

Abstract
A major challenge faced by screening centers developing image-based assays is the wide range of assays needed compared to the limited resources that are available to effectively analyze and manage them. To overcome this limitation, we have developed the web-based myImageAnalysis (mIA) application, integrated with an open database connectivity compliant database and powered by Pipeline Pilot (PLP) that incorporates dataset tracking, scheduling and archiving, image analysis, and data reporting. For system administrators, mIA provides automated methods for managing and archiving data. For the biologist, this application allows those without any programming or image analysis experience to quickly develop, validate, and share results of complex image-based assays. Further, the structure of the application within PLP allows those with experience in PLP programming to easily add additional analysis tools as required. The tools within mIA allow users to assess basic (cell count, protein per cell, protein subcellular localization) and more advanced (engineered cell lines analysis, cell toxicity) biological image-based assays that employ advanced statistics and provides key assay performance metrics.
Introduction
High-content analysis (HCA), the automated collection and analysis of imaging data, is poised to make a dramatic impact on both academic and commercial scientific research.1 The direct examination of cells, through the use of microscopes or other technologies, is a powerful method of understanding cellular function.2,3 When combined with appropriate stains and antibody labels, a single image offers a wealth of information about the state of biological mechanisms in the cell. However, a significant limitation in the wide spread application of image-based methods has been the time and effort required to capture, analyze, and manage the large image data sets acquired during screening.4
Recently, the development of advanced microscopes that have the ability to capture many thousands of auto-focused images per day has removed one of the major limitations to the wide adoption of image-based screening. Using multiwell plates in combination with compounds, RNAi, or expression libraries generates large image data sets that capture information from a vast biological landscape, comparable in scale to the first genome projects.5,6 However, in contrast to the concerted genomic efforts, data from image-based screens often comprise a number of discrete projects performed by researchers acting independently to address specific questions. Therefore, the resulting image data and its associated experimental metadata are often stored in a distinct, poorly linked fashion.5 These disparate collections of data can severely limit collaborative efforts, the ability to retrospectively mine existing image data sets, and ultimately limit the amount of information learned from each experiment. These limitations have been a driving force behind the development of the Open Microscopy Environment (OME) Remote Objects (referenced as OMERO) software platform, a large collaborative effort involving an international collection of academic and commercial laboratories started nearly 10 years ago, which continues to mature.5
In addition to the data management and storage problem, large HCA image data sets also pose an analysis conundrum as well. Although some large-scale image-based screens have been analyzed by eye, this process is time-consuming, requires expert biologist(s), and is prone to bias due to lack of quantitation.7,8 As discussed by Carpenter et al., automated image analysis results in a greater number of features being quantified on a cell-by-cell rather than per image basis. This can result in a higher degree of sensitivity that is impossible with the human eye and is far less labor intensive.4 Although a growing number of commercial software solutions have been developed for automated image analysis (Cellomics, GE Healthcare, TTP LabTech, Molecular Devices, Nikon, Vala Sciences, etc.), these tend to be expensive and focused on a limited number of applications.9 Furthermore, these solutions are often proprietary, which can stymie the development of new assays requiring addition of new tools and/or modified algorithms.
A number of open source image analysis tools are available. The ImageJ platform has been successfully used by a number of laboratories; however, it is optimized for the deep analysis of single images and does not handle large multi-image data sets without additional macros that require some degree of programming knowledge.9 Alternatively, the CellProfiler project, available from and supported by the Broad Institute, was developed to offer the ability to develop custom analysis algorithms that can be applied to large datasets without extensive programming or image analysis experience.4,10,11 This platform offers an expanding library of modular and powerful image and data analysis tools (via KNIME implementation) and served as an excellent model for our work using the Pipeline Pilot (PLP) software platform. However, CellProfiler relies upon external data management support and often requires the user to provide significant computer processing power, preferably with a computer cluster, to efficiently analyze large complex datasets.
The Accelrys PLP platform is an enterprise level software solution for the automation of data analysis that is widely distributed in both commercial and academic environments. This platform utilizes a visual programming experience allowing users to build tools that integrate and catalog multiple data types and perform complex analysis, data mining, informatics, and reporting. This can be performed across numerical, textual, structural and, with the tools available in the Imaging Collection, image-based data types. The Imaging Collection tool box contains a library of configurable integrated performance primitives and ITK (www.itk.org) code-based components that cover a large spectrum of image manipulation and measurement functions. Importantly, the library of components is validated and designed in a scientifically aware manner to provide a complete set of tools needed for the robust analysis of imaging data.
In this study, we describe the myImageAnalysis (mIA) project, our effort to develop a web-based application that provides the tools for high-throughput/high-content image data management and analysis to the scientific community with access to the PLP platform. This application takes advantage of PLP capabilities to integrate multiple data types and software tools to facilitate handling the diverse range image data and experimental metadata produced by high-content screening. Importantly, the mIA application allows users to design new analysis algorithms to adapt new assays without the requirement of an on-site image analysis/programming expert. In this study, we provide an overview of the mIA application, and demonstrate its features on several image data sets to seed the collaborative interest in the scientific community leading toward additional application development.
Materials and Methods
Sample Preparation
Samples for assay performance studies were prepared as previously described.12–14 Briefly, for the transcription factor (TF) assay, HeLa cells stably expressing the green fluorescent protein (GFP)-tagged androgen receptor (AR) were plated into 384-well plates in the DMEM/F12 medium supplemented with 5% charcoal stripped fetal bovine serum (S-FBS). After 48 h, either dihydrotestosterone (DHT) or DMSO was added to wells to a final concentration (10 nM, 0.5%), and plates were incubated for an additional 24 h. For the nuclear spot (NS) assay, HeLa cells containing integrated prolactin promoter elements and stably expressing the GFP-tagged estrogen receptor (ER) were plated into 384-well plates in the phenol red-free DMEM supplemented with 5% charcoal stripped and dialyzed FBS. After 48 h, estradiol, tamoxifen, or DMSO were added to selected wells to final concentrations (10 nM, 10 nM, 0.5%), and plates were incubated for an additional 24 h. For the cell toxicity (CT) assay, HeLa cells were seeded into 384-well plates in the DMEM supplemented with 5% regular FBS. After 16 h, m-chlorophenyl hydrazone (CCCP) or DMSO was added to selected wells to a final concentration (50 μM, 0.5%), and plates were incubated for an additional 7 h. For all assays, cells were plated into wells at a target density of 2,000 cells per well using a Thermo Scientific Multi-drop cell dispenser.
