

Abstract
The dentate granule cells (DGCs) form the most numerous neuron population of the hippocampal memory system, and its gateway for cortical input. Yet, we have only limited knowledge of the intrinsic membrane properties that shape their responses. Since SK and Kv7/M potassium channels are key mechanisms of neuronal spiking and excitability control, afterhyperpolarizations (AHPs) and synaptic integration, we studied their functions in DGCs. The specific SK channel blockers apamin or scyllatoxin increased spike frequency (excitability), reduced early spike frequency adaptation, fully blocked the medium-duration AHP (mAHP) after a single spike or spike train, and increased postsynaptic EPSP summation after spiking, but had no effect on input resistance (Rinput) or spike threshold. In contrast, blockade of Kv7/M channels by XE991 increased Rinput, lowered the spike threshold, and increased excitability, postsynaptic EPSP summation, and EPSP–spike coupling, but only slightly reduced mAHP after spike trains (and not after single spikes). The SK and Kv7/M channel openers 1-EBIO and retigabine, respectively, had effects opposite to the blockers. Computational modelling reproduced many of these effects. We conclude that SK and Kv7/M channels have complementary roles in DGCs. These mechanisms may be important for the dentate network function, as CA3 neurons can be activated or inhibition recruited depending on DGC firing rate.
Introduction
The dentate gyrus (DG) is the gateway of the hippocampal memory system, receiving cortical input from all sensory modalities, for forming episodic and spatial memories (Scharfman, 2007). Its granule cells (DGCs) constitute the most numerous hippocampal cell population. During spatial exploration and memory tasks, the DGCs fire sparsely within multiple place fields (Jung & McNaughton, 1993) and perform pattern separation (Leutgeb et al. 2007). Thus, the dispersal of cortical input onto the numerous, sparsely firing DGCs, which in turn connect sparsely to CA3, favours segregation of input entering the CA3 auto-associative network (Treves et al. 2008). The DG is also where long-term potentiation (LTP) was first discovered (Lømo, 1968; Bliss & Lømo 1973), and its output synapses are the site of a distinct form of LTP (Nicoll & Schmitz, 2005).
Despite the central role of the DGCs in the hippocampal memory system, their intrinsic membrane properties have only been partly determined and are often neglected in studies of hippocampal functions. In particular, afterhyperpolarizations (AHPs) and their underlying conductances following single action potentials (APs) or spike bursts have been shown to be important for controlling intrinsic neuronal excitability, firing frequency and pattern, including spike frequency adaptation, in a variety of central neurons (Kernell, 1968; Madison & Nicoll, 1982, 1984; Pedarzani et al. 1998; Lancaster et al. 2001; Sah & Faber, 2002; Vogalis et al. 2003; Peters et al. 2005). Many vertebrate neurons typically show three AHP components: a ‘fast AHP’ (fAHP, lasting 2–10 ms), a ‘medium AHP’ (mAHP) lasting 20–200 ms, and a ‘slow AHP’ (sAHP) lasting 1–5 s (Storm, 1987, 1990; Bean, 2007). Whereas the enigmatic nature of the sAHP has received considerable attention in recent years (Tzingounis & Nicoll, 2008; Andrade et al. 2012), the mAHP of DGCs remains poorly characterized. The mechanisms controlling both early and late spike frequency adaptation have particular relevance in DGCs, since there is evidence that different DGC discharge frequencies can (1) differentially impact the output of CA3 pyramidal cells and interneurons through the strong mossy fibre (MF) synapses (Henze et al. 2002), (2) recruit different network-related mechanisms (Mori et al. 2004, 2007), and (3) trigger somatic protein synthesis and subsequent transport along the axon to stabilize MF-LTP (Barnes et al. 2010).
In CA1 pyramidal cells, the mAHP is caused by dual voltage-dependent mechanisms (Storm, 1989, 1990): the M-current (IM, Kv7/M channels) generates the mAHP at depolarized membrane potentials, whereas Ih (HCN channels) causes a similarly looking mAHP at more negative potentials. Later, small-conductance calcium-activated potassium (SK) channels were found to underlie the mAHP in many other brain regions and neuron types (for review, see: Bond et al. 1999; Sah & Faber 2002; Vogalis et al. 2003), while convergent evidence has confirmed that the mAHP of CA1 pyramidal cells is generated by the dual IM and Ih mechanisms with little or no contribution from SK channels (Storm, 1989, 1990; Williamson & Alger, 1990; Gu et al. 2005, 2008; Peters et al. 2005). These findings suggest that different channel types generate AHPs of medium duration (mAHP; ∼20–200 ms) in different cell types.
Among the candidate AHP mechanisms, there is prior evidence for the expression of both SK and Kv7/M channels in DGCs. Thus, a small apamin-sensitive SK current, ISK, was identified in rat (Sailer et al. 2002) and human DGCs (Beck et al. 1997), in agreement with the low or moderate levels of SK1–3 mRNA (Stocker & Pedarzani, 2000) and SK1–3 protein (Sailer et al. 2002, 2004) in the DG. There are low levels of Kv7.2 and Kv7.3 immunostaining in the granule cell layer, whereas the MF axons of the DGCs show strong expression of these Kv7/M channel subunits (Cooper et al. 2001; Devaux et al. 2004; Klinger et al. 2011). Furthermore, it was reported that Kv7/M channels generate about 50% of the so-called ‘mAHP’ and ‘sAHP’ currents (ImAHP, IsAHP) in mouse DGCs, suggesting that IM contributes to their mAHP (Storm 1989; Tzingounis & Nicoll, 2008). However, to our knowledge, the mechanisms underlying the mAHP in DGCs have so far not been directly examined. Thus, although the roles of perisomatic Kv7/M channels in generating mAHPs, subthreshold resonance and control of neural excitability have been described in CA1 pyramidal cells (Storm, 1989; Hu et al. 2002; Yue & Yaari, 2004, 2006; Gu et al. 2005; Hu et al. 2007; Shah et al. 2008), little is known about their function in DGCs.
