmill through short, stationary MreB filaments by polymerizing at one end of the filament and depolymerizing at the opposite end (FIG. 2b).
The tracking experiments were complemented by a single-molecule super-resolution imaging study in C. crescentus that exploited the fact that, using long frame acquisition times of 100 ms, slowly diffusing eYFP–MreB molecules could be preferentially distinguished from the background of faster moving free MreB monomers38. Consistent with results from diffraction-limited imaging21,25,27,28, the super-resolution reconstructions from the fitted single-molecule positions revealed an approximately helical ultrastructure that spanned the entire cell (FIG. 2a).
The question of why the helical pitch that was observed in the super-resolution images did not exactly match the direction of the single-molecule tracks prompted further research. Experimental evidence from single-particle tracking of fluorescent foci using total internal reflection fluorescence microscopy (TIRF microscopy)39–41 and images from cryo-electron tomography42,43 challenged the helical structure model of MreB. The foci of N-terminal GFP–MreB fusion constructs in B. subtilis moved in directions that were perpendicular to the long axis of the cell39–41 (FIG. 2c), which is similar to the eYFP–MreB single-molecule motions that were observed in C. crescentus37. However, the velocities that were observed in B. subtilis were faster than those detected in C. crescentus and ranged from 20 nm to 60 nm per second.
A third single-particle tracking study41 used GFP fused to an internal MreB site, which was independently shown to be a better functional complement than the previously used N-terminal fusion constructs44,45. When this internal fusion construct was expressed in E. coli, the fluorescent foci moved at 6.7 nm per second, and the orientations of the trajectories showed less than 3-degree deviation from the perpendicular angles relative to the long axis of the cell. Importantly, in all three single- particle tracking studies39–41, MreB movement was shown to be driven by the process of peptidoglycan synthesis itself, and the motion of the individual fluorescent MreB foci was observed to be uncorrelated and bidirectional. This has led to the suggestion of an alternative model, in which MreB is proposed to form short, actin-like filaments that move as independent units in directions that are perpendicular to the long axis of the cell39–41 (FIG. 2b).
Interestingly, in cryo-electron tomograms of unlabelled wild-type cells, a cell-spanning MreB helix is not observed42; however, when the N-terminal eYFP–MreB fusion construct is expressed in E. coli, the cryo-electron tomograms clearly show a helical cell-spanning MreB filament43. Together, these results show that, in the case of MreB, the observed localization patterns are highly dependent on the placement and possibly the identity of the fluorescent protein tag. This highlights the need for robust control experiments to reduce artefacts due to labelling as well as the need for systematic variation in the method of attachment of the fluorescent protein to the target protein to minimize functional perturbation.
At present, the mechanistic details of MreB function at the molecular level still remain unclear. As the role of MreB in the maintenance of a rod shape is undisputed and as multiple copies of MreB molecules are observed travelling together as a unit, it is now hypothesized that short polymeric filaments of MreB fulfill a scaffolding function to organize the peptidoglycan synthetic complex17,46. However, the architecture of such a complex, as well as the existence of filaments that would be too small or too close to the cell membrane to be observed in cryo-electron tomography, has yet to be experimentally verified.
FtsZ
FtsZ is a highly conserved bacterial homologue of tubulin that is essential for bacterial cell division47,48. After binding to GTP, FtsZ monomers polymerize to form a ring-like structure (which is called the Z ring) at the site of the division septum. The Z ring recruits components of the cell division machinery and also provides the constrictive forces that are necessary for cell division49. Cryo-electron tomograms of C. crescentus have shown that FtsZ monomers polymerize into short protofilaments that are 40–160 nm in length and 4–5 nm wide50 (FIG. 2b). These protofilaments localize to the site of constriction immediately before cell division, and there is no discernible coordination between the filaments that constitute the Z ring.
A super-resolution imaging study showed that, in non-constricting E. coli cells, FtsZ assembles into more extended helical conformations of variable lengths and pitch (FIG. 2c,d) in addition to the closed ring structure that is formed in constricting cells51. Owing to the high spatial resolution that was attained, these images confirmed earlier indications from diffraction-limited imaging that FtsZ has a helical structure in E. coli, B. subtilis and C. crescentus52–55. Consistent with the observed overlapping protofilament arrangement in the cryo-electron tomograms of C. crescentus50, the apparent 110 nm thickness of the condensed Z ring in the super-resolution fluorescence images was interpreted to arise from a loose bundle of overlapping FtsZ protofilaments that were not individually resolved owing to the estimated image resolution of 35 nm.
The carboxy-terminal FtsZ–mEos2 (mEosFP variant) fusion construct that was used in this study51 could not be expressed as a genomic replacement of wild-type FtsZ, probably because the bulky fluorescent protein label interfered with binding to membrane-anchoring proteins. However, the protofilaments of a C-terminal FtsZ–YFP–membrane-targeting sequence fusion protein that were reconstituted into liposomes had a morphology that was indistinguishable from that of wild-type FtsZ proto-filaments and were able to constrict the liposomes in the presence of GTP49. Therefore, the authors concluded that, although FtsZ–mEos2 was incapable of binding to the membrane, it still provided a reliable label of the native FtsZ structure when it was expressed in addition to wild-type FtsZ, as tagging of the C terminus did not interfere with the polymerization and constriction activity of FtsZ.
Single-molecule imaging and tracking has also made it possible to quantify the highly dynamic nature of polymeric FtsZ protofilaments. It was already known from fluorescence recovery after photobleaching (FRAP) experiments that the fluorescence signal from the Z-ring structure is recovered with a half-time of 8–9 seconds56. A single-molecule tracking study in E. coli, using an FtsZ–Dendra2 fusion construct, identified two distinct populations of FtsZ8: a group of mostly stationary molecules that were localized close to the centre of the cell, belonging to the Z ring ultrastructure; and a second population that exhibited faster motion and diffused throughout the cell at speeds that were similar to those of monomeric and/or polymeric entities moving on the cell membrane (diffusion coefficient (D) ≈ 0.1 μm2 per second). Intriguingly, diffusion of the second population seemed to be spatially restricted to an approximately helical pattern8, which is consistent with the helical patterns that have been observed in traditional epifluorescence images52–55 but further illustrates that these helical patterns are not part of a static structure.
Another super-resolution imaging study characterized the localization and polymerization–depolymerization dynamics of FtsZ in C. crescentus57. Using short exposure times of 15 ms, FtsZ–Dendra2 was only detectable when it was incorporated into a compact Z ring near mid-cell and became or remained undetectable when it rapidly diffused as a monomer. The average visibility times of polymerized FtsZ–Dendra2 molecules were 58 ms in live cells and 128 ms in fixed cells. In fixed cells, the visibility time is limited only by the time it takes for a single Dendra2 molecule to photobleach; however, in live cells, the visibility time also incorporates the time it takes for a single molecule to be released from the proto-filament. Thus, the reduction in visibility time (by a factor of approximately two) in live cells shows that single FtsZ molecules depolymerize at a rate that is about two orders of magnitude faster than the turnover half-time of all the protofilaments in the Z ring (8–9 seconds, as measured in E. coli and B. subtilis56). This suggests that individual FtsZ molecules can associate and dissociate with the Z ring on a subsecond timescale, whereas complete exchange with the cytoplasmic population occurs on a slower timescale of seconds.
The utilization of 3D wide-field super-resolution imaging using an astigmatic point spread function (astigmatic PSF) further showed that the Z rings in C. crescentus span the cell diameter and have a considerable thickness (of 70–100 nm), such that central openings with diameters of only ~150–300 nm are present57 (FIG. 2b). These results are consistent with the model of a loose bundle of overlapping protofilaments that extends into the cytoplasm51, but this model seems to be at odds with the close membrane association of the FtsZ protofilaments that was observed using cryo-electron tomography in C. crescentus50. To be detectable in cryo-electron tomography, the proteins must form a relatively coherent long-range structure (such as a protofilament); however, FtsZ is a highly dynamic protein, and the single-molecule fluorescence method may be detecting those FtsZ molecules that are in the additional locations in the cell interior, where they can transiently localize within the ~15 ms frame exposure times.