After incubation was complete, using a Biomek FX robot, plates were washed with phosphate-buffered saline (PBS) and fixed for 20 min at RT in 4% formaldehyde prepared in the CSK buffer (80 mM potassium PIPES, pH 6.8, 5 mM EGTA, 2 mM MgCl2). After fixation, cells were briefly permeabilized (5 min) with 0.5% Triton-X and prepared for imaging by washing in PBS, aspirating the washed solution, and adding a 1 ng/mL 4′,6-diamidino-2-phenylindole (DAPI) solution. Cells were imaged in PBS.
Automated Imaging
All cells were imaged using the IN Cell 6000 Analyzer (GE Healthcare). For the TF assay, a two color (DAPI, GFP) 2D image set was captured using a 40×/0.95NA objective with a total of nine fields captured per well, resulting in >500 cells imaged per well. For the NS assay, a two color (DAPI, GFP) 3D image set was captured using a 40×/0.95NA objective with a total of nine fields captured per well, resulting in >500 cells imaged per well. For each field, a total of 11 vertical z-planes were captured at 1 μm increments. For the CT assay, a single color (DAPI) 2D image set was captured using a 20×/0.4NA objective with a total of two fields captured per well, resulting in >800 cells imaged per well. For all assays, channel laser intensity and channel exposure times were set to achieve a minimum signal to a background ratio of 5:1. All images collected were 16-bit depth tiff images.
Software
Enterprise Platform (version 8.5) that includes PLP was obtained from Accelrys (San Diego, CA).*
Results
Overview of the Web Application
The mIA web application is a server-based, web accessible tool for high-content data management and image analysis built within the Apache web service provided by the Accelrys PLP platform (Fig. 1). To enhance functionality, the mIA server runs the freely available GNU R project software for statistical analysis and Bio-formats standalone Java library for converting nonstandard image formats into the Tiff format used within the mIA analysis protocols.15,16 These solutions are integrated within the PLP software directly (Fig. 1B). Although based on a Microsoft Windows server, the mIA application utilizes a number of Linux-like commands through the command line options available within PLP and the Cygwin (www.cygwin.org) collection of tools. Due to its internet browser-based interface, the mIA application is platform independent (Fig. 1A), can be accessed remotely, and requires minimal resources from the user to run. Finally, when the mIA application is initialized, connections with the computers/instruments that will generate the raw image sets are established (Fig. 1C), facilitating a direct upload of the imaging data and log files from the instrument to the system, which minimizes the time that data remain associated with the acquiring microscope.
Fig. 1.

Architecture of the mIA application. (A) Users of the mIA application use any java enabled web browser (reader) to interact with the web server running the mIA application protocols developed within Accelrys Pipeline Pilot (PLP). (B) Server integration of mIA. Through Accelrys PLP service, the server runs PLP protocols that manage integration of publically available software tools such as GNU R Stats and Bio-Formats that provide statistical functionality and support for a large number of image formats, respectively. All key metadata, file indexing, and results are stored on a mysql relational database server/service, while images are stored and archived using DAS and NAS storage server solutions. (C) Through the standard PLP file browser and network drive mapping, users can upload data produced and stored locally by a variety of instrument types from within the mIA interface. DAS, directly attached storage; mIA, myImageAnalysis; NAS, network-attached storage.
The mIA application is able to store, manage, and analyze hundreds of thousands of images that are typically generated in a high-throughput/high-content experiment. Importantly, all data uploaded to mIA is fully annotated down to the individual well level and include instrument collection parameters extracted from log files, eliminating the dataset confusion often observed in the multiperson laboratory and/or a screening center setting. Furthermore, since all members of a research group have access to all datasets belonging to that group, mIA facilitates sharing of datasets and results among collaborators.
The mIA application provides a number of automatic and manual tools for curating the thousands of images stored on the central server. Upon initial image set upload, mIA automatically archives the image set and stores backup copies on multiple distinct network-attached storage (NAS) locations (Fig. 1B), greatly increasing the protection against image data loss. Image data that are actively being analyzed are stored on directly attached storage (DAS), allowing for high data transfer rates required during the image processing stage (Fig. 1B). Administrative tools monitor available storage space on the DAS file server and upon reaching critical levels, automatically remove image sets based upon (1) upload date, (2) last time accessed, and (3) completed image analysis. After removal from active storage space, the mIA interface allows simple one-click reactivation of archived image sets for reanalysis, as needed. These tools work to keep the active storage space dynamic and minimize the risk of lack of usable storage space commonly encountered in active high-content screening centers.
Data Structure Within mIA
All data within the mIA application are organized in a project-centered manner (Fig. 2). All users who belong to the same research group (determined during the initial user login process) as the creator of the project are able to view all the content within the shared project. Each project contains six primary types of data: (1) general information describing project scope, (2) event log data recording each event that occurs with the project, (3) image set data, (4) numerical image analysis results, (5) grouped analysis data sets, and (6) reports generated from processed images and result data.
Fig. 2.
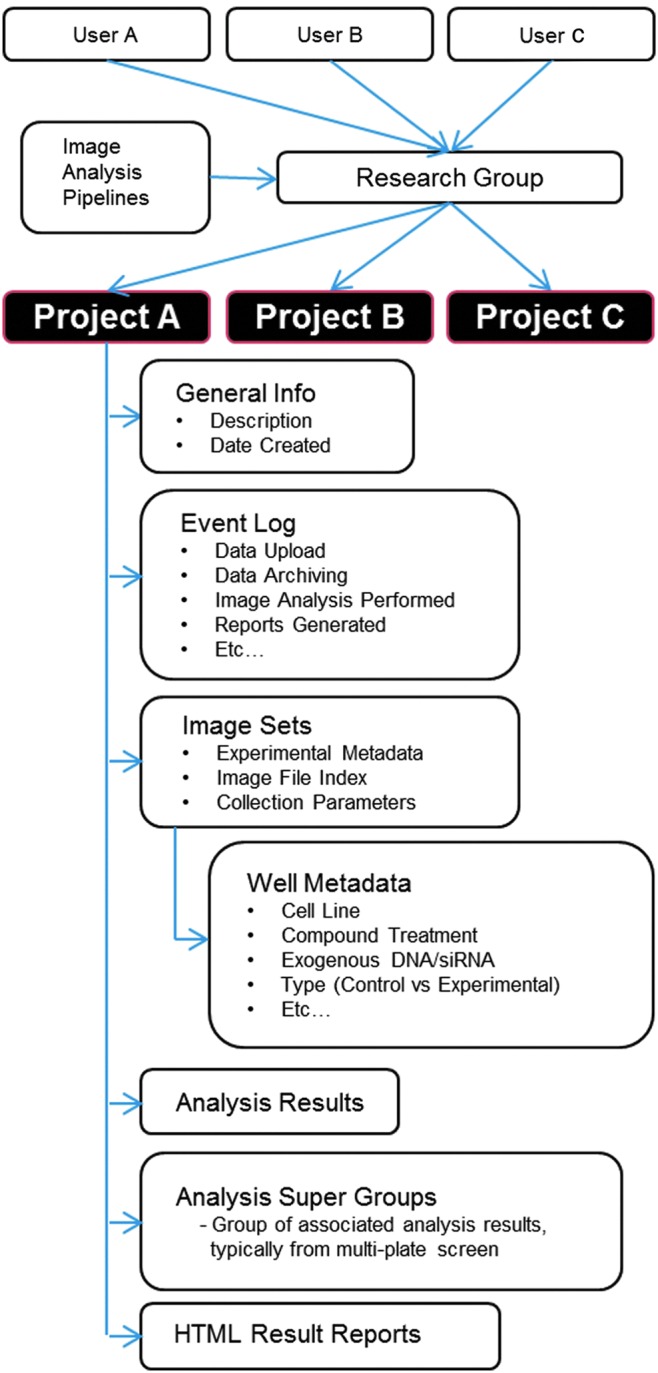
Data structure within the relational database used by the mIA application.