Therefore, we have now examined the functions of SK and Kv7/M channels in rat DGCs, including their roles in AHPs, adaptation, spike threshold regulation, and synaptic integration, by combining whole-cell patch clamp recordings, pharmacological tests and modelling. Surprisingly, we find that SK channels are the main generator of the mAHP and early spike frequency adaptation, although this previously appeared unlikely based on the comparatively small ISK amplitude in this cell type (Sailer et al. 2002). This result is intriguing also because blockade or knock-out of SK channels seems to facilitate hippocampus-dependent learning and memory (Messier et al. 1991; Deschaux et al. 1997; Ikonen & Riekkinen, 1999; Stackman et al. 2002; Deschaux & Bizot, 2005; Hammond et al. 2006; Brennan et al. 2008). We also find complementary effects of Kv7/M and SK channels on synaptic integration. Two preliminary reports of these results have already been presented in abstract form (Mateos-Aparicio et al. 2010a,b2010b).
Methods
Ethical approval
All procedures were approved by the responsible veterinarian of the Institute, in accordance with the statute regulating animal experimentation (Norwegian Ministry of Agriculture, 1996). The experiments reported in this work comply with the policies and regulations of The Journal of Physiology (Drummond, 2009). The animals were deeply anaesthetized with isoflurane inhalation before rapid decapitation.
Hippocampal slice preparation
Horizontal hippocampal slices (300–400 μm) were cut from young male Wistar rats (3–5 weeks of age). Following decapitation, the brain was quickly removed and submerged in ice-cold artificial cerebrospinal fluid (ACSF) with the following content (in mm): 87 NaCl, 75 sucrose, 25 NaHCO3, 16 d-glucose, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2 and 0.5 CaCl2.
Slices were prepared using a vibratome (either Vibratome, MO, USA or Leica VT1200S, Heidelberg, Germany) and incubated for 30 min at 35°C in the ACSF solution mentioned above. Before recording, slices were stored at room temperature and then transferred to the recording chamber superfused with ACSF: 125 NaCl, 25 NaHCO3, 25 d-glucose, 1 MgCl2, 2.5 KCl, 1.25 NaH2PO4 and 1.6 CaCl2. All solutions were saturated with 95% O2 and 5% CO2.
Electrophysiological recordings
Somatic whole-cell patch clamp recordings were obtained from DGCs visually identified under infrared differential interference contrast (IR-DIC) video microscopy. All recordings were made in the suprapyramidal blade of the DGC layer. Slices were superfused with ACSF (2–3 ml min−1) and the temperature was maintained between 31 and 33°C (less than 0.5°C variation during each recording). Patch pipettes (4–7 MΩ) were pulled from borosilicate glass tubing (Sutter Instruments, CA, USA) and filled with a solution containing (in mm): 120 KMeSO4, 10 KCl, 10 phosphocreatine disodium salt, 10 Hepes, 10 inositol, 4 MgATP and 0.3 NaGTP, adjusted to pH 7.2 with KOH, and with an osmolarity of 280–290 mosmol l−1. For somatic current clamp recordings, a Dagan BVC 700A amplifier (Dagan Corporation, MN, USA) was used, and signals were low-pass filtered at 5 or 10 kHz (–3dB) and digitized at 10 or 20 kHz, respectively. In order to block spontaneous synaptic transmission, 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 μm), dl-2-amino-5-phosphonopentanoic acid (dl-AP5, 50 μm) and gabazine (5 μm) were routinely added to the ACSF during current clamp experiments. In some experiments, the adenylyl cyclase activator forskolin (50 μm) was added to the bath in order to block the sAHP and measure the mAHP in isolation. Whole-cell voltage clamp recordings were obtained using an Axopatch 1-D amplifier (Axon Instruments, CA, USA), filtered at 5 kHz (–3 dB), and digitized at 10 kHz. To record SK-mediated currents in relative isolation, TTX (1 μm) and TEA (5 mm) were routinely added to the ACSF, blocking Na+ and some K+ channels (including BK, Kv7/M, and delayed rectifier channels), respectively (Sailer et al. 2002). The cell was voltage clamped at a holding potential of –50 mV, and currents were elicited by a brief (100 ms) depolarizing voltage step to +10 mV once every 60 s. Under these conditions, the depolarizing step triggered a robust, unclamped calcium spike followed by a brief inward tail current and slower outward tail currents.
The access resistance was typically 15–30 MΩ for current clamp recordings and 10–20 MΩ for voltage clamp experiments. In current clamp, recordings in which the access resistance changed >25% were discarded. Series resistance compensation was not used in voltage clamp experiments; neurons with >10% variation in series resistance were discarded. Potentials are corrected for the liquid junction potential error (7 mV).
Data acquisition, storage and analysis
The data were acquired using pCLAMP 10 (Axon Instruments) and stored in a computer for further analysis with Clampfit 10. Statistical analysis was performed using Origin 8.0 (Microcal). Values are expressed as means ± SEM, two-tailed Student's t test was used for statistical significance (α = 0.05), and the P values are given in the figure legends. During whole-cell current clamp recordings of AHPs, each cell was kept constantly depolarized at –62 mV by injection of a positive holding current in order to unmask the AHPs and measure them at the same potential in all cells. The mAHP and sAHP values were measured by averaging a time window (30 and 100 ms, respectively) around the peak of each one (20–50 ms and 0.3–0.4 s after a spike train, respectively). Spike frequency was measured by injecting depolarizing pulses (1 s) at different intensities, keeping the background membrane potential at –77 mV. The cell input resistance (Rinput) was determined from the voltage at the end of 500 ms small hyperpolarizing current pulses (0.1 nA), by dividing the steady-state voltage response by the current pulse amplitude (Rinput = ΔV/ΔI). For time course analysis, the input resistance was measured by injecting small (0.05 nA) hyperpolarizing pulses (1 or 0.5 s long) once every minute, while the cell's membrane potential was kept depolarized at –62 mV. In the experiments shown in Figs 9 and 10, we injected (through the somatic patch pipette) trains of artificial excitatory postsynaptic currents (‘αEPSCs’) defined by a double exponential function:
Figure 9.