Although FtsZ is a highly conserved cell division protein, there is a high degree of variation in the proteins that interact with FtsZ in different bacterial species48. This may explain the apparent differences in single-molecule localization patterns and dynamics that have been observed thus far, and it also highlights the importance of carrying out live-cell measurements in different species. Other super-resolution imaging techniques that do not rely on single-molecule detection (BOX 1) have also been applied to study FtsZ58–60. As more high-resolution data on FtsZ and its interacting proteins become available, it will be interesting to see whether it is possible to obtain a general model of FtsZ architecture and function.
PopZ
PopZ is conserved in Alphaproteobacteria and forms dense clusters at the cell poles61–63 (FIG. 2b). In C. crescentus, PopZ has polar organizing and anchoring functions, and recruits several proteins to the cell pole, including proteins that are essential for faithful chromosome segregation and partitioning in replicating cells. Electron micrographs of PopZ filaments have shown that they form a connected polymeric network in vitro61. Following the observation of a ribosome exclusion zone at the cell pole of intact cells62,63, it was proposed that PopZ forms a similar network in vivo that prevents large macromolecules from occupying the polar regions (FIG. 2b).
Super-resolution 3D imaging with a double-helix point spread function (PSF) using a photoactivatable mCherry1 (PAmCherry1)–PopZ fusion construct showed that PopZ does indeed form nanodomains at the cell poles, which occupy the entire polar space64 (FIG. 2e). Importantly, although the size and volume of the PopZ nanodomains (as well as the number of localized single molecules within each nanodomain) varied significantly at the population level, the density of PopZ proteins was uniform in the nanodomains. This property was repeatedly observed for dozens of cells in different fields-of-view, which indicates that it is an invariant structural feature of PopZ. Thus, these data further support the hypothesis that PopZ forms an architecturally conserved 3D polymeric network at the C. crescentus cell poles. We speculate that these PopZ ultrastructures, which are dense enough to exclude large particles (such as ribosomes or polymeric biomolecules), might form a specialized sub-cellular scaffold or environment for specific biochemical binding events and reactions. Further experiments that probe the precise role of PopZ in the organization and regulation of other proteins at the cell poles are required to test this hypothesis.
In summary, single-molecule imaging studies have revealed features of the cytoskeletal network that are not accessible using other technologies, including the time-scale of movement of MreB and FtsZ and the uniform density of the PopZ polymeric network. These studies also demonstrate the potential for sample-induced artefacts to be introduced in current fluorescent-protein labelling methods (as shown by the MreB studies42,43), which supports the need for the development of improved labelling approaches.
Nucleoid organization and partitioning
Understanding the organization, processing and maintenance of the bacterial nucleoid during the cell cycle is another area that has progressed owing to single-molecule imaging in live cells. As has already been shown using diffraction-limited imaging of stained DNA, the shapes of the nucleoid boundaries differ considerably between C. crescentus and E. coli65,66 (FIG. 3a,b). Super-resolution imaging of individual nucleoid-associated proteins (NAPs) is one approach that has been used to probe nucleoid organization on a scale that is otherwise obfuscated by the diffraction limit of conventional fluorescence imaging. Insights can be gained by measuring the spatial distribution of isolated single proteins and by tracking their motion.
Figure 3. Nucleoid organization in model organisms observed by single-molecule methods.

a | In Caulobacter crescentus, the nucleoid fills most of the volume of the cell at all stages of the cell cycle66. The spatial distribution of the DNA-binding protein HU2 shows little clustering in swarmer cells and stalked cells, whereas considerable clustering is observed in pre-divisional cells, which is suggestive of local chromosome compaction (left)69. b | In Escherichia coli, the nucleoid is divided into distinct volumes depending on the stage of the cell cycle88. Surprisingly, the nucleoid-associated protein histone-like nucleoid-structuring protein (H-NS) forms two, three or four smaller clusters per cell depending on the length of the cell (and, correspondingly, the stage of the cell cycle), which is thought to spatially sequester regulated operons71. c | MukB is a large complex that has two heads connected by coiled-coil regions (green helices) that are assembled by a dimerization domain (indicated as a MukB hinge). In the presence of bound ATP, the two head domains of MukB are engaged, forming a closed structure that loops DNA. On ATP hydrolysis, the head domains disengage and the structure opens, which enables DNA to be captured or released. d | In E. coli, the chromosome-partitioning protein MukB either rapidly diffuses as independent molecules or forms stationary clusters when incorporated into the MukBEF structural maintenance of chromosomes (SMC) complex73. A minimal functional MukB/E/F complex that has 4/4/2 subunit stoichiometry is observed in the stationary clusters. The super-resolution image in part a (right panel) is reproduced, with permission, from REF. 69 © (2011) Elsevier. The image in part b (right panel) is reproduced, with permission, from REF. 71 © (2011) American Association for the Advancement of Science. The images in part d are reproduced, with permission, from REF. 73 © (2012) American Association for the Advancement of Science.
Structural organization of the chromosome
Imaging of NAPs provides information about chromosome organization, as these proteins bind to DNA and have organizational and regulatory roles. The spatial distribution of DNA-binding protein HU, which is the most abundant NAP67,68, was investigated in fixed C. crescentus cells using photoinduced blinking of the HU2–eYFP fusion protein69. HU is typically present as a heterodimer, consisting of HU1 and HU2 in C. crescentus (or HU1α and HUβ in E. coli), that nonspecifically binds to both double-stranded and single-stranded DNA67,68. Access to high-resolution positional information of more than 1,000 single HU2–eYFP molecules per cell enabled a quantitative assessment of whether HU2 is uniformly distributed throughout the nucleoid or whether local clusters are formed, using spatial point statistical methods70 (FIG. 3a). In the case of swarmer cells and stalked cells (which are two different developmental stages in the C. crescentus cell cycle), only a very slight degree of HU2 clustering was observed, whereas pre-divisional cells exhibited considerable HU2 clustering. Because the interaction between HU and DNA is nonspecific, this clustering suggests that chromosome reorganization and compaction occur before cell division.
In a second study in live E. coli cells, 3D super-resolution images of five mEos2-labelled NAPs were obtained71. Four of these proteins — HU, Fis (factor-for-inversion stimulation), IHF (integration host factor) and StpA (stalk protein A) — were randomly positioned throughout the nucleoid. However, the transcriptional silencer histone-like nucleoid-structuring protein (H-NS) formed a pair of compact clusters on each chromosome; thus, depending on cell length, two, three or four clusters were observed (FIG. 3b). A mutant variant of H-NS that contained a point mutation in the N-terminal domain failed to induce clustering, which indicates that the N-terminal domain is required for cluster formation in vivo. A point mutation in the C-terminal domain, which is responsible for DNA binding, resulted in a ~20-fold decrease in the number of single molecules per cluster, which indicates that there is a strong association between DNA and individual H-NS proteins within the clusters. In addition, colocalization imaging of H-NS and tetracycline repressor protein (TetR)–eYFP-labelled gene loci and a chromosome conformation capture (3C) assay72 were used to investigate the pairwise proximity with nine H-NS-regulated genes. Both approaches showed that H-NS clusters tend to bring regulated operons into close spatial proximity and thereby sequester those regions of the chromosome that are not undergoing active transcription and translation to a common location in the nucleoid.