Two primary types of images are used within the mIA application. A raw image set typically consists of all images from a single multiwell plate or from a set of imaged coverslips during assay development or simply in very small experiments. The original plate location of each image is retained allowing a quick association with well level annotations. This annotation includes the cell line imaged, treatment conditions, and how the cells were labeled for imaging. Processed images are those generated by image analysis and consist of the illumination-corrected images and mask images corresponding to each identified subcellular region. All image data are stored within the file system of the DAS file server with file locations indexed for rapid access. Because the exact method used to create the processed images is recorded and therefore can be replicated, processed images are not archived and are removed from active storage as space needs dictate.
The numerical data associated with a project are handled in a singular manner. The numerical metrics extracted from each image set reside in an independent database from that of the main mIA application. Each analysis generates a unique metric table ranging from 50 to >500 parameters recorded per cell. After each analysis, mIA automatically generates additional tables describing the mean, median, standard deviation, mode, and number of samples from each well for each measure collected. Furthermore, if the analysis filters the total cell population to a subset of interest, mIA also records the members of that subpopulation and generates the summary statistics for those cell populations. Since high-throughput/high-content screens often span multiple multiwell plates, mIA includes functionality to group analysis results from multiple plates together into a single grouped analysis data set. This allows all data from a single screen to be manipulated at once, ensuring consistent normalization and interpretation across all plates in a screen.
The final data type associated with a project is a web-based report specific to the type of assay or analysis being performed. This report allows users to view overall responses, check assay quality metrics, and view processed images associated with the observed response. Furthermore, users have the ability to generate custom reports examining nonstandard metrics and save them to the project to share with collaborators.
Image Processing/Analysis
One of the primary goals of the mIA project was to provide the biologist a set of powerful, yet simple to use tools to analyze the complex image sets that are now readily obtainable with the latest generation of image cytometers. This is accomplished by using the array of image analysis/manipulation tools available in the Imaging Collection of PLP. These tools are organized into components within the mIA interface that are combined to accomplish the steps typically involved in image analysis: (1) loading images, (2) image correction, (3) primary object detection, (4) secondary object detection, (5) measurement and quantification, and (6) result classification. Importantly, end users do not need to understand PLP programming; however, for those users who are experienced with PLP, they can easily add new functionality due to the modular nature of the components used within the mIA interface, including the incorporation of CellProfiler/MatLab or ImageJ image analysis tools. To increase flexibility, custom reporting can incorporate images and other data generated on platforms external to the mIA application.
Developing an image analysis algorithm within mIA involves the selection of a number of point-and-click options from the interface (Fig. 3). Initially, users must select the project and image set to develop the method (Fig. 3A). Within this image set, users can choose to use images from selected wells or images from a random set of wells across the image set. This is useful during the initial algorithm development where positive and negative control wells are typically selected. In contrast, when assessing the performance of the final algorithm, one typically uses a larger set of random wells. This approach allows the user to be confident in the algorithm performance across the entire image set and mitigates concern for result skewing due to unknown algorithm bias.
Fig. 3.

mIA image analysis pipeline editor. (A) Top half of interface that allows users to select dataset and wells to use during method development. (B) Bottom half of the interface with partial algorithm displayed along with component option panel.
Actual image analysis algorithm development occurs within the algorithm editor, which contains an extension of the pipeline strategy used within PLP (Fig. 3B). Users locate each component by the desired task they wish to accomplish and place them into the algorithm in the order they wish to perform the tasks. This process is facilitated by a number of predefined algorithms designed to accomplish many of the more common high-content assays performed. Currently, there are over 40 modules and over 10 predefined algorithms (Table 1) for users to select from. Three of these algorithms are described in further detail in the following sections.
Table 1.
Predefined Analysis Pipelines
| Type | Regions identified | Report content |
|---|---|---|
| Basic | Cell | Reports cell count and signal intensity for each image type in dataset per well. |
| Cytoplasm | ||
| Nucleus | ||
| Transcription factor | Cell | Reports cell count, four key metrics quantifying the transcription factor, and reporter accumulation if present. |
| Cytoplasm | ||
| Nucleus | ||
| Nuclear spot | Cell | Reports cell count, percent cells with spots, and metrics describing spot size and intensity. |
| Cytoplasm | ||
| Nucleus | ||
| Spots | ||
| Cell toxicity | Cell | Reports a calculated toxicity index based on markers present. Provides access to detailed toxicity and cell cycle reports. |
| Cytoplasm | ||
| Mitochondria | ||
| Nucleus | ||
| Pattern and intensity | Cell | Reports cell count, signal intensity, results clustered into those with distinct signal patterns. |
| Cytoplasm | ||
| Nucleus | ||
| Multinuclear | Cell | Identifies multinucleated cells based on nuclear position and reports cell count and basic signal intensity metrics. |
| Cytoplasm | ||
| Nucleus | ||
| Two-color correlation | Cell | Reports cell count, signal intensities, degree of pixel intensity correlation between two distinct signals. |
| Cytoplasm | ||
| Nucleus | ||
| Bacteria intensity | Bacteria | Reports bacteria count and multiple signal intensity metrics. |
| Yeast cell wall | Yeast | Reports yeast count, intensity metrics, and percent cells with septum. |
| Septum | ||
| Cell wall |
Whereas some components are fully automatic, many have settings that require interactive input. Because the settings in each component are autosaved and sharable, the mIA application is able to provide users with most commonly used values for each setting from all users, users within the research group, and the individual user. This not only expedites algorithm development, but limits the algorithm drift that can occur between individual users within a research group. Once individual settings for components are adjusted, the effect of these changes can be previewed using images from the selected wells.