A, representative examples showing the effect of SK channel blockade on subthreshold summation recorded at 36°C. αEPSPs were injected at 10 and 40 Hz into the somatic compartment while keeping the neuron depolarized near AP threshold. B, SK channel blockade significantly increased the number of spikes during a 10 αEPSP train both at 10 and 40 Hz (n = 5, P < 0.01 (**)). C, examples shown in A at higher magnification clearly show the reduced summation of αEPSPs during the mAHP and the dramatic increase after apamin application. Top arrows show αEPSPs before and during the mAHP. Note the undershoot due to the mAHP (open triangle). D, E and F, summary plots comparing the absolute peak level, amplitude and decay time constant (τ) of the αEPSP before the first AP (‘Pre’) and the first αEPSP after the first AP and associated mAHP (‘mAHP’) in control and apamin conditions. The mAHP significantly reduced the absolute αEPSP peak (D) and decay time constant (τ) (F) (n = 5, P < 0.05 (*)) under control conditions but apamin abolished this difference.
Figure 10.

A, typical examples of αEPSP trains at 10 (upper traces) and 40 Hz (bottom traces) before (black) and after application of 10 μm XE991 (green). B, summary plots of the number of spikes elicited by the αEPSP trains. XE991 significantly increased the number of action potentials both at 10 (n = 7, P < 0.001 (***)) and 40 Hz (n = 7, P < 0.01 (**)). C, examples shown in A at higher magnification show the reduced αEPSP absolute peak level during the mAHP (control, arrows, black traces) and the increased number of APs after XE991 (green). D, typical examples of subthreshold αEPSP trains evoked at 10 and 40 Hz during the control period (black) and after XE991 application (green). E, XE991 significantly increased the subthreshold αEPSP amplitude (n = 6, P < 0.05 (*); left panel) while having no clear effect on the αEPSP decay time constant (τ) (n = 6, P > 0.05; right panel).
In both figures, τon was set to 0.5 ms and τoff to 10 ms.
In voltage clamp, the apamin-sensitive current (IaAHP) was obtained by subtracting traces recorded after apamin application from those recorded before, and the peak of the apamin-sensitive current was calculated by averaging points within a 20 ms time window at the peak of the subtracted trace. Due to the small size of IaAHP in the DGCs, digital subtraction was required to distinguish this component from the entire IAHP.
Chemicals and drugs
Apamin, gabazine and forskolin were purchased from Sigma-Aldrich Norway AS (Oslo, Norway). A different batch of apamin and scyllatoxin (leiurotoxin I) was purchased from Latoxan (Valence, France). DNQX, dl-AP5, 1-EBIO, TTX and XE991 were obtained from Tocris Bioscience (Bristol, UK). Retigabine was obtained from Alomone Labs (Jerusalem, Israel). Potassium methyl sulphate (KMeSO4) was obtained from Fluka Chemie AG (Switzerland) and MP Biomedicals (USA). Other chemicals used in this study were from Sigma-Aldrich Norway AS. All drugs were bath-applied and superfused to the slices (2–3 ml min−1).
Modelling
We used a compartmental model of a DGC based on Aradi & Holmes (1999). Simulations were performed using NEURON 7.2 (Carnevale & Hines, 2006). As described in the online Supporting information (section S1), a number of adjustments to their model were required in order to account for our experimental data. In particular we included an axonal Hodgkin–Huxley-type M-conductance (gM) based on the analysis of voltage clamp data in Main et al. (2000). The model also included a somatic sAHP-conductance (gsAHP) but this is set to zero in Fig. 6 to mimic the situation in which the sAHP was blocked with forskolin (Fig. 4) or is minimal (Fig. 5). Details of the modelling procedures are included in the online Supporting information.
Figure 6.

A and B show plots of V(t) with ‘+’ and ‘−’ indicating ‘presence’ and ‘absence’ of the M-and SK-conductances. These plots should be compared with the experimental data in Figs 4 and 5. C and D show plots of the currents ISK(t) and IM(t) (both integrated over the model neuron) for the +M, +SK regime.
Figure 4.

A, typical traces showing the effect of apamin (100 nm, top traces) on the isolated mAHP after sAHP suppression by forskolin (50 μm); bottom traces show representative examples of the effect of XE991 (10 μm) on the isolated mAHP. B, averaged time plot (n = 5, left panel) showing the effect of apamin on the time course of the isolated mAHP. Summary plot (right panel) showing the individual and mean values before and after application of apamin (n = 5, P < 0.001 (***)). C, similar plots summarizing the effect of XE991 on the isolated mAHP (n = 7, P < 0.05 (*)). Note the smaller mAHP reduction after Kv7/M channel blockade compared to SK channel blockade.
Figure 5.

A, typical examples of single AP elicited by steady depolarization near the spike threshold. In control conditions (black trace), the spontaneous AP was followed by a fAHP and mAHP. Bath application of 100 nm apamin (red) effectively blocked the mAHP, while 10 μm XE991 (green) had little or no effect on the mAHP amplitude. Right panel shows individual and averaged values for the mAHP under control and apamin (n = 6, P < 0.01 (**)) or XE991 (n = 6, P > 0.05 (NS)) conditions. B, left, overlay of averaged single APs under control (black) and apamin (red) or XE991 (green) conditions. Right, phase plots showing the dV/dt of the AP waveform plotted against the voltage. Upper panel shows that application of apamin (red) did not affect the AP threshold while XE991 (bottom panel, green) had a hyperpolarizing effect.
Results
In this study, whole-cell patch clamp recordings were obtained from 77 mature DGCs from the suprapyramidal blade of the DG in hippocampal slices from young male rats (3–5 weeks postnatal). The mean resting membrane potential (RMP) was –82.4 ± 0.5 mV. Because neurons in the DGC layer are known to be heterogeneous, all of the DGCs used in this study were identified using the criteria defined by Lübke et al. (1998). In particular, the DGC layer contains subpopulations of neurons at different stages of maturation (Liu et al. 2000), due to ongoing neurogenesis of DGCs throughout the life of the animal (van Praag et al. 2002). These maturational stages can be distinguished, among other parameters, by the Rinput, which is known to be markedly higher in young than in mature neurons, as well as by the RMP (Liu et al. 2000; but see Schmidt-Hieber et al. 2004) and AP threshold (Schmidt-Hieber et al. 2004). Therefore, to record from a uniform population of mature DGCs, we selected for this study only cells having RMP more negative than –75 mV, AP threshold between –45 and –50 mV, stable AP amplitude higher than 70 mV, and Rinput < 350 MΩ.