Another NAP that is involved in the structural maintenance of chromosomes (SMC), is the MukBEF complex in E. coli73 and B. subtilis74. MukBEF is responsible for chromosome condensation and chromosome partitioning by ‘grabbing’ and bringing together DNA loop regions75–77 (FIG. 3C). Similarly to H-NS, single-molecule imaging has shown that most MukB monomers rapidly diffuse throughout the E. coli cell, whereas ~20% of the MukB population is in a stationary fraction of MukB dimers within MukBEF complexes that form several punctuate clusters in the nucleoids. In B. subtilis, this was shown to be mediated by recruitment of MukBEF by the stably positioned segregation and condensation protein (Scp) complex ScpAB74, which together form the previously identified condensation centres78–80. Previous biochemical studies reported MukB/MukE/MukF subunit stoichiometries of 2/4/2 or 2/2/1 for ATP-unbound and ATP-bound states, respectively. By comparing the initial fluorescence intensity of the clusters with the intensity decrease of a digital single-molecule photobleaching step5,81 in multiple E. coli cells (see also Transcription and translation section below), the minimal functional unit in live cells was found to be an ATP-bound dimeric complex that had a MukB/MukE/MukF stoichiometry of 4/4/2. Furthermore, on the basis of the pre-bleach brightness of the clusters, it was estimated that each cluster contained about eight to ten of these dimeric complexes. By varying the imaging speed (BOX 1), freely diffusing MukB molecules could be distinguished from immobile MukB molecules in the clusters (FIG. 3c). On the basis of reaction diffusion modelling, the dwell time of MukBEF in the clusters was ~50 seconds. This relatively slow turnover indicates that almost simultaneous hydrolysis of four ATP molecules is needed for the complete release of a minimal functional 4/4/2 unit to occur. These 4/4/2 dimeric complexes could therefore continually try to grab and release additional DNA loops without losing tight association with the already condensed portions of the chromosome (FIG. 3d).
Chromosome integrity and partitioning
In addition to structural organization of the chromosome, the integrity of double-stranded DNA must be maintained to avoid the accumulation of DNA damage. The process of base excision repair (BER) by DNA polymerase I and DNA ligase has been investigated in live E. coli cells82. On binding to DNA, both of these enzymes show a distinct change in their diffusive behaviour. Thus, by using a combination of single-molecule tracking (using short 15 ms exposure times) and single-molecule imaging (using long 500–1000 ms exposure times), it was possible to determine the repair rates of these enzymes in vivo (FIG. 4a). Under physiological conditions, DNA polymerase I and DNA ligase molecules require just ~2 seconds to carry out a repair event and spend ~80 seconds and ~60 seconds diffusing along the DNA in search of additional substrates, respectively. Consequently, the fraction of bound DNA polymerase I and DNA ligase molecules in a given cell is very small (<5%). Under conditions of increased DNA damage, the single-molecule repair rates remain unchanged, whereas the fraction of bound molecules increases and eventually saturates at <20%. These data indicate that the activity of E. coli DNA polymerase I and DNA ligase is limited by substrate availability owing to upstream rate-limiting steps in the BER pathway. Further experiments are now needed to characterize the entire BER pathway in order to identify these upstream reactions.
Figure 4. Chromosome integrity and partitioning observed by single-molecule approaches.
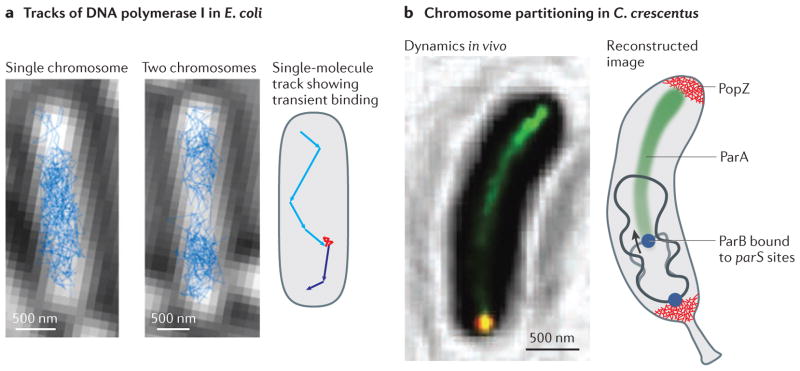
a | Most DNA polymerase I (and DNA ligase) molecules rapidly diffuse within the Escherichia coli nucleoid (left and middle panels) 82. After binding to stationary DNA during base excision repair events, a clear change in motion is observed. Shown is a schematic single-molecule track (as observed for both enzymes) that features a transient binding event (red) that is flanked by periods of fast motion (cyan and blue). b | DNA-bound ParA molecules (green) localize along a narrow structure that runs along the centre of Caulobacter crescentus cells while the newly replicated ParB–parS centromeric complex (blue, in the right panel), which is located close to the origin of replication, follows the receding end of this structure to move towards the opposite cell pole during chromosome segregation83. The cell poles are populated by PopZ molecules that form uniform polymeric networks. The single-molecule tracking images in part a are reproduced, with permission, from REF. 82 © (2013) Proceedings of the National Academy of Sciences USA (PNAS). The super-resolution image in part b is reproduced, with permission, from REF. 83 © (2010) Macmillan Publishers Ltd. All rights reserved.
During cell division, a considerable reorganization of the nucleoid occurs to ensure that duplicated chromosomes are partitioned into the daughter cells. This process has been studied using super-resolution imaging in C. crescentus83 (FIG. 4b). The key proteins that are involved are components of the chromosome-partitioning ParABS system. ParB binds to parS sites, which are located in close proximity to the origin of replication on the chromosome, and it spreads along neighbouring DNA, thus forming a centromere-like ParB–parS complex. ParA is a Walker-type ATPase that can polymerize into filaments in vitro and interacts with ParB. By localizing ParA (labelled with eYFP) in living cells, high-resolution insights into chromosome segregation were obtained. ParA–eYFP, which was probably bound to the nucleoid, localized in a narrow, linear structure running along the central axis of the cell (FIG. 4b). Furthermore, super-resolution and traditional epifluorescence images at different stages of the cell cycle showed that, after duplication of the chromosomal origin at the cell pole, the new copy of the mCherry-labelled ParB–parS centromere follows the receding end of the narrow ParA structure until it ultimately arrives at the opposite cell pole. The original ParB–parS centromere remains at the old pole, which ensures correct partitioning and positioning of the replicated chromosomes in each daughter cell. These data indicate that ParA and ParB are in close spatial proximity and that ParB destabilizes DNA-bound ParA complexes. This has led to the suggestion that ParB-stimulated ParA depolymerization activity is responsible for moving the centromere to the opposite cell pole.
Transcription and translation
Coupling of transcription and translation
Central to the processes of transcription and translation are questions about where they occur within bacterial cells and how they are regulated. Given that the nucleoids of E. coli and B. subtilis do not occupy the entire cell volume, a more general question arises: to what extent are the transcription and translation machineries physically coupled owing to the simultaneous synthesis of mRNA and polypeptide chains? Early electron microscopy of E. coli extracts showed that some RNA transcripts are anchored to DNA via RNA polymerase (RNAP) while also being translated by multiple ribosomes84. By contrast, electron micrographs of E. coli have shown that ribosomes are predominantly localized to the cell poles and to the lateral periphery of the nucleoid, which suggests that transcription and translation are spatially segregated85. On the basis of the possible coupling between transcription and translation, several mechanistic hypotheses have been proposed; for example, for membrane proteins, it has been suggested that transcription–translation coupling, and concurrent insertion of the encoded product into the membrane (known as transertion) might function as a mechanism to anchor DNA segments to the cell envelope86,87. Single-molecule experiments in live cells are well suited to test the extent and the validity of specific mechanisms, such as transertion, and several studies to address the differing hypotheses have now begun to emerge.