Once development is complete, the algorithm is saved. Access to the saved algorithm is restricted to the research group, cannot be changed by any user, and can only be used within the algorithm editor as template for future algorithms. Complete image sets are analyzed by associating an image set and an analysis algorithm to create an analysis job within mIA that is then submitted to the server. By utilizing the parallel processing options available within PLP, each image analysis job is divided among the available resources to minimize the processing time required. This allows mIA to function on a wide range of systems ranging from those containing a single processor, to multiprocessor servers, and finally to large computational clusters containing hundreds of processors.
The combination of a simple graphical interface and predefined analysis pipelines results in a typical initial learning curve of less than 2 h. Once an algorithm is developed, it takes less than 1 min to generate new analysis jobs on newly collected image sets. This is in contrast to programming within PLP itself, which has an initial learning curve on the order of multiple weeks if a previous programming experience is present. To further explain the analysis features of mIA, we use a TF assay in the following section.
mIA: TF Assay
TFs, proteins that bind DNA (directly or indirectly) and regulate gene expression, play a key role in a number of biological responses and in human disease.17 Therefore, it is of little surprise that significant effort has gone into the understanding of the mechanisms that regulate these proteins. It has become clear that controlling the expression level, subcellular distribution, and even the subnuclear distribution of TFs are all key regulatory mechanisms to control gene expression.17,18 Traditional biochemical methods for understanding each of these mechanisms are often time-consuming, fail to capture the cellular heterogeneity in a population, and are not easily amendable to high-throughput screening.
To overcome these limitations, a number of investigators have developed image-based methods to quantify the levels and subcellular distribution of a TF. These algorithms have been used to successfully analyze a vast number of proteins, such as NF-kB, STAT3 alpha and beta, and FOXO1.19–21 We have used these methods to extensively characterize a number of nuclear receptors and their coactivators in both traditional cultured cancer cell lines and patient-derived primary cell lines.12,22–24 The tools that we have used to simultaneously study the multiple aspects regulating a transcriptional factor have been incorporated into the predefined “Transcription Factor” algorithm within mIA's editor (Fig. 4A).
Fig. 4.

mIA transcription factor (TF) analysis pipeline. (A) Schematic of the steps included in the TF pipeline. (B) Although many image cytometers apply proprietary image correction algorithms, source images (left) often contain uneven illumination artifacts observed in a profile plot (top right) examining pixel intensity (y-axis) versus position (x-axis) across the image that are eliminated by algorithms incorporated into mIA (bottom right). (C) Colored masks show primary (nuclei) and secondary (cells) object detection in HeLa cells. Each analysis produces a HTML report that includes (D) plate heat maps, (E) well response (y-axis) versus sample ID (x-axis), (F) replicate A (y-axis) versus replicate B (x-axis) response, and (G) waterfall Z-score plots that allows rapid visualization of plate results. (H) Dose–response plots and EC50 values are generated for each dose–response series indicated by well annotations.
Image loader
The image analysis tools integrated into mIA are optimized for two-dimensional (2D) images, the most common image format used in high-content screening. Loading and using this image format is straightforward in the mIA interface. mIA also supports the uploading of more complex, three-dimensional (3D) image sets. To utilize these images, the mIA image loader component provides multiple methods for automatically converting 3D images into 2D, including projection methods (mean, maximum), middle plane selection, and best focal plane selection. Furthermore, for more complex analysis such as the NSs detection described in the next section, mIA supports loading and manipulating true 3D image sets within the analysis pipeline.
Image correction
Quantification of a biological response relies upon an accurate measurement of the signal present in the image. A key factor in limiting the accuracy of these measurements is uneven illumination and/or background across a single field that can vary by as much as 1.5-fold (unpublished observations). This issue has become more prominent with the implementation of the larger scientific-grade CMOS cameras recently being incorporated into numerous imaging systems. These cameras allow the latest generation of image cytometers to capture much larger areas per field (four- to fivefold); however, when retrofitted onto existing platforms, these cameras can result in significant illumination issues due to limitations of the light path originally designed for a smaller detector. Furthermore, illumination issues can remain even after correction by commercial software and/or algorithms provided by the image cytometers themselves (Fig. 4B). The mIA application addresses this issue by providing multiple options for correcting images, ranging from image pixel intensity minimum or mode-based background subtraction to a pixel-by-pixel flat field correction that ensures equal potential minimum and maximum intensity of each pixel across an image (Fig. 4B, bottom plot). These image correction components are included in all predefined pipelines as use of raw images will introduce noise and degrade the quality of measurements collected. In image sets, where appropriate correction across the entire image is not possible, mIA provides components to allow image cropping as necessary.
Primary object identification
The greatest obstacle to an accurate analysis of image data is identifying primary objects, the initial objects identified in an image. Typically, these objects are DNA-labeled nuclei; however, they can be cells, speckles, whole yeast, whole bacteria, cell clusters, and so on. The ability of an algorithm to perform this task has important implications on the degree of accuracy in the resulting measurements. The imaging collection within PLP has several published and validated methods for object detection. These threshold methods include, but are not limited to, OTSU, k-means, mean/median-based adaptive, and Canny edge detection. Furthermore, PLP is able to integrate tools developed within MatLab, CellProfiler, and ImageJ to further expand the tools available to users. Whereas simple threshold-based approaches function well on simple, well-dispersed samples, they fail to accurately separate dense or clustered samples. To overcome this limitation, several threshold techniques can be combined to produce robust methods that are able to segment crowded, complex samples. For example, mIA contains additional two identification algorithms based upon the basic components within PLP. Both algorithms use a combination of methods to identify either dispersed or clustered cells. Initially, a global OTSU threshold approach is used to identify where objects are within an image. Next, a local adaptive threshold based on neighborhood mean pixel intensity is used to identify potential separations between objects; finally, a distance transformation, shape filter, and watershed from marker operation is used to define the final identified objects (Fig. 4C, left). The two algorithms differ in the shape of the final objects to be identified, with a circular filter working best for most mammalian cells and an oval filter working best for bacteria and yeast. Importantly, limits are set on object size and intensity, preventing the algorithm from falsely identifying debris in the image as true objects.