SK channels contribute to the mAHP but not to the sAHP in DG granule cells
SK channels are known to show cumulative activation during and after a train of APs, due to accumulation of calcium in the cytoplasm (Pennefather et al. 1985; Lancaster & Adams, 1986; Sah & Faber, 2002). To compare the mAHP and sAHP amplitudes following a constant amount of Ca2+ influx, the cells were kept depolarized at –62 mV by steady current injection, and spike trains of 7–8 spikes were evoked by applying depolarizing current steps (100 ms duration). Under these conditions, all of the recorded DGCs (n = 54) showed a fAHP after each spike during the train, as well as the classical sequence of AHPs after each spike train: a mAHP followed by a sAHP. In order to test whether SK channels contribute to the mAHP or sAHP we bath-applied apamin (100 nm), a bee venom toxin that selectively blocks SK channels.
Figure 1A shows typical recordings from a rat DGC under control conditions and after bath application of 100 nm apamin. The mAHP was strongly reduced after apamin application (Fig. 1A, Overlay, and Fig. 1B), while the sAHP was not significantly affected (Fig. 1C). When the depolarizing current pulse was kept constant, apamin caused an increase in spike frequency, indicating that SK channels control excitability (see online Supporting information Fig. S2A, Apamin). To keep the evoked number of APs constant, for comparison of AHPs, we reduced the current pulse amplitude after apamin application (the mean ± SEM was reduced from 0.25 ± 0.03 nA to 0.18 ± 0.03 nA; Fig. 1A, bottom traces; n = 8, P < 0.01), and always recorded the mAHP from the ‘holding potential’ of –62 mV. Under these conditions, the peak amplitude of the mAHP was rapidly suppressed by more than 80% by apamin (from 4.0 ± 0.6 mV to 0.6 ± 0.2 mV; Fig. 1B; n = 8, P < 0.001).
Figure 1.

A, typical examples of whole-cell current clamp recordings from a DGC showing repetitive spiking in response to a depolarizing current pulse (100 ms long) before (black) and after (red) bath application of 100 nm apamin. The APs are followed by fAHP, mAHP and sAHP. The background membrane potential prior to stimulation was kept at –62 mV (dashed line) by depolarizing holding current injection, and the current pulse amplitude was adjusted to produce a train of seven APs. The insets show expanded traces from the marked periods (dashed rectangles). Note that apamin suppressed the mAHP, but had essentially no effect on the sAHP. B, left, time course of the effect of apamin on the mAHP peak amplitude (n = 8). The summary graph (right) shows the effect of apamin (n = 8, P < 0.001 (***)) on the mAHP in all cells tested (○) and the mean value (▪). C shows that apamin had no significant effect on the sAHP peak amplitude (n = 8, P > 0.05 (NS)). For the analysis, the mean AHP amplitudes during the last 3 min (1) before apamin application were compared to the last 3 min after full effect of apamin (2).
In contrast to its effect on the mAHP, apamin did not significantly reduce the sAHP amplitude, in agreement with results from CA1 pyramidal and other neurons (Lancaster & Nicoll, 1987; Storm, 1989; Stocker et al. 1999; Gu et al. 2005, 2008). Thus, the peak amplitude of the sAHP (4.8 ± 1.6 mV in control medium, before apamin) did not change significantly after apamin application (4.2 ± 1.5 mV; Fig. 1A and C; n = 8, P > 0.05). Under control conditions (without apamin in the bath), the sAHP amplitude was more variable between cells (range: 0–15.8 mV; Fig. 1C, right) than the mAHP amplitude (range: 1.5–6.5 mV; Fig. 1B, right).
Although non-specific, SK channel-independent effects of 100 nm apamin have to our knowledge not been reported, such effects are conceivable. Therefore, to further test our hypothesis about the roles of SK channels in the mAHP and excitability control, we also used a different SK channel blocker: the scorpion toxin scyllatoxin (leiurotoxin I), which has been reported to block SK channels in dorsal vagal neurons (Pedarzani et al. 2000) and CA1 pyramidal neurons (Stocker et al. 1999). Scyllatoxin has been shown to compete with 125I-apamin binding sites in the rat brain (Auguste et al. 1992). In a subset of five DGCs, we recorded the mAHP after a train of eight action potentials (Fig. 2A, Control). Bath application of 100 nm scyllatoxin strongly reduced the mAHP (Fig. 2A, Scyllatoxin, Overlay). As we saw with apamin, scyllatoxin also increased the spike frequency in response to a constant, depolarizing step current, so we had to reduce the step current to evoke a constant number of spikes (Fig. 2A, overlay, bottom traces). Figure 2C (left) shows the average time course of the mAHP amplitude before and after application of scyllatoxin. The effect of scyllatoxin was significant for all the five cells tested (Fig. 2C, right; n = 5, P < 0.01).
Figure 2.

A, typical examples showing the mAHP under control (black) and 100 nm scyllatoxin application (red). The overlay shows the mAHP effectively blocked after toxin application. B, the mAHP was enhanced by 500 μm 1-EBIO application. Both the mAHP amplitude and duration were increased after 1-EBIO application (red) compared to control traces (black). Note the increased positive current step injected to evoke a train of seven APs after 1-EBIO application. C, time course showing the effect of 100 nm scyllatoxin application during the recording time (n = 5, left panel). Summary graph of the five cells tested with this toxin (right panel) showing the comparison between control period (1) and values with full effect (2), measured in the time plot (n = 5, P < 0.01 (**)). D, similar analysis of the data in B, showing enhancement of the mAHP after 1-EBIO application (n = 5, P < 0.001 (***)).