To assess transcription–translation coupling, the time-averaged spatial distribution and trajectories of individual ribosomes and RNAP were measured in live E. coli cells using C-terminal eYFP fusions to the 30S ribosomal protein S2 and yGFP fusions to the β-subunit of RNAP, respectively, in two different E. coli strains88. The super-resolution images (FIG. 5a) showed that ~15% of ribosomes were located within the nucleoids, whereas the larger remaining fraction populated the periphery around the nucleoids and the cell poles. In an independent study, similar ribosome distributions were found by labelling the 30S ribosomal subunit protein S22 with mEos2 (REF. 71). By contrast, RNAP was found primarily in the nucleoid; thus, these findings quantitatively show that the spatial distributions of ribosomes and RNAPs are mostly anti-correlated in E. coli.
Figure 5. Transcription and translation are spatially uncoupled in Escherichia coli.

a | Most RNA polymerase (RNAP) molecules localize within the nucleoid, whereas ribosomes cluster around the nucleoid periphery and at the cell poles (spatial distributions of RNAP and ribosomes in the lower two panels are shown in yellow)88. b | The motion tracks of ribosomes show clear avoidance of the nucleoid region88. c | Binding events of single Venus-labelled LacI molecules can be detected after rapid removal of the inducer from the cell medium (right panel). At long exposure times, dissociated molecules of LacI–Venus only contribute to the background autofluorescence, whereas the emergence of bright diffraction-limited single-molecule images over time is indicative of transcription factors binding to their operators on stationary DNA (right)93. d | According to the facilitated diffusion model, individual transcription factors search for their target DNA sequence using a combination of sliding on DNA strands (which is known as one-dimensional (1D) diffusion) and hopping between strands through the cytoplasm (which is known as three-dimensional (3D) diffusion)141. Images in part a and b are reproduced, with permission, from REF. 88 © (2012) John Wiley and Sons. Part d is modified, with permission, from REF. 141 © (2009) Elsevier.
The apparent diffusion coefficient of the labelled S2–eYFP construct (Dapp ≈ 0.04 μm2 per second) was consistent with computationally modelled free, but confined, diffusion rates of fully assembled 70S monomers and polysomes in the ribosome-rich regions of the cell (FIG. 5b). These results suggest that the vast majority of ribosomes (at least 85%) are not coupled to transcription in E. coli. Of the remaining ~15% of ribosomes, only the ribosomes that are near the lateral cell membrane could potentially be involved in transertion of membrane proteins, because transertion is unlikely to occur at the cell poles owing to the long distances between DNA and polar cell membranes (300–500 nm).
Intriguingly, in C. crescentus, the available data from diffraction-limited imaging seem to support the possibility that transcription and translation are strongly coupled, as ribosomes and DNA seem to be homogeneously mixed rather than anti-correlated66. Furthermore, data from fluorescence in situ hybridization (FISH) — in which a large probe that consists of 120 tandem lac operator sequences is used to assess mRNA dispersion — together with FRAP experiments to probe ribosome mobility, seem to suggest that mRNA and the majority of ribosomes do not freely diffuse. The FISH-labelled mRNA construct was found to remain close to the DNA locus from which it originated. By contrast, the existence of a translation-independent transport mechanism for mRNA has recently been demonstrated in E. coli, in which labelled transcripts quickly reached different subcellular regions (possibly by active transport) in a sequence-dependent manner89. The underlying reasons for these apparent differences between C. crescentus and E. coli are currently unknown, and the identification of possible mRNA transport mechanisms awaits further experimental characterization.
Transcription factor dynamics
Single-molecule techniques have also been applied to the investigation of transcription factor-mediated regulation of gene expression. One of the earliest experiments quantified gene expression by using the appearance and subsequent photobleaching of a localized fluorescence signal (known as detection by localization) to count the number of single eYFP-labelled membrane proteins that were controlled by the lac repressor90. A related method, in which total fluorescence from many fluorescent labels is normalized to the fluorescence intensity of a single molecule, has also been used to estimate the copy number of proteins5,81. This method has been used to determine the permease expression threshold that is required to cause E. coli to switch from one phenotype to another91 and to quantify parts of the proteome and transcriptome in the same organism92.
Detection by localization has been used to determine the rate of transcription factor binding events at the single-molecule level (FIG. 5c). This is possible because transcriptional regulation relies on dynamic interactions between individual transcription factor molecules and the mostly stationary polymeric DNA structure. In combination with strategic manipulation of the DNA sequence in the vicinity of the operator sequence (which is the site to which transcription factors bind), these experiments have revealed the mechanism by which a single transcription factor searches for its operator sequence. The search process of LacI for the lac operator in live E. coli cells was characterized by monitoring the diffusive behaviour of individual LacI molecules that were labelled with the YFP variant Venus93. To achieve the low expression of labelled LacI (three LacI molecules per cell) that is necessary for unambiguous single- molecule detection, the expression of LacI was repressed to below wild-type levels (~20 LacI molecules per cell). In the first set of experiments, the exposure time was set to 1,000 ms to minimize the detection of nonspecifi-cally bound, and thus rapidly diffusing, LacI molecules. LacI molecules that were bound to operator sequences produced localized diffraction-limited single-molecule images (FIG. 5c). Measurement of the binding kinetics at the population level in response to chemical treatment revealed that a single LacI molecule requires a search time of less than ~350 seconds to locate its specific operator sequence.
In a second set of experiments, which used stroboscopic laser illumination with 10 ms laser pulses (BOX 1), the diffusive behaviour of nonspecifically bound LacI molecules was characterized. The apparent diffusion coefficient (Dapp= 0.4 ± 0.02 μm2 per second), which was obtained by pooling all of the observed single-molecule tracks that were measured for the entire population of cells, was found to contain contributions from LacI dimers that were sliding along DNA and free LacI proteins that were diffusing in the cytoplasm. By measuring the diffusion constants of both of these populations in separate experiments, the LacI dimer was found to spend ~87% of the time nonspecifically bound to, and diffusing along, DNA. These in vivo results provide strong and quantitative support to the long-standing hypothesis that individual transcription factors search for their target location by a combination of one-dimensional diffusion along DNA segments and 3D diffusion between DNA segments (known as the facilitated diffusion model)94. A follow-up study revealed that LacI repeatedly slides over a distance of ~45 bp when it is nonspecifically bound to DNA95. The facilitated diffusion model was thus refined to include the possibility that a single transcription factor can slide over its specific operator sequence several times before binding (FIG. 5d). These findings may reflect the conflicting challenge of transcription factor function, which results from a compromise between the ability to rapidly search nonspecific DNA segments and the ability to rapidly bind to specific target sites.
Summary and outlook
The examples that are described in this Review illustrate that single-molecule tracking and super-resolution imaging in bacteria are useful tools for determining biologically relevant information at unprecedented spatial scales and with unsurpassed sensitivity and specificity. In addition, optical fluorescence imaging provides this information in a relatively non-invasive manner that is compatible with live-cell imaging and thus serves as a critical complement to in vitro experiments. These advantages undoubtedly contribute to the wide applicability of these methods to the study of structural and cytoskeletal proteins, the organization of the nucleoid and the segregation and partitioning of the chromosome, as well as the mechanistic details of transcription and translation. The single-molecule imaging techniques that are described in this Review are most beneficial in circumstances in which the problem that is being studied involves spatial dimensions or distance scales of 200–300 nm. By contrast, single-molecule tracking methods provide information about the precise motion of individual bacterial proteins and therefore are more useful for investigating binding interactions and protein dynamics.