Secondary object identification
After the primary objects (nuclei) are defined, the TF pipeline implements a secondary object detection module. Secondary objects are those that surround each primary object. In this case, the secondary objects are cells. The mIA application currently has three methods for secondary object detection: (1) tessellation, (2) signal limited tessellation, and (3) a watershed from markers. By default, the TF pipeline uses tessellation to define cell limits, as we have found this method the most straightforward and is sufficient in most cases (Fig. 4C, right). Additional cell regions can be identified by manipulating both primary and secondary objects, such as the cytoplasm (cell region minus nuclear region).
Object filtering
An important advantage of image-based approaches over biochemical methods is the ability to select which cells to analyze on a cell-by-cell basis. This allows an investigator the ability to remove cells that may artificially skew the final results. For example, dead, dying, or mitotic cells often contain protein levels and distributions that differ greatly from nonmitotic healthy cells. Furthermore, if transient transfections are used, filters can be placed to remove either nontransfected or grossly overexpressing cells. The TF pipeline contains filters to remove cells based upon the nuclear area, nuclear circularity, and the expression level of the protein of interest, typically the TF being examined. Furthermore, we have incorporated a filter that compares the signal intensity at the extreme edge of the cell region against the overall signal within the cell. This edge ratio filter is useful in eliminating cells that contain errors in either primary or secondary object detection.
Object quantification
The PLP Imaging collection includes a large number of basic and advanced shape and intensity measurements that are available through the mIA application. Measurements are collected for primary, secondary, and derived cell regions identified for each cell. Within the TF algorithm, this results in greater than 100 measurements per cell. All measurements are recorded in an image set-specific database table for later access. Furthermore, the mIA interface allows users to define new measurements that consist of a ratio, addition, or subtraction of any existing two measurements. The vast selection of available measures provides the researcher with flexibility as new biological questions arise and the ability to perform high-level statistics to define previously unappreciated responses.
Reporting
For each image set that is analyzed, the mIA application generates an HTML report allowing for easy visualization of the metrics collected. The mIA algorithm editor allows the selection of a report type that is customized to the type of assay and analysis being performed. Each report contains up to four types of elements: (1) heat map and bar chart visualizations, (2) Z score and robust Z-score calculations, (3) dose–response calculations, and (4) data links (Fig. 4D–H). The TF pipeline report displays per well metrics that describe cell count and effects on the TF protein level, nuclear accumulation, nuclear translocation, and nuclear hyperspeckling pattern formation. Furthermore, if a transcriptional reporter gene is in use, a report on reporter protein accumulation is displayed.
Assessment of algorithm performance
For basic assessment of the TF pipeline performance, we utilized an engineered HeLa cell line that stably expresses GFP-tagged AR, and also contains a stable integration of a dsRED2 transcriptional reporter driven by the AR responsive probasin promoter. Data shown (Supplementary Fig. S1A–E; Supplementary Data are available online at www.liebertpub.com/adt) are from a single 384-well plate containing wells treated with either 10 nM dihydrotesterone (192 wells, DHT, positive control) or DMSO (192 wells, negative control). Samples were imaged as described in Materials and Methods, generating an image set containing over 10,000 images. This image set was uploaded into the mIA application and analyzed using a single multiprocessor server at a rate of 9.3 s per field.
Using the data analysis tools contained within the standard TF report, we are able to visualize and quickly (<30 s) calculate the assay performance for each of the five standard metrics of interest across the dataset. Using the assay quality tool associated with each heat map within a mIA report, we determined the Z-factor value for total AR level, AR nuclear accumulation, percent AR in the nucleus, nuclear AR hyperspeckling, and dsRED transcriptional reporter accumulation. Whereas assay quality for each measure is assay and cell-line specific, we were able to routinely achieve Z-factor values ranging from 0.59 to 0.78 when using the engineered HeLa cell line.
mIA: NS Assay
Although a great deal can be learned from studying TF expression levels and subcellular distribution, direct visualization of DNA binding at a promoter is not possible using standard light microscopy. To overcome this limitation, several research groups have developed cell lines possessing multicopy gene units integrated at a single genomic locus.25,26 The high number of TF-binding sites per square micron of nucleoplasm allows the direct detection of a TF targeting a gene locus, frequently termed in our group as visual ChIP due to the similarities of biochemically detecting TF-chromatin interactions commonly used in chromatin immunoprecipitation assays.2,26
Our group has developed engineered HeLa cell lines that contain multiple integrated copies of an ER-regulated prolactin promoter/enhancer-regulated dsRED2 reporter gene. We have validated that these cells allow us to characterize physiological responses to hormones and their effect upon ER DNA binding, promoter/chromatin remodeling, coregulator recruitment, and mRNA production.14,26,27 We have also used these cell lines to characterize how environmental estrogens differentially affect the ER response.13
The image analysis tools to perform these studies have been incorporated into the mIA algorithm editor, creating the predefined “Nuclear Spot” analysis pipeline. The NS algorithm is largely similar to the TF pipeline, with nuclear and cell segmentation being performed in an identical manner (Fig. 5A). The NS algorithm differs in that it also contains components that allow for the selection of the best focus plane for each cell in an image field Z stack and the detection of NSs that correspond to the integrated promoter elements. Although we have used the NS algorithm for the analysis of engineered cell lines, the same algorithm can be used for the detection of multiple spots throughout the cell allowing for the analysis of biological processes such as DNA repair foci, cytoplasmic vesicle formation, peroxisomes, and mitochondria.
Fig. 5.

mIA nuclear spot (NS) analysis pipeline. (A) Schematic of the steps included in the NS pipeline. (B) Maximum projection of 3D image stack results in increased blur in 2D not seen in (C) single best focus plane image. (D) Best plane for each cell selected by defining plane with maximal variance. (E) Best focus plane projection results in 2D image with focused spot in each nucleus. (F) Spot segmentation minimizes the effect of dark pixels by normalizing pixel intensity before (G) local threshold is applied to maximize the signal to background ratio. (H) Final spot mask. 2D, two dimensional; 3D, three dimensional.
Cell-by-cell best plane selection
The challenge of identifying a single spot representing the TF-bound integrated locus within a nucleus is that the vertical position of the locus can vary from nucleus to nucleus. This natural biologic variation combined with the narrow depth of field associated with high-quality/high-magnification objectives results in many transcription arrays/spots not being adequately captured in the single plane of a 2D image set. This lowers the quality of the measurements collected. Therefore, to acquire maximal quality data sets, more complex 3D image sets containing multiple z planes must be captured; however, due to larger dataset sizes, this solution leads to new challenges. The image analysis tools inherited by mIA from PLP are optimized for 2D image sets, therefore, the 3D dataset must be converted to 2D. Since by definition the optimal plane of focus for the small NS (∼1 μm) consists of only one Z position per cell in the 3D dataset, all other Z planes contain lesser quality, out-of-focus information. Therefore, when projected using the standard methods in the image loader components, blur from the out-of-focus planes is included, decreasing the quality of the 2D image set to be analyzed (Fig. 5B, C).