To further elucidate the contribution of SK channels to the mAHP and excitability control, we also applied the SK and IK (SK4) channel opener 1-EBIO, a small organic (benzimidazolinone) compound (Pedarzani et al. 2001, 2005). 1-EBIO is known to increase the apparent sensitivity of SK channels to calcium, therefore increasing the amplitude and duration of SK channel-mediated currents in a variety of neurons, including CA1 pyramidal cells (Pedarzani et al. 2001, 2005). Figure 2B shows that 500 μm 1-EBIO enhanced both the amplitude and duration of the mAHP (overlay). In the subset of cells that we used for 1-EBIO experiments, the sAHP was small or absent (due to the cell-to-cell variation in sAHP), thus making the effect on the mAHP clearly visible. The mAHP amplitude was 4.8 ± 0.2 mV during the control period and increased to 6.9 ± 0.2 mV after bath application of 1-EBIO (Fig. 2D, right; n = 5, P < 0.001). The left panel in Fig. 1D shows the averaged time course of the effect. In addition, it was necessary to apply a stronger depolarizing current step to maintain a constant number of seven spikes within each spike train, showing that 1-EBIO reduced the cell's excitability (Fig. 2B, Overlay, bottom). Overall, the results indicate that, unlike CA1 pyramidal cells (Gu et al. 2005, 2008), SK channels make a major contribution to the mAHP in DGCs.
The mAHP increased with the number of spikes
In order to control the exact number and timing of spikes, we applied a train of brief (2 ms) depolarizing pulses within 100 ms, each pulse evoking only a single AP (Supporting information Fig. S1). We thus applied spike trains at different frequencies: 50, 70, 100 and 120 Hz, while maintaining a depolarized background membrane potential of –62 mV (Fig. S1A). As expected, and as observed in CA1 pyramidal cells (Lancaster & Adams, 1986), both the mAHP and the sAHP increased monotonically in amplitude with the number of spikes in the train (Fig. S1A). As shown in the time course plots, apamin again strongly suppressed the mAHP in all cases (Fig. S1B), but had no detectable effect on the sAHP amplitude (Fig. S1C). The effect of apamin was significant for all four frequencies (Fig. S1D, n = 6, P < 0.001).
Following trains of only five spikes, when the mAHP and sAHP had relatively small amplitudes, a slow afterdepolarization (sADP) appeared after apamin had abolished the mAHP (‘sADP’ in Supporting information Fig. S1A). This suggests that the spike train evokes a long-lasting inward tail current that may cause a sADP, but that this effect is normally prevented (masked) by the mAHP and sAHP. Taken together, these results indicate that SK channels underlie the mAHP but not the sAHP following spike trains in DGCs.
Feedback regulation of Ca2+ influx and sAHP by SK channels
We next asked whether SK channels mediate negative feedback regulation of calcium influx in DGCs, by regulating the spike frequency and spike number. As mentioned above, apamin increased the AP frequency and thus spike number in response to a constant depolarizing current pulse, and this is expected to enhance the Ca2+ influx. Since the sAHP is highly calcium sensitive, we used the sAHP amplitude as an indicator of the intracellular calcium level (Supporting information Fig. S2A). We used for this particular purpose a subset of cells (n = 6) which had a sAHP amplitude ranging from 3.1 to 13.6 mV. Apamin application increased the number of spikes during the pulse (Fig. S2A, Control, Apamin), while the mAHP was reduced (although partly masked by the overlapping sAHP, Fig. S2A, Overlay, arrow). In parallel, the sAHP amplitude increased from 5.7 ± 1.6 mV (control) to 7.4 ± 2.2 mV after apamin (Fig. S2B and C; n = 6, P < 0.05). Apamin often also increased the duration of the sAHP (Fig. S2A). These results support the idea that the SK channels mediate negative feedback regulation of Ca2+ influx in DGCs, and that this can be detected indirectly through modulation of the sAHP.
To measure the apamin-sensitive current, IaAHP, we used somatic whole-cell voltage clamp (see Methods). Following a depolarizing voltage step, there was an outward tail current that showed no obvious early component, but a prominent slow component lasting about 3 s (Fig. S3A, Control), in agreement with previous results (Sailer et al. 2002). Bath application of 100 nm apamin uncovered an early inward transient and delayed the peak of the total outward current (Fig. S3A, Apamin). Digital subtraction of the traces recorded before and after full effect of apamin revealed an early, outward, IaAHP, lasting about 200 ms, with a fast rising phase and a slower decay, also in agreement with Sailer et al. 2002. The apamin-sensitive current peaked within 30–40 ms after the onset of the depolarizing step (n = 5), with a mean amplitude of 46.9 ± 6.0 pA (Fig. S3C, Subtracted). Fig. S3D shows the time course of the effect of the apamin on the tail current by plotting the subtracted current recorded once every minute after bath application of 100 nm apamin, for all cells tested. The effect of apamin was significant for all cells tested (Fig. S3E; n = 5, P < 0.001).
Do Kv7/M channels also contribute to AHPs in DGCs?
Results from whole-cell voltage clamp recordings in DGCs from transgenic mice lacking KCNQ2 and 3 subunits indicate that an M-current component contributes about 50% of both the AHP-related tail currents ImAHP and IsAHP (Tzingounis & Nicoll, 2008). However, the relative contribution of the M-current to the mAHP and sAHP under current clamp conditions might be different, and this has not been examined previously. For example, in CA1 pyramidal cells, a prominent apamin-sensitive ImAHP was found following calcium spikes in voltage clamp (Stocker et al. 1999; Sailer et al. 2002), but apamin had little or no effect on the mAHP after a normal spike train in current clamp (Storm, 1989; Gu et al. 2005, 2008). In order to examine the mechanisms of mAHP and sAHP, we applied both the specific Kv7/M channel blocker XE991 (10 μm) and the Kv7/M channel opener retigabine (10 μm) separately and recorded their effects on DGCs in current clamp (Fig. 3), using the same protocol as shown in Figs 1 and 2. XE991 application was followed by a reduction of both the mAHP and sAHP amplitudes (Fig. 3A, top traces). Thus, the mAHP peak was significantly reduced from 5.5 ± 0.7 mV (control) to 2.6 ± 0.7 mV in XE991 (Fig. 3D, top left; n = 7, P < 0.05). XE991 application also reduced the peak sAHP amplitude from 5.2 ± 0.7 mV during the control period to 2.6 ± 0.5 mV in XE991 (Fig. 3C and D, top right; n = 7, P < 0.01). Next, we tested the effects of retigabine. The mAHP was significantly increased after retigabine application, from 5.9 ± 0.2 mV (control) to 7.6 ± 0.3 mV in retigabine (Fig. 3C and D, bottom left; n = 6, P < 0.001). In contrast, the sAHP amplitude was significantly reduced, from 4.5 ± 0.8 mV to 2.0 ± 0.4 mV (Fig. 3A, bottom; Fig. 3C and D bottom right; n = 6, P < 0.01). For comparison, to test whether the changes observed in Fig. 3C and D were caused by the added blockers or were merely time dependent, we also show (Fig. 3B) the time courses of mAHP and sAHP amplitudes for five cells that were recorded in the absence of blockers. These plots show that the AHPs were quite stable for a comparable time period under control conditions (n = 5).