As the examples described in this Review illustrate, access to high spatial resolution brings some of the general challenges for fluorescence microscopy to the fore-front. Fluorescent fusion proteins are by far the preferred labelling technique despite their apparent shortcomings. Among these shortcomings, the possible structural perturbation of native protein function is perhaps the most challenging to solve and, not surprisingly, seems to be more of an issue for protein structures that rely on close intermolecular packing. Novel bio-orthogonal strategies for in vivo fluorescent labelling using smaller and brighter fluorescent probes are therefore urgently needed to improve not only the achievable resolution but also the faithfulness of the obtained images95,96. Several methods that provide high specificity and quantitative labelling capabilities continue to be refined and should eventually provide a means to avoid the steric hindrance effects of bulky fluorescent proteins. These include the introduction of single unnatural amino acids in conjunction with fast and efficient labelling reactions97–102, probe incorporation mediated by enzymes (PRIME)103–106 or fluorescent -amino acid substrates107,108 and fluorogen-activating peptides109,110. Besides the labelling issues, a further challenge is that most of the exogenous fluorescent dyes that are typically used for super-resolution imaging cannot permeate live cells and can only be used for intracellular labelling of fixed and permeabilized cells111. Consequently, exogenous labelling methods have not been used for single-molecule experiments in live bacterial cells, with the notable exception of live-cell-permeable azido 2-dicyanomethylene-3-cyano-2,5-dihydrofuran (DCDHF) dyes112.
As these two primary challenges are being overcome and the technology begins to mature, single-molecule tracking and super-resolution imaging are likely to become indispensable tools for bacterial cell biology. Particularly when combined with strategic manipulation of the organism and/or its environment, as well as with complementary information from in vitro biochemical assays, the ability to explore bacterial cells at high resolution ‘opens the door’ to quantitative imaging-based research.
Acknowledgments
The authors gratefully acknowledge fruitful collaborations and stimulating discussions with L. Shapiro and members of her laboratory over the years, as well as many current and former members of the Moerner laboratory. The authors specifically thank M. K. Lee, A. R. von Diezmann and J. L. Ptacin for critical reading of the manuscript. This work was supported in part by the US National Institute of General Medical Sciences Grant No. R01GM086196 (W.E.M) and a Swiss National Science Foundation Postdoctoral Fellowship PA00P2_145310 (A.G.).
Glossary
- Centroid estimation
In the context of localization microscopy, a method to determine the location of a fluorescent emitter or a group of several closely packed emitters (referred to as a single-particle) by calculating the spatial arithmetic mean of all pixel positions, which are weighted by their intensity
- Photoactivation
The process by which fluorophores that are initially in a non-fluorescent (dark) state can be converted to a fluorescent (bright) state by illumination with short-wavelength light
- Photoswitching
The process by which photoswitchable fluorophores can be turned ‘on’ or ‘off’ by an active control mechanism, such as illumination with a specific wavelength of light
- Photoinduced blinking
Using certain illumination intensities (or, in some cases, by adding chemical additives), fluorophores can reversibly enter a non-fluorescent (dark) state. From this state, they can spontaneously recover and become fluorescent (bright) again, which gives the appearance of blinking
- Epifluorescence microscopy
A standard wide-field fluorescence microscopy technique, in which the same objective lens is used to illuminate the entire specimen and to collect emitted fluorescence
- Cryo-electron tomography
(CET). A technique in transmission electron microscopy, in which a vitrified specimen is imaged from different angles at cryogenic temperatures. From the resulting electron micrograph tilt series, a three- dimensional tomogram can be computationally reconstructed
- Total internal reflection fluorescence microscopy
(TIRF microscopy). A technique in which only fluorophores that are in close proximity to the glass–water interface are excited by an evanescent wave that is generated by total internal reflection of the excitation light at this interface. The large reduction of the excitation volume in the axial direction (from ~700 nm to ~100 nm) results in more selective excitation of the sample and lower background fluorescence compared with epifluorescence illumination
- Fluorescence recovery after photobleaching
(FRAP). An optical technique that is used to estimate the diffusion of fluorescently labelled molecules by determining the timescale of fluorescence recovery after high-intensity light has been applied to a well-defined region of the specimen to photobleach many of the fluorophores in its footprint
- Astigmatic point spread function
(Astigmatic PSF). A cylindrical lens can be inserted in the fluorescence collection path to offset the x and y focus position along the optical axis. An imaging system that has an astigmatic PSF can be used to determine the z-position of a single-molecule emitter by calibrating the change in elliptical shape of the PSF as a function of defocus
- Double-helix point spread function
(Double-helix PSF). Optical phase manipulation in the Fourier plane of the fluorescence emission path can be used to produce a double-helix PSF. An imaging system that has a double-helix PSF can be used to determine the z-position of a single-molecule emitter by calibrating the amount of angular rotation of the PSF as a function of defocus
- Chromosome conformation capture
(3C). A molecular biology technique that is based on crosslinking and analysis of which DNA segments are closely associated, to determine the spatial organization of the chromosome
- Fluorescence in situ hybridization
(FISH). An optical technique that is used to detect and locate specific DNA (or RNA) sequences. A FISH probe, which consists of a fluorophore linked to a single-stranded DNA (or RNA) sequence, binds to its complementary target DNA (or RNA) site after being introduced into fixed and permeabilized cells
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Moerner WE, Kador L. Optical detection and spectroscopy of single molecules in a solid. Phys Rev Lett. 1989;62:2535–2538. doi: 10.1103/PhysRevLett.62.2535. [DOI] [PubMed] [Google Scholar]
- 2.Orrit M, Bernard J. Single pentacene molecules detected by fluorescence excitation in a p-terphenyl crystal. Phys Rev Lett. 1990;65:2716–2719. doi: 10.1103/PhysRevLett.65.2716. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose WP, Moerner WE. Fluorescence spectroscopy and spectral diffusion of single impurity molecules in a crystal. Nature. 1991;349:225–227. [Google Scholar]
- 4.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. On/off blinking and switching behavior of single molecules of green fluorescent protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 5.Deich J, Judd EM, McAdams HH, Moerner WE. Visualization of the movement of single histidine kinase molecules in live Caulobacter cells. Proc Natl Acad Sci USA. 2004;101:15921–15926. doi: 10.1073/pnas.0404200101. This paper provides the first evidence that single copies of fluorescent protein fusions can be imaged and analysed in bacteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapiro L, McAdams H, Losick R. Generating and exploiting polarity in bacteria. Science. 2002;298:1942–1946. doi: 10.1126/science.1072163. [DOI] [PubMed] [Google Scholar]
- 7.Shapiro L, McAdams HH, Losick R. Why and how bacteria localize proteins. Science. 2009;326:1225–1228. doi: 10.1126/science.1175685. This review describes the central role that protein localization patterns have in bacterial cell biology, which should be investigated using advanced imaging methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niu L, Yu P. Investigating intracellular dynamics of FtsZ cytoskeleton with photoactivation single-molecule tracking. Biophys J. 2008;95:2009–2016. doi: 10.1529/biophysj.108.128751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manley S, et al. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nature Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 10.Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 11.Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nature Methods. 2006;3:793–796. doi: 10.1038/nmeth929. References 10, 11, and 12 are the first reports of super-resolution imaging by single-molecule localization and active control of emitter concentrations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie XS, Choi PJ, Li GW, Lee NK, Lia G. Single-molecule approach to molecular biology in living bacterial cells. Annu Rev Biophys. 2008;37:417–444. doi: 10.1146/annurev.biophys.37.092607.174640. This review covers bacterial single-molecule studies up to 2008. [DOI] [PubMed] [Google Scholar]