To overcome this limitation, we have developed a component that selects the best focus plane on a cell-by-cell basis. This is accomplished in a three-step process. First, following cell segmentation, the pixel intensity variance within the nuclear region for each cell and each vertical z plane is measured. Next, a mask for each z plane is generated representing those cells that have maximal variance in that plane (Fig. 5D). Finally, these masks are used to generate a final composite 2D image that contains only data from the single best focus plane from each cell (Fig. 5E).
Spot segmentation
Identification and segmentation of the NS that corresponds to the integration site is an example of secondary object detection since information from primary objects (nuclei) is used. In this instance, detection is complicated by the relatively small spot size, and the ratio of spot signal-to-nucleoplasm that ranges between 1.1- and 3-fold. The small signal-to-nucleoplasm ratio is further complicated by the normal exclusion of ER from the nucleolus, which results in dark regions in the nucleus with a mean intensity two to threefold less than the surrounding nucleoplasm. Finally, the ER, like many other nuclear receptors, forms a hyperspeckled pattern within the nucleus when bound to a ligand. Therefore, the algorithm must distinguish between these speckles and the integration site.
The algorithm overcomes these issues by utilizing three processing steps. First, the influence of the dark nucleolus is removed by identifying the 75th percentile nuclear pixel intensity and correcting all pixels below this value up to this value (Fig. 5F). Next, a local background subtraction method using a spot size radius that is slightly larger than expected increases the signal-to-nucleoplasm ratio from 5- to 20-fold (Fig. 5G). Finally, all potential spots are ranked based upon the loading index (the ratio between spot intensity and surrounding nucleoplasm intensity), maximum intensity, and size to select the spot most likely to represent the integration site (Fig. 5H). It is important to note that the original ER composite image is not altered, allowing this image to be used for measurement collection.
Assessment of assay performance
To demonstrate the performance of the NS algorithm, we used the engineered prolactin (PRL) array HeLa cell line (PRL-HeLa) variant stably expressing GFP-tagged ER treated with saturating concentrations of 10 nM 17- estradiol, 10 nM 4-OH-tamoxifen, and DMSO for 24 h. A total of 120 wells were used for each treatment condition. In total, there were over 77,000 images from the large data set. Due to the complexity of the image set and analysis, the image processing time is slower compared with the TF algorithm at 31.1 s per field when a single server is used.
Using the results from this dataset, we calculated assay quality measures for each of the five key metrics describing the ER binding the integrated promoter elements (Supplementary Fig. S2). For the percent cells with a spot measurement, which is an indicator of the ER ability to bind DNA, wells treated with either estradiol (Supplementary Fig. S2A) or 4-OH-tamoxifen (Supplementary Fig. S2B) were used as positive controls, while wells treated with DMSO were used as negative controls for both. As expected, the NS pipeline analysis resulted in excellent Z-factor value for detecting percent of cells with arrays in estradiol-treated (0.91) and tamoxifen-treated (0.89) wells compared with DMSO control. When comparing the loading index (Supplementary Fig. S2C), the ratio between the spot mean signal and the surrounding nucleoplasm, using estradiol (ER agonist) wells as the positive control and tamoxifen (ER antagonist) wells as the negative control, we achieved a good Z-factor value of 0.55. DMSO was unable to be used as a negative control due to the absence of arrays preventing the determination of loading index in these wells. A slightly lower Z-factor value of 0.41 was achieved when the spot size (Supplementary Fig. S2E) was compared between the estradiol-treated (positive control) and tamoxifen-treated (negative control) wells. At these concentrations of agonist and antagonist, we were unable to observe a difference in the total signal (Supplementary Fig. S2D) at the integration site between estradiol and tamoxifen treatment conditions.
mIA: CT Assay
Traditional biochemical assays for CT have been shown to have low sensitivity.28 This is likely due to these assays measuring a single endpoint that involves late stages of toxicity and fails to account for the involvement of multiple mechanisms. Furthermore, due to the indirect nature of many biochemical assays, they are prone to false results due to specific alterations in cell physiology not related to cell death.29,30 An image-based approach allows for the simultaneous analysis of multiple aspects of CT with minimal resource consumption, utilizing only economical DNA and protein dyes. This method is compatible with most image-based assays allowing for analysis of toxicity in addition to other biologically significant responses in the screen, maximizing available new information. Multiple well-established cellular markers for toxicity involving multiple cytotoxic mechanisms include cell number, nuclear size, nuclear morphology, formation of micronuclei, DNA content/cell cycle progression, cell size, and cell morphology.29 For a more advanced analysis of toxicity, additional markers can be added such as antibodies specific to proteins involved in apoptosis. The CT pipeline supports the analysis of both basic toxicity based on DNA dyes and also when using additional markers (Table 2).
Table 2.
Cell Toxicity Markers Supported
| Label | Measures |
|---|---|
| DNA dye | Cell count |
| Nuclear shape | |
| Nuclear size | |
| DNA content | |
| DNA pattern | |
| Phospho-histone 3 | Mitotic index |
| Cell cycle | |
| Click-IT EdU (Invitrogen) | S-phase index |
| Cell cycle | |
| Cell mask (Molecular Probes) | Cell area |
| Cell shape | |
| Mito-Tracker (Molecular Probes) | Mitochondria mass |
| JC-1 | Mitochondria potential |
| Annexin V | Early apoptosis |
| c-PARP | Caspace target |
| HSP-70 | Cell stress |
| HSF-1 | Cell stress |
Toxicity index
Although many features of the CT pipeline are similar to other analysis pipelines available within mIA (Fig. 6A), a key feature is the generation of a HTML report that contains a toxicity index. The toxicity index consists of a single composite score ranging from 0 (minimal toxicity) to 1 (maximum toxicity) based on each of the toxicity markers present in the sample (Fig. 6B). For the data shown in Figure 6 that are derived from a sample containing only a DNA marker dye (DAPI), the toxicity index is calculated as follows.
Fig. 6.