Figure 3.

A, representative examples showing the effect of Kv7/M channel blockade (top traces) by XE991 (10 μm). Bottom traces show the effect of the Kv7/M channel opener retigabine (10 μm) on both the mAHP and sAHP. Note the reduction in holding current and depolarizing step after application of XE991 and the increase in the depolarizing step after application of retigabine. B, time plots for both the mAHP and sAHP amplitudes in a subset of cells (n = 5) recorded under control conditions, to test the stability of AHPs in our experimental conditions. Dashed lines represent the mean values during the ‘control’ period (first 5 min), in the case of drug application. C, averaged time plots of the experiments shown in A for both the mAHP and sAHP. D, summary graphs showing the effect of both XE991 (n = 7, P < 0.05 (*), P < 0.01 (**), top panels) and retigabine (n = 6, P < 0.001 (***), P < 0.01 (**), bottom panels) on the mAHP (left) and the sAHP (right).
XE991 and retigabine seemed to have quite clear, opposite effects on the mAHP as judged from the time courses (Fig. 3C), and both effects support the conclusion that Kv7/M channels contribute to the mAHP. However, the effects of XE991 and retigabine on the sAHP seem at first glance contradictory: the apparent reduction by XE991 seems to suggest that Kv7/M channels contribute to the sAHP, but retigabine did not produce the expected sAHP enhancement but rather the opposite. As discussed below, this apparent paradox may be resolved if retigabine reduces the calcium influx, or if retigabine opens Kv7/M channels already before the sAHP occurs, thus causing occlusion or shunting. Thus, it is possible that XE991 and retigabine may have indirect effects on the sAHP. The recent proposal that sAHPs may be generated by different channel types (Andrade et al. 2012) must also be considered.
In summary, our results support the conclusion that Kv7/M channels contribute to the mAHP of DGCs, although the full effect of this channel type on the mAHP may be partly masked by the overlapping sAHP. This may lead to an underestimation of the real contribution of Kv7/M channels to the mAHP (Gu et al. 2005; Kaczorowski et al. 2007). To prevent the mAHP from being contaminated by the sAHP, we next studied the mAHP in isolation, after suppressing the sAHP.
SK and KCNQ channel contribution to the isolated mAHP
As reported for CA1 pyramidal cells (Madison & Nicoll, 1986; Pedarzani & Storm, 1993; Gu et al. 2005), the adenylyl cyclase activator forskolin strongly suppressed the sAHP in DGCs (Fig. 4A, Forskolin). We exploited this to study the mAHP in isolation (Gu et al. 2005). Application of 100 nm apamin almost fully suppressed the isolated mAHP, reducing the peak amplitude from 3.7 ± 0.3 mV (control) to 0.5 ± 0.3 mV in apamin (Fig. 4A, top traces, and B; n = 5, P < 0.001). Next, we tested the effect of Kv7/M channel blockade on the isolated mAHP (Fig. 4A, bottom traces), and found that XE991 (10 μm) reduced the isolated mAHP from 5.6 ± 0.5 mV to 4.0 ± 0.3 mV (Fig. 4C; n = 7, P < 0.05). Thus, in the absence of the sAHP, XE991 had apparently a weaker effect on the mAHP than shown in Fig. 3, probably because of the apparent XE991 effect on the overlapping sAHP in Fig. 3A (top traces). In summary, these results suggest that, in contrast to other hippocampal principal neurons, both SK and Kv7/M channels differentially contribute to the mAHP in DGCs, with SK channels being the main generators of the mAHP in this cell type.
SK channels generate the mAHP after single action potentials
Previous studies have suggested that mAHPs after single and multiple APs can contribute to the refractory period and control interspike intervals (ISIs) and spike frequency (Lanthorn et al. 1984; Storm, 1989, 1990). In addition, a recent network model of stellate neurons in the medial entorhinal cortex (MEC) suggests that after-spike dynamics such as mAHP and ADP may be crucial for both phase precession and the change in scale of grid fields along the dorso-ventral axis of MEC (Navratilova et al. 2012).
To study mAHPs after single APs, we evoked slow repetitive firing (∼0.5–3 Hz) by injecting weak, steady depolarizing current at just suprathreshold intensity (Fig. 5A). Each of these APs was followed by a prominent fAHP, a mAHP (Fig. 5A, Control), and in some cells also an appreciable sAHP, as found in CA1 pyramidal cells (Storm, 1987). Bath application of apamin abolished (n = 6) the single spike mAHP (Fig. 5A, Apamin, Overlay), suggesting that SK channels activated by Ca2+ influx from a single AP generate the mAHP. In contrast, blockade of Kv7/M channels by XE991 did not significantly (n = 6) reduce the mAHPs after single, low frequency APs, although there was a slight reduction in the mean mAHP amplitude, from 4.4 ± 0.5 mV to 4.1 ± 0.5 mV (Fig. 5A, bottom; n = 6, P > 0.05). Unexpectedly, it thus appears that, whereas SK channels are strongly activated by a single AP, generating the somatic mAHP, Kv7/M channels are not or only minimally activated by single spikes in DGCs, in contrast to hippocampal CA1 pyramidal cells, where it is exactly the opposite (Storm, 1989; Gu et al. 2005, 2008).