- 14.Reyes-Lamothe R, Sherratt DJ, Leake MC. Stoichiometry and architecture of active, DNA replication machinery in Escherichia coli. Science. 2010;328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lia G, Michel B, Allemand J. Polymerase exchange during Okazaki fragment synthesis observed in living cells. Science. 2012;335:328–331. doi: 10.1126/science.1210400. [DOI] [PubMed] [Google Scholar]
- 16.Robinson A, van Oijen AM. Bacterial replication, transcription and translation: mechanistic insights from single-molecule biochemical studies. Nature Rev Microbiol. 2013;11:303–315. doi: 10.1038/nrmicro2994. [DOI] [PubMed] [Google Scholar]
- 17.Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nature Rev Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. This review describes the dynamics of MreB and FtsZ and their role in cell wall synthesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams DW, Errington J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nature Rev Microbiol. 2009;7:642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- 19.Gerdes K, Howard M, Szardenings F. Pushing and pulling in prokaryotic DNA segregation. Cell. 2010;141:927–942. doi: 10.1016/j.cell.2010.05.033. [DOI] [PubMed] [Google Scholar]
- 20.Bi E, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia Coli. Nature. 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 21.Jones LJF, Carballido-López R, Errington J. Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell. 2001;104:913–922. doi: 10.1016/s0092-8674(01)00287-2. [DOI] [PubMed] [Google Scholar]
- 22.van den Ent E, Amos LA, Loewe J. Prokaryotic origin of the actin cytoskelecton. Nature. 2001;413:39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- 23.Celler K, Koning RI, Koster AJ, van Wezel GP. Multidimensional view of the bacterial cytoskeleton. J Bacteriol. 2013;195:1627–1636. doi: 10.1128/JB.02194-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabeen MT, Jacobs-Wagner C. The bacterial cytoskeleton. Annu Rev Genet. 2010;44:365–392. doi: 10.1146/annurev-genet-102108-134845. [DOI] [PubMed] [Google Scholar]
- 25.Figge RM, Divakaruni AV, Gober JW. MreB, the cell shape-determining bacterial actin homologue, co-ordinates cell wall morphogenesis in Caulobacter crescentus. Mol Microbiol. 2004;51:1321–1332. doi: 10.1111/j.1365-2958.2003.03936.x. [DOI] [PubMed] [Google Scholar]
- 26.Gitai Z, Dye N, Shapiro L. An actin-like gene can determine cell polarity in bacteria. Proc Natl Acad Sci USA. 2004;101:8643–8648. doi: 10.1073/pnas.0402638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gitai Z, Dye NA, Reisenauer A, Wachi M, Shapiro L. MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell. 2005;120:329–341. doi: 10.1016/j.cell.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 28.Divakaruni AV, Loo RRO, Xie Y, Loo JA, Gober JW. The cell-shape protein MreC interacts with extracytoplasmic proteins including cell wall assembly complexes in Caulobacter cresentus. Proc Natl Acad Sci USA. 2005;102:18602–18607. doi: 10.1073/pnas.0507937102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwai N, et al. Structure–activity relationship of S-benzylisothiourea derivatives to induce spherical cells in Escherichia coli. Biosci Biotechnol Biochem. 2004;68:2265–2269. doi: 10.1271/bbb.68.2265. [DOI] [PubMed] [Google Scholar]
- 30.Bean GJ, et al. A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry. 2009;48:4852–4857. doi: 10.1021/bi900014d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carballido-Lopez R. The bacterial actin-like cytoskeleton. Microbiol Mol Biol Rev. 2006;70:888–909. doi: 10.1128/MMBR.00014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaevitz JW, Gitai Z. The structure and function of bacterial actin homologs. Cold Spring Harb Perspect Biol. 2010;2:a000364. doi: 10.1101/cshperspect.a000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takacs CN, et al. MreB drives de novo rod morphogenesis in Caulobacter crescentus via remodeling of the cell wall. J Bacteriol. 2010;192:1671–1684. doi: 10.1128/JB.01311-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dye NA, Pincus Z, Theriot JA, Shapiro L, Gitai Z. Two independent spiral structures control cell shape in Caulobacter. Proc Natl Acad Sci USA. 2005;102:18608–18613. doi: 10.1073/pnas.0507708102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Divakaruni AV, Baida C, White CL, Gober JW. The cell shape proteins MreB and MreC control cell morphogenesis by positioning cell wall synthetic complexes. Mol Microbiol. 2007;66:174–188. doi: 10.1111/j.1365-2958.2007.05910.x. [DOI] [PubMed] [Google Scholar]
- 36.den Blaauwen T, de Pedro MA, Nguyen-Disteche M, Ayala JA. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev. 2008;32:321–344. doi: 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- 37.Kim SY, Gitai Z, Kinkhabwala A, Shapiro L, Moerner WE. Single molecules of the bacterial actin MreB undergo directed treadmilling motion in. Caulobacter crescentus Proc Natl Acad Sci USA. 2006;103:10929–10934. doi: 10.1073/pnas.0604503103. This study shows that single copies of MreB move in a slow, directed, circumferential fashion around the cell periphery in C. crescentus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biteen JS, et al. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nature Methods. 2008;5:947–949. doi: 10.1038/NMETH.1258. This study introduces eYFP protein fusion constructs as a suitable label for super-resolution imaging in live bacteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garner EC, et al. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science. 2011;333:222–225. doi: 10.1126/science.1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez-Escobar J, et al. Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science. 2011;333:225–228. doi: 10.1126/science.1203466. [DOI] [PubMed] [Google Scholar]
- 41.van Teeffelen S, et al. The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc Natl Acad Sci USA. 2011;108:15822–15827. doi: 10.1073/pnas.1108999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swulius MT, et al. Long helical filaments are not seen encircling cells in electron cryotomograms of rod-shaped bacteria. Biochem Biophys Res Commun. 2011;407:650–655. doi: 10.1016/j.bbrc.2011.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swulius MT, Jensen GJ. The helical MreB cytoskeleton in Escherichia coli MC1000/pLE7 is an artifact of the N-terminal yellow fluorescent protein tag. J Bacteriol. 2012;194:6382–6386. doi: 10.1128/JB.00505-12. This study shows that the helical shape of MreB superstructure is an artefact of the fluorescent protein fusion construct that was used. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bendezu FO, Hale CA, Bernhardt TG, de Boer PAJ. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 2009;28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salje J, van den Ent F, de Boer P, Loewe J. Direct membrane binding by bacterial actin MreB. Mol Cell. 2011;43:478–487. doi: 10.1016/j.molcel.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White CL, Kitich A, Gober JW. Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol Microbiol. 2010;76:616–633. doi: 10.1111/j.1365-2958.2010.07108.x. [DOI] [PubMed] [Google Scholar]
- 47.Erickson HP, Anderson DE, Osawa M. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev. 2010;74:504–528. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang K, Durand-Heredia J, Janakiraman A. FtsZ ring stability: of bundles, tubules, crosslinks, and curves. J Bacteriol. 2013;195:1859–1868. doi: 10.1128/JB.02157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osawa M, Anderson DE, Erickson HP. Reconstitution of contractile FtsZ rings in liposomes. Science. 2008;320:792–794. doi: 10.1126/science.1154520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Trimble MJ, Brun YV, Jensen GJ. The structure of FtsZ filaments in vivo suggests a force-generating role in cell division. EMBO J. 2007;26:4694–4708. doi: 10.1038/sj.emboj.7601895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu G, et al. In vivo structure of the E. coli FtsZ-ring revealed by photoactivated localization microscopy (PALM) PLoS ONE. 2010;5:e12680. doi: 10.1371/journal.pone.0012680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ben-Yehuda S, Losick R. Asymmetric cell division in B. subtilis involves a spiral-like intermediate of the cytokinetic protein FtsZ. Cell. 2002;109:257–266. doi: 10.1016/s0092-8674(02)00698-0. [DOI] [PubMed] [Google Scholar]