mIA cell toxicity analysis pipeline and output. (A) Schematic of the steps included in the CT pipeline. (B) Toxicity index, a composite measurement, heat map (left) as well as heat maps for each measurement used in the index (right). (C) Summary bar chart indicating toxicity index (x-axis) for each sample type (y-axis) in each cell line (solid vs. open bars) indicated by well annotations. (D) K-Means Cluster analysis of defining toxicity patterns observed within dataset with an example detail available describing each cluster (inset). (E) HeLa cells were treated with 50 μM CCCP or DMSO for 7 h. A total of 120 wells were used for each treatment condition. Toxicity index calculated by examining cell count, nuclear size, nuclear shape, DNA content, and DNA pattern. CCCP, m-chlorophenyl hydrazone; DMSO, dimethyl sulfoxide.
- 1. Number of cells per well normalized to 0 to 1 using Equation 1 with a minimum allowed value of 0.
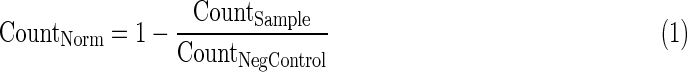
- 2. Average nuclear area per well normalized to 0 to 1 using Equation 2 if the well value is above the negative control value or Equation 3 if below.

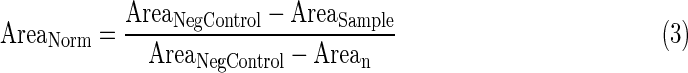
- 3. Average nuclear circularity per well normalized to 0 to 1 using Equation 4, wherein the higher value for circularity reflects a more circular object.
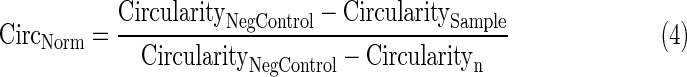
- 4. Average DAPI signal per cell (DNA content) per well normalized to 0 to 1 using Equation 5 if the well value is above the negative control value or Equation 6 if below.
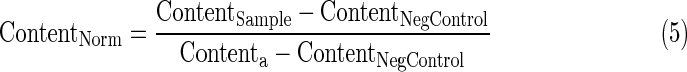
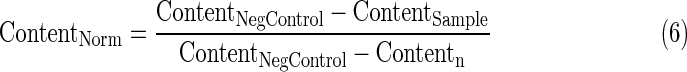
- 5. Average DAPI coefficient of variance (DNA patterning) per well normalized to 0 to 1 using Equation 7.
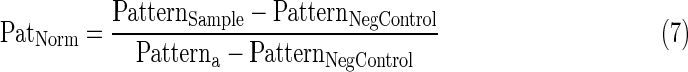
- 6. Toxicity index is calculated using Equation 8.
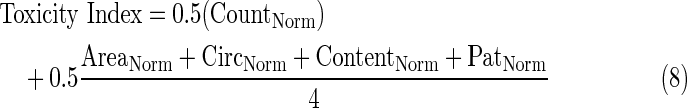
Due to the normalization steps involved, the toxicity index is relative to the specific sample being examined and the exact equation used will vary depending on the toxicity markers present. The coefficients or weight of each marker can be empirically determined or based on kinetic studies examining the correlation between each marker and future decrease in cell count. An example using those markers present with only a DNA dye was chosen since most HCA samples will have these markers regardless of the specific goal of the experiment. We have found that the toxicity index simplifies analysis and speeds progression to secondary screens; however, a heat map of each individual toxicity marker is also provided for complete and rapid data review (Fig. 6B). An index range for each feature is determined by the range of metrics that are present in the individual image sets for each cell line if multiple cell types are present in the sample. As toxicity can be cell line specific, the HTML report also contains a summary bar chart displaying the toxicity index for each unique treatment condition relative to each cell line indicated by well annotations (Fig. 6C). The CT report also uses tools made available from the integration of the R-stats platform to identify unique toxicity phenotypes by k-means-based clustering of responses across all toxicity markers (Fig. 6D) and allows the user to view the mean value for each measure for all samples grouped into any one cluster (Fig. 6D, inset).
Assay performance
To demonstrate the performance of the CT algorithm, we used HeLa cells treated with CCCP and DMSO. A total of 120 wells were used for each treatment condition. The multiwell plate was labeled with the DNA-binding Hoechst dye and imaged. The CT algorithm analyzed the collected image set at a rate of 3.1 s per field when a single server was used. Using metrics corresponding to the cell count, nuclear shape, nuclear area, DNA patterning, and DNA content, the CT pipeline generated a toxicity index for each well (Fig. 6E). Assigning CCCP-treated wells as positive control and DMSO-treated wells as negative control, the calculated Z- factor score within this dataset was 0.61, indicating the ability to reliably detect toxicity related to the CCCP compound.
Discussion
In this study, we report the development and validation of the mIA application, which has been shown to be a powerful tool for the management and analysis of the image sets that are generated during assay development and screening by HCA. We have shown the three most commonly used analysis algorithms available, although there are numerous additional tools already functioning within the application (Table 1), with many more in development. Indeed, the algorithm editor enables scientists with little background in image analysis to generate new algorithms as new biological questions arise.
Currently, the mIA application is utilized by several laboratories within the Texas Medical Center community, showing broad application on a growing number of different cell types. In total, over 30 terabytes of imaging data have been successfully managed and analyzed. Importantly, with the obstacle of the tedious analysis of large image sets minimized, investigators are now planning and executing large image-based screens that were not previously possible.
We are working with academic collaborators to further enhance the functionality of the mIA platform. These enhancements are focused on true 3D image set analysis and incorporation of tools to directly analyze the data stored within the growing OMERO project for data standardization and management. mIA is already amenable to the analysis of increasingly popular 3D culture-based assays and screens, and OMERO integration will facilitate the sharing and analysis of high-content data across a global HCS community. By making the mIA application protocols freely available and an open source to the PLP user community, we hope that the continual development of the mIA application will facilitate new discoveries in the many data-rich image-based screens that are currently being performed in both the pharmaceutical and academic environments.
Supplementary Material
Abbreviations Used
- 2D
two dimensional
- 3D
three dimensional
- AR
androgen receptor
- CCCP
m-chlorophenyl hydrazone
- CT
cell toxicity
- DAPI
4′,6-diamidino-2-phenylindole
- DAS
directly attached storage
- DHT
dihydrotestosterone
- DMSO
dimethyl sulfoxide
- ER
estrogen receptor
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HCA
high-content analysis
- mIA
myImageAnalysis
- NAS
network attached storage
- NS
nuclear spot
- OME
Open Microscopy Environment
- PLP
Pipeline Pilot
- TF
transcription factor
Footnotes
For access to these resources, the reader may contact the Director of Imaging at Accelrys, Tim Moran (tim.moran@accelrys.com).