In many neurons, Kv7/M channels are known to be concentrated in the axon and/or axon initial segment (AIS), and can modulate the spike threshold (Shah et al. 2008). Since both immunocytochemical (Cooper et al. 2001) and patch clamp data (Alle et al. 2009) indicate that there is a high density of Kv7/M channels in the MF axons of the DGCs, we wished to test whether they affect the spike threshold of these cells. We used phase–plane plots to accurately determine the threshold of evoked low frequency spikes like those shown in Fig. 5A, and found that 10 μm XE991 shifted the AP threshold to more negative potentials, by 3.6 ± 0.2 mV. This indicates that Kv7/M channels regulate the AP threshold in these cells. In contrast, apamin application caused no significant change in the spike threshold (Fig. 5B, top right) nor in the AP waveform (Fig. 5B, top left), indicating that SK channels have little or no influence on these parameters. This is not surprising, since SK channel activation depends on calcium influx, which is thought to mainly occur during APs in DGCs.
In conclusion, these results indicate that SK channels are the main generators of mAHPs after each single action potential during low frequency firing, whereas the Kv7/M channels regulate the spike threshold, but contribute little to the single-spike mAHP.
Modelling the contribution of SK and Kv7/M channels to the mAHP
In order to better understand the contributions of SK and Kv7/M channels to the mAHP, we constructed a computational compartmental model of a DGC based on the model of Aradi & Holmes, 1999 (see Methods and online Supporting information). Figure 6A and B shows that the DGC model gives a semiquantitative account of the isolated mAHP following seven APs (panel A) or one AP (panel B). The continuous black curves show the control situation in which both the SK-and M-conductances (gSK and gM, respectively) are present. Omitting gSK (‘−SK’ in the figure legend) essentially abolished the mAHP following both seven APs and one AP (red curves in Fig. 6A and B). In contrast, omitting gM (‘−M’) resulted in only a small reduction in the mAHP following seven APs and virtually no effect following one AP (green curves). Because the sAHP was not included in the model, these simulation results should be compared with the experimental data obtained in the presence of forskolin (Fig. 4, 7 APs) and the single AP results where the sAHP was minimal (Fig. 5). Figure 6C and D shows the total SK-and M-currents (i.e. integrated over the model neuron). It can be seen that following seven APs, ISK predominates for the first ∼100 ms and is the main cause of the peak mAHP (Fig. 6C). Note that at the end of the current pulse, ISK is quite similar to the peak apamin-sensitive current measured under voltage clamp (Supporting information Fig. S3). IM is initially smaller than ISK but lasts longer and so produces a more prolonged but small AHP (cf. black and green curves in Fig. 6A). IM is hardly activated by a single AP (Fig. 6D) and consequently has little effect on the mAHP which is essentially due to ISK (cf. black and green curves in Fig. 6B). In the model, the greater effect of gM activation on the mAHP following seven APs as compared with one AP reflects a progressive recruitment of proximal axonal Kv7/M channels during the AP train (Supporting information, section S2, Fig. S8A and B).
In the above simulations, gSK was inserted into the soma, the granule cell layer dendrites and the proximal dendrites, as described by Aradi & Holmes, 1999. However, the spatial distribution of gSK was not critical. Thus, virtually identical mAHPs could be obtained with gSK inserted uniformly in the dendrites only, provided the total gSK remained the same, or with gSK in the distal dendrites only, with the total SK-conductance increased 3-fold. The latter increase was necessary to compensate for a reduced Ca2+ influx through voltage-gated Ca2+ channels associated with AP attenuation; without this adjustment the mAHP was greatly reduced (Supporting information Fig. S11A, grey curve). It should also be noted that Kv7/M channel kinetics and the maximum specific conductance (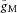 = 20 pS μm−2) were spatially uniform in the axon. In particular, the voltage for half-maximal conductance (Vhalf) was −38 mV and the activation time constants were 18-to 30-fold higher than those expected from Main et al. (2000). By contrast, the data of Alle et al. (2009) suggest a Vhalf of around −50 mV and considerably lower τ values in MF boutons. These considerations raise the possibility that gM kinetics in the proximal axon are different from those in the rest of the axon, specifically that in the proximal axon gM activates/deactivates more slowly and at a more depolarized voltage than in the distal axon. To investigate this possibility we included a 20 μm-long AIS in the model. Within the AIS gM parameters were as before, but in the rest of the axon Vhalf was set to −50 mV and the activation time constants were set according to the temperature-adjusted data of Main et al. (2000). Suitable adjustments of
= 20 pS μm−2) were spatially uniform in the axon. In particular, the voltage for half-maximal conductance (Vhalf) was −38 mV and the activation time constants were 18-to 30-fold higher than those expected from Main et al. (2000). By contrast, the data of Alle et al. (2009) suggest a Vhalf of around −50 mV and considerably lower τ values in MF boutons. These considerations raise the possibility that gM kinetics in the proximal axon are different from those in the rest of the axon, specifically that in the proximal axon gM activates/deactivates more slowly and at a more depolarized voltage than in the distal axon. To investigate this possibility we included a 20 μm-long AIS in the model. Within the AIS gM parameters were as before, but in the rest of the axon Vhalf was set to −50 mV and the activation time constants were set according to the temperature-adjusted data of Main et al. (2000). Suitable adjustments of 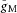 in the AIS and distal axon yielded similar results to those obtained previously. For example, virtually identical results to those in Fig. 6 were obtained with
in the AIS and distal axon yielded similar results to those obtained previously. For example, virtually identical results to those in Fig. 6 were obtained with 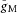 = 100 pS μm−2 in the AIS and 10 pS μm−2 in the distal axon and with no change in the injected I pulse. This proposed difference in gM kinetics between the AIS and distal axon represents a prediction of the model that is in principle experimentally testable.
= 100 pS μm−2 in the AIS and 10 pS μm−2 in the distal axon and with no change in the injected I pulse. This proposed difference in gM kinetics between the AIS and distal axon represents a prediction of the model that is in principle experimentally testable.
In the above simulations gsAHP was set to zero. Since the sAHP is not the focus of this paper, simulation results for the sAHP are presented in the Supporting information, section S3. The main results are: (a) the simulated effect of retigabine on the sAHP is consistent with observation; and (b) contrary to the observed effect of XE991, setting gM = 0 causes a slight increase in the sAHP following seven APs (see Supporting information Fig. S9C). Accordingly, it is suggested that either the XE991-sensitive component of the sAHP is not due to gM or that when contributing to the sAHP, gM is operating in a mode different from that associated with the mAHP.
Differential control of excitability mediated by SK and Kv7/M channels
To test how SK and Kv7/M channels control DGC excitability, we evoked repetitive firing by applying 1 s-long depolarizing current pulses starting from a membrane potential of –77 mV, which is close to the RMP of these cells. Apamin clearly enhanced the excitability: it increased the number of spikes (Fig. 7A, red) and increased the slope of the f–I plot from 48.2 ± 1.9 Hz nA−1 to 103.2 ± 6.1 Hz nA−1 (Fig. 7C, top panel, where the average spike frequency (f) during the 1 s-long pulse is plotted against the injected current intensity (I); n = 6, P < 0.001). Apamin did not, however, change the neuronal Rinput, as shown in Fig. 7B (red). To see how SK channels affected the spike frequency adaptation, and thus how the SK channel impact changed during a spike train, we plotted the average spike frequency for 100 ms time windows during a 1 s-long current pulse of 0.15 nA (Fig. 7D). Apamin significantly increased the spike frequency only within the first 200 ms of the response (for the interval 0–100 ms: 6.7 ± 0.8 Hz in control, 23.3 ± 0.8 Hz after apamin, n = 6, P < 0.001; for the interval 100–200 ms: 6.7 ± 0.8 Hz control, 16.6 ± 0.8 Hz apamin, n = 6, P < 0.001), for all cells tested (Fig. 7D, top panel). Conversely, 1-EBIO (500 μm) shifted the slope of the f–I plot from 44.6 ± 10.1 Hz nA−1 in control conditions to 31.8 ± 8.8 Hz nA−1 (Supporting information Fig. S12A and B; n = 5, P = 0.054). 1-EBIO decreased the number of spikes during a 1 s-long current injection of 0.25 nA (Fig. S12B, n = 5, P < 0.05), most notably during the first 200 ms of the response (Fig. S12C). Thus, SK channels are important only for the early spike frequency adaptation, but apparently less for the late adaptation under these conditions. However, as discussed below, it is likely that an enhancement of the sAHP due to increased Ca2+ influx caused by enhanced firing in the beginning of the train, partly masked an effect of the SK channels during the last part of the train.
Figure 7.

A, typical traces from two DGCs firing in response to 1 s-long depolarizing current injection (0.15 nA) at a clamped membrane potential of –77 mV. Compared to control conditions (black), 100 nm apamin (red) and 10 μm XE991 (green) increased the firing frequency response of each cell. B, representative examples comparing the voltage responses to –50 pA steps at subthreshold potential of –62 mV between control (black) and apamin (red) or XE991 (green). C, f–I plots of the DGC responses in experiments as shown in A (n = 6, Apamin; n = 6, XE991). The firing frequencies were augmented after 100 nm apamin or 10 μm XE991. D, averaged firing frequencies measured within 100 ms time windows during 1 s, 0.15 nA current pulses. Apamin (top panel) significantly increased the firing frequency during the first 200 ms of the pulse without affecting the late phase of the response. However, XE991 (bottom panel) increased the frequency during the whole 1 s response, including the late phase. E, time course of the Rinput measured in B showing no significant changes over time and after apamin (top) application. However, XE991 increased the Rinput (bottom).
Next, we tested the role of Kv7/M channels in excitability control. XE991 (10 μm) also increased the number of spikes (Fig. 7A, green) and thus also the slope of the f–I plot from 46.3 ± 2.1 Hz nA−1 to 87.4 ± 3.2 Hz nA−1 (Fig. 7C, bottom; n = 6, P < 0.001). In contrast to apamin, XE991 also increased the Rinput measured at subthreshold potentials (Fig. 7B, green). The plot of the spike frequency within 100 ms time windows (Fig. 7D, bottom) revealed several differences compared to SK channel blockade. While apamin selectively and sharply increased the early spike frequency only during the first 200 ms, XE991 produced a uniform and more moderate increase in spike frequency throughout the entire spike train. Thus, unlike apamin, XE991 enhanced also the steady firing rate towards the end of the 1 s-long train.
Finally, we examined in detail the early spike frequency adaptation under conditions of constant initial spike frequency (Fig. 8). Using a stimulation protocol similar to the one used in Figs 4, we injected 100 ms-long depolarizing current pulses to evoke a train of six action potentials per pulse. After apamin application, we reduced the current pulse intensity in order to obtain the same number of spikes before and during apamin. By further adjusting the strength of the current pulse, we also made sure that the first ISI duration was very similar before and during apamin (Fig. 8A, Overlay; Fig. 8B, top panel, first ISI), in order to ensure that the rate of spike adaptation could be compared under similar conditions. This is important because both calcium-dependent (e.g. SK, BK and sAHP channels) and voltage-dependent (e.g. Kv7/M channel activation and Nav channel inactivation) mechanisms of adaptation are spike-frequency dependent. Spike trains should therefore be compared at similar initial spike frequencies to avoid interference between different mechanisms, e.g. an AP frequency-dependent increase in the sAHP masking the effects of SK or Kv7/M channel blockade (Storm, 1989; Gu et al. 2005).
Figure 8.

A, apamin (100 nm) reduced the early spike frequency adaptation at depolarized membrane potentials (–62 mV) in DGCs (top traces, red). 100 ms long depolarizing current pulses (see Methods) evoked bursts of six APs once every minute. The first to fifth ISIs were measured before and after apamin application. In order to obtain comparable responses with six APs, the current pulse amplitude was reduced to compensate for the increased excitability due to the mAHP blockade. However, XE991 (bottom traces, green) had little effect on early spike frequency adaptation. B, summary data of ISI duration in control conditions and after apamin (n = 8; top red plot) and XE991 (n = 5; bottom green plot) applications. Note the reduction in the third to fifth ISI duration after SK channel blockade. C, normalized adaptation index (defined as the slope of the line that fitted best to the plot of the first to fifth ISI, as shown in B) in control conditions (black) and after