- 53.Thanedar S, Margolin W. FtsZ exhibits rapid movement and oscillation waves in helix-like patterns in Escherichia coli. Curr Biol. 2004;14:1167–1173. doi: 10.1016/j.cub.2004.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thanbichler M, Shapiro L. MipZ, a spatial regulator coordinating chromosome segregation with cell division in Caulobacter. Cell. 2006;126:1–16. doi: 10.1016/j.cell.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 55.Peters PC, Migocki MD, Thoni C, Harry EJ. A new assembly pathway for the cytokinetic Z ring from a dynamic helical structure in vegetatively growing cells of Bacillus subtilis. Mol Microbiol. 2007;64:487–499. doi: 10.1111/j.1365-2958.2007.05673.x. [DOI] [PubMed] [Google Scholar]
- 56.Anderson DE, Guieros-Filho FJ, Erickson HP. Assembly dynamics of FtsZ rings in Bacillus subtilis and Escherichia coli and effects of FtsZ-regulating proteins. J Bacteriol. 2004;186:5775–5781. doi: 10.1128/JB.186.17.5775-5781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biteen JS, Goley ED, Shapiro L, Moerner WE. Three-dimensional super-resolution imaging of the midplane protein FtsZ in live Caulobacter crescentus cells using astigmatism. ChemPhysChem. 2012;13:1007–1012. doi: 10.1002/cphc.201100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eswaramoorthy P, et al. Cellular architecture mediates DivIVA ultrastructure and regulates Min activity in Bacillus subtilis. mBio. 2011;2:e00257–11. doi: 10.1128/mBio.00257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strauss MP, et al. 3D-SIM super resolution microscopy reveals a bead-like arrangement for FtsZ and the division machinery: implications for triggering cytokinesis. PLoS Biol. 2012;10:e1001389. doi: 10.1371/journal.pbio.1001389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jennings PC, Cox GC, Monahan LG, Harry EJ. Super-resolution imaging of the bacterial cytokinetic protein FtsZ. Micron. 2011;42:336–341. doi: 10.1016/j.micron.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 61.Bowman GR, et al. A polymeric protein anchors the chromosomal origin/ParB complex at a bacterial cell pole. Cell. 2008;134:945–955. doi: 10.1016/j.cell.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bowman GR, et al. Caulobacter PopZ forms a polar subdomain dictating sequential changes in pole composition and function. Mol Microbiol. 2010;76:173–189. doi: 10.1111/j.1365-2958.2010.07088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ebersbach G, Briegel A, Jensen GJ, Jacobs-Wagner C. A self-associating protein critical for chromosome attachment, division, and polar organization in Caulobacter. Cell. 2008;134:956–968. doi: 10.1016/j.cell.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gahlmann A, et al. Quantitative multicolor subdiffraction imaging of bacterial protein ultrastructures in 3D. Nano Lett. 2013;13:987–993. doi: 10.1021/nl304071h. This paper introduces a method of two-colour, super-resolution imaging to accurately colocalize single molecules that are labelled with different fluorophores. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wery M, Woldringh CL, Rouvière-Yaniv J. HU-GFP and DAPI co-localize on the Escherichia coli nucleoid. Biochimie. 2001;83:193–200. doi: 10.1016/s0300-9084(01)01254-8. [DOI] [PubMed] [Google Scholar]
- 66.Llopis PM, et al. Spatial organization of the flow of genetic information in bacteria. Nature. 2010;466:77–81. doi: 10.1038/nature09152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dillon SC, Dorman CJ. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nature Rev Microbiol. 2010;8:185–195. doi: 10.1038/nrmicro2261. [DOI] [PubMed] [Google Scholar]
- 68.Sarkar T, Vitoc I, Mukerji I, Hud NV. Bacterial protein HU dictates the morphology of DNA condensates produced by crowding agents and polyamines. Nucleic Acids Res. 2007;35:951–961. doi: 10.1093/nar/gkl1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee SF, Thompson MA, Schwartz MA, Shapiro L, Moerner WE. Super-resolution imaging of the nucleoid-associated protein HU in Caulobacter crescentus. Biophys J. 2011;100:L31–L33. doi: 10.1016/j.bpj.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Illian J, Penttinen A, Stoyan H, Stoyan D. Statistical Analysis and Modelling of Spatial Point Patterns. John Wiley & Sons; 2008. [Google Scholar]
- 71.Wang W, Li G, Chen C, Xie XS, Zhuang X. Chromosome organization by a nucleoid-associated protein in live bacteria. Science. 2011;333:1445–1449. doi: 10.1126/science.1204697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 73.Badrinarayanan A, Reyes-Lamothe R, Uphoff S, Leake MC, Sherratt DJ. In vivo architecture and action of bacterial structural maintenance of chromosome proteins. Science. 2012;338:528–531. doi: 10.1126/science.1227126. This paper determines the minimal functional complex of MukBEF in living cells using single-molecule imaging and stoichimetry analysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kleine Borgmann LA, Ries J, Ewers H, Ulbrich MH, Graumann PL. The bacterial SMC complex displays two distinct modes of interaction with the chromosome. Cell Rep. 2013;3:1483–1492. doi: 10.1016/j.celrep.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 75.Niki H, et al. Escherichia coli MukB protein involved in chromosome partition forms a homodimer with a rod-and-hinge structure having DNA-binding and ATP/GTP binding activities. EMBO J. 1992;11:5101–5109. doi: 10.1002/j.1460-2075.1992.tb05617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamanaka K, Ogura T, Niki H, Hiraga S. Identification of two new genes, mukE and mukF, involved in chromosome partitioning in Escherichia coli. Mol Gen Genet. 1996;250:241–251. doi: 10.1007/BF02174381. [DOI] [PubMed] [Google Scholar]
- 77.Wang X, Llopis PM, Rudner DZ. Organization and segregation of bacterial chromosomes. Nature Rev Genet. 2013;14:191–203. doi: 10.1038/nrg3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.den Blaauwen T, Lindqvist A, Lowe J, Nanninga N. Distribution of the Escherichia coli structural maintenance of chromosomes (SMC)-like protein MukB in the cell. Mol Microbiol. 2001;42:1179–1188. doi: 10.1046/j.1365-2958.2001.02691.x. [DOI] [PubMed] [Google Scholar]
- 79.Lindow J, Kuwano M, Moriya S, Grossman A. Subcellular localization of the Bacillus subtilis structural maintenance of chromosomes (SMC) protein. Mol Microbiol. 2002;46:997–1009. doi: 10.1046/j.1365-2958.2002.03235.x. [DOI] [PubMed] [Google Scholar]
- 80.Mascarenhas J, Soppa J, Strunnikov A, Graumann P. Cell cycle-dependent localization of two novel prokaryotic chromosome segregation and condensation proteins in Bacillus subtilis that interact with SMC protein. EMBO J. 2002;21:3108–3118. doi: 10.1093/emboj/cdf314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leake MC, et al. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 2006;443:355–358. doi: 10.1038/nature05135. [DOI] [PubMed] [Google Scholar]
- 82.Uphoff S, Reyes-Lamothe R, Garza de Leon F, Sherratt DJ, Kapanidis AN. Single-molecule DNA repair in live bacteria. Proc Natl Acad Sci USA. 2013;110:8063–8068. doi: 10.1073/pnas.1301804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ptacin JL, et al. A spindle-like apparatus guides bacterial chromosome segregation. Nature Cell Biol. 2010;12:791–798. doi: 10.1038/ncb2083. This study shows that the chromosome-partitioning protein ParB in C. crescentus moves along a receding ParA structure during chromosome segregation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller OL, Hamkalo BA, Thomas CA. Visualization of bacterial genes in action. Science. 1970;169:392–395. doi: 10.1126/science.169.3943.392. [DOI] [PubMed] [Google Scholar]
- 85.Robinow C, Kellenberger E. The bacterial nucleoid revisited. Microbiol Rev. 1994;58:211–232. doi: 10.1128/mr.58.2.211-232.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woldringh CL. The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol Microbiol. 2002;45:17–29. doi: 10.1046/j.1365-2958.2002.02993.x. [DOI] [PubMed] [Google Scholar]
- 87.Norris V, Madsen M. Autocatalytic gene expression occurs via transertion and membrane domain formation and underlies differentiation in bacteria: a model. J Mol Biol. 1995;253:739–748. doi: 10.1006/jmbi.1995.0587. [DOI] [PubMed] [Google Scholar]
- 88.Bakshi S, Siryaporn A, Goulian M, Weisshaar JC. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol Microbiol. 2012;85:21–38. doi: 10.1111/j.1365-2958.2012.08081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nevo-Dinur K, Nussbaum-Shochat A, Ben-Yehuda S, Amster-Choder O. Translation-independent localization of mRNA in E. coli. Science. 2011;331:1081–1084. doi: 10.1126/science.1195691. [DOI] [PubMed] [Google Scholar]
- 90.Yu J, Xiao J, Ren X, Lao K, Xie XS. Probing gene expression in live cells, one protein molecule at a time. Science. 2006;311:1600–1603. doi: 10.1126/science.1119623. This paper uses single-molecule imaging to directly observe gene expression dynamics in live cells. [DOI] [PubMed] [Google Scholar]
- 91.Choi PJ, Cai L, Frieda K, Xie XSA. Stochastic single-molecule event triggers phenotype switching of a bacterial cell. Science. 2008;322:442–446. doi: 10.1126/science.1161427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.von Hippel PH, Berg OG. Facilitated target location in biological systems. J Biol Chem. 1989;264:675–678. [PubMed] [Google Scholar]
- 95.Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Edn Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sauer M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci. 2013;126:3505–3513. doi: 10.1242/jcs.123612. This paper provides a comprehensive review of single-molecule localization microscopy, with a thorough discussion of current challenges. [DOI] [PubMed] [Google Scholar]
- 97.Plass T, et al. Amino acids for Diels–Alder reactions in living cells. Angew Chem Int Edn Engl. 2012;51:4166–4170. doi: 10.1002/anie.201108231. [DOI] [PubMed] [Google Scholar]
- 98.Plass T, Milles S, Koehler C, Schultz C, Lemke EA. Genetically encoded copper-free click chemistry. Angew Chem Int Edn Engl. 2011;50:3878–3881. doi: 10.1002/anie.201008178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lang K, et al. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nature Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lang K, et al. Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels–Alder reactions. J Am Chem Soc. 2012;134:10317–10320. doi: 10.1021/ja302832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Charbon G, et al. Subcellular protein localization by using a genetically encoded fluorescent amino acid. ChemBioChem. 2011;12:1818–1821. doi: 10.1002/cbic.201100282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lukinavicius G, et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nature Chem. 2013;5:132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- 103.Puthenveetil S, Liu DS, White KA, Thompson S, Ting AY. Yeast display evolution of a kinetically efficient 13-amino acid substrate for lipoic acid ligase. J Am Chem Soc. 2009;131:16430–16438. doi: 10.1021/ja904596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fernandez-Suarez M, Ting AY. Fluorescent probes for super-resolution imaging in living cells. Nature Rev Mol Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. This paper reviews fluorescent probes for super-resolution imaging. [DOI] [PubMed] [Google Scholar]
- 105.Uttamapinant C, et al. A fluorophore ligase for site-specific protein labeling inside living cells. Proc Natl Acad Sci USA. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu DS, et al. Diels–Alder cycloaddition for fluorophore targeting to specific proteins inside living cells. J Am Chem Soc. 2012;134:792–795. doi: 10.1021/ja209325n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Siegrist MS, et al. D-amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol. 2013;8:500–505. doi: 10.1021/cb3004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kuru E, et al. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Edn Engl. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Szent-Gyorgyi C, et al. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nature Biotech. 2008;26:235–240. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 110.Yushchenko DA, Zhang M, Yan Q, Waggoner AS, Bruchez MP. Genetically targetable and color-switching fluorescent probe. Chem Bio Chem. 2012;13:1564–1568. doi: 10.1002/cbic.201200334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Benke A, Olivier N, Gunzenhaeuser J, Manley S. Multicolor single molecule tracking of stochastically active synthetic dyes. Nano Lett. 2012;12:2619–2624. doi: 10.1021/nl301018r. [DOI] [PubMed] [Google Scholar]
- 112.Lee HD, et al. Superresolution imaging of targeted proteins in fixed and living cells using photoactivatable organic fluorophores. J Am Chem Soc. 2010;132:15099–15101. doi: 10.1021/ja1044192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 114.Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci USA. 2005;102:17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gustafsson MGL. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198:82–87. doi: 10.1046/j.1365-2818.2000.00710.x. [DOI] [PubMed] [Google Scholar]
- 116.Gustafsson MGL. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moerner WE. New directions in single-molecule imaging and analysis. Proc Natl Acad Sci USA. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hell SW. Microscopy and its focal switch. Nature Methods. 2009;6:24–32. doi: 10.1038/nmeth.1291. [DOI] [PubMed] [Google Scholar]
- 119.Coltharp C, Xiao J. Superresolution microscopy for microbiology. Cell Microbiol. 2012;14:1808–1818. doi: 10.1111/cmi.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lippincott-Schwartz J, Patterson GH. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heilemann M, Dedecker P, Hofkens J, Sauer M. Photoswitches: key molecules for subdiffraction-resolution fluorescence imaging and molecular quantification. Laser Photon Rev. 2009;3:180–202. [Google Scholar]
- 122.Moerner WE. Microscopy beyond the diffraction limit using actively controlled single molecules. J Microsc. 2012;246:213–220. doi: 10.1111/j.1365-2818.2012.03600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cordes T, et al. Resolving single-molecule assembled patterns with superresolution blink-microscopy. Nano Lett. 2010;10:645–651. doi: 10.1021/nl903730r. [DOI] [PubMed] [Google Scholar]
- 124.Schoen I, Ries J, Klotzsch E, Ewers H, Vogel V. Binding-activated localization microscopy of DNA structures. Nano Lett. 2011;11:4008–4011. doi: 10.1021/nl2025954. [DOI] [PubMed] [Google Scholar]
- 125.Lee MK, Williams J, Twieg RJ, Rao J, Moerner WE. Enzymatic activation of nitro-aryl fluorogens in live bacterial cells for enzymatic turnover-activated localization microscopy. Chem Sci. 2013;4:220–225. doi: 10.1039/C2SC21074F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci USA. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sahl SJ, Moerner WE. Super-resolution fluorescence imaging with single molecules. Curr Opin Struct Biol. 2013;23:778–787. doi: 10.1016/j.sbi.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang F, et al. Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms. Nature Methods. 2013;10:653–658. doi: 10.1038/nmeth.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ondrus AE, et al. Fluorescent saxitoxins for live cell imaging of single voltage-gated sodium ion channels beyond the optical diffraction limit. Chem Biol. 2012;19:902–912. doi: 10.1016/j.chembiol.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jones SA, Shim S, He J, Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nature Methods. 2011;8:499–505. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lill Y, et al. Single-molecule study of molecular mobility in the cytoplasm of Escherichia coli. Phys Rev E Stat Nonlin Soft Matter Phys. 2012;86:021907. doi: 10.1103/PhysRevE.86.021907. [DOI] [PubMed] [Google Scholar]
- 132.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nature Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 133.Subach FV, et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nature Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 135.Gurskaya NG, et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nature Biotech. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- 136.Chudakov DM, Lukyanov S, Lukyanov KA. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nature Protoc. 2007;2:2024–2032. doi: 10.1038/nprot.2007.291. [DOI] [PubMed] [Google Scholar]
- 137.McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nature Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Biteen JS, Thompson MA, Tselentis NK, Shapiro L, Moerner WE. Superresolution imaging in live Caulobacter crescentus cells using photoswitchable enhanced yellow fluorescent protein. Nature Methods. 2009;5:947–949. doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gunewardene M, et al. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys J. 2011;101:1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. Segregation of molecules at cell division reveals native protein localization. Nature Methods. 2012;9:480–482. doi: 10.1038/nmeth.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li G, Elf J. Single molecule approaches to transcription factor kinetics in living cells. FEBS Lett. 2009;583:3979–3983. doi: 10.1016/j.febslet.2009.11.035. [DOI] [PubMed] [Google Scholar]