Acknowledgments
The authors would like to thank the members of the Baylor College of Medicine Integrated Microscopy Core, Tim Moran, Ivana Mikic, and other Accelrys members for their help and input during the development of the mIA project, Maureen Mancini for help in development of the cell lines used, and Jeannie Zhong for the preparation of all samples used. This work was supported by the Diana Helis Henry Medical Research Foundation (M.A.M.) through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine and the Cancer Program. A.T.S. is a K12 scholar supported by the NIH grant K12DK0083014, the multidisciplinary K12 Urologic Research (KURe) Career Development Program awarded to Dr. Dolores J. Lamb.
Disclosure Statement
No competing financial interests exist.
References
- 1.Westwick JK, Lamerdin JE: Improving drug discovery with contextual assays and cellular systems analysis. Methods Mol Biol 2011;756:61–73 [DOI] [PubMed] [Google Scholar]
- 2.Berno V, Hinojos CA, Amazit L, Szafran AT, Mancini MA: High-resolution, high-throughput microscopy analyses of nuclear receptor and coregulator function. Methods Enzymol 2006;414:188–210 [DOI] [PubMed] [Google Scholar]
- 3.Hartig S, Newberg J, Bolt M, Szafran AT, Marcelli M, Mancini MA: Automated microscopy and image analysis for androgen receptor function. Methods Mol Biol 2011;776:313–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter AE, Jones TR, Lamprecht MR, et al. : CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006;7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allan C, Burel J, Moore J, et al. : OME remote objects (OMERO): a flexible, model-driven data management system for experimental biology. Nat Methods 2012;9:245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng H, Bateman A, Valencia A, Wren JD: Bioimage informatics: a new category in bioinformatics. Bioinformatics 2012;28:1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perrimon N, Hughes H: A functional genomic analysis of cell morphology using RNA interference. J Biol 2003;2:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JK, Gabel HW, Kamath RS, et al. : Functional genomic analysis of RNA interference in C. elegans. Science 2005;308:1164–1167 [DOI] [PubMed] [Google Scholar]
- 9.Prasad K, Prabhu GK: Image analysis tools for evaluation of microscopic views of immunohistochemically stained specimen in medical research-a review. J Med Syst 2012;36:2621–2631 [DOI] [PubMed] [Google Scholar]
- 10.Jones TR, Kang IH, Wheeler DB, et al. : CellProfiler Analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics 2008;9:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stöter M, Niederlein A, Barsacchi R, Meyenhofer F, Brandl H, Bickle M: CellProfiler and KNIME: open source tools for high content screening. Methods Mol Biol 2013;986:105–122 [DOI] [PubMed] [Google Scholar]
- 12.Szafran AT, Szwarc M, Marcelli M, Mancini MA: Androgen receptor functional analyses by high throughput imaging: determination of ligand, cell cycle, and mutation-specific effects. PLoS One 2008;3:e3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashcroft FJ, Newberg JY, Jones ED, Mikic I, Mancini MA: High content imaging-based assay to classify estrogen receptor-α ligands based on defined mechanistic outcomes. Gene 2011;477:42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolt MJ, Stossi F, Newberg JY, Orjalo A, Johansson HE, Mancini MA: Coactivators enable glucocorticoid receptor recruitment to fine-tune estrogen receptor transcriptional responses. Nucleic Acids Res 2013;41:4036–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tabelow K, Clayden JD, de Micheaux PL, Polzehl J, Schmid VJ, Whitcher B: Image analysis and statistical inference in neuroimaging with R. Neuroimage 2011;55:1686–1693 [DOI] [PubMed] [Google Scholar]
- 16.Linkert M, Rueden CT, Allan C, et al. : Metadata matters: access to image data in the real world. J Cell Biol 2010;189:777–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lonard DM, O'Malley BW: Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol 2012;8:598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imai Y, Youn M-Y, Inoue K, Takada I, Kouzmenko A, Kato S: Nuclear receptors in bone physiology and diseases. Physiol Rev 2013;93:481–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y, Qiu J, Dong S, et al. : Stat3 isoforms, alpha and beta, demonstrate distinct intracellular dynamics with prolonged nuclear retention of Stat3beta mapping to its unique C-terminal end. J Biol Chem 2007;282:34958–34967 [DOI] [PubMed] [Google Scholar]
- 20.Morelock MM, Hunter EA, Moran TJ, et al. : Statistics of assay validation in high throughput cell imaging of nuclear factor kappaB nuclear translocation. Assay Drug Dev Technol 2005;3:483–499 [DOI] [PubMed] [Google Scholar]
- 21.Mediwala SN, Sun H, Szafran AT, et al. : The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate 2013;73:267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amazit L, Pasini L, Szafran AT, et al. : Regulation of SRC-3 intercompartmental dynamics by estrogen receptor and phosphorylation. Mol Cell Biol 2007;27:6913–6932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szafran AT, Hartig S, Sun H, et al. : Androgen receptor mutations associated with androgen insensitivity syndrome: a high content analysis approach leading to personalized medicine. PLoS One 2009;4:e8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartig SM, He B, Long W, Buehrer BM, Mancini MA: Homeostatic levels of SRC-2 and SRC-3 promote early human adipogenesis. J Cell Biol 2011;192:55–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller WG: Large-scale chromatin decondensation and recondensation regulated by transcription from a natural promoter. J Cell Biol 2001;154:33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharp ZD, Mancini MG, Hinojos CA., et al. : Estrogen-receptor-alpha exchange and chromatin dynamics are ligand- and domain-dependent. J Cell Sci 2006;119:4365. [DOI] [PubMed] [Google Scholar]
- 27.Berno V, Amazit L, Hinojos C, et al. : Activation of estrogen receptor-alpha by E2 or EGF induces temporally distinct patterns of large-scale chromatin modification and mRNA transcription. PLoS One 2008;3:e2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight AW, Little S, Houck K, et al. : Evaluation of high-throughput genotoxicity assays used in profiling the US EPA ToxCast chemicals. Regul Toxicol Pharmacol 2009;55:188–199 [DOI] [PubMed] [Google Scholar]
- 29.Giuliano KA, Gough AH, Taylor DL, Vernetti LA, Johnston PA: Early safety assessment using cellular systems biology yields insights into mechanisms of action. J Biomol Screen 2010;15:783–797 [DOI] [PubMed] [Google Scholar]
- 30.Marqués-Gallego P, den Dulk H, Backendorf C, Brouwer J, Reedijk J, Burke JF: Accurate non-invasive image-based cytotoxicity assays for cultured cells. BMC Biotechnol 2010;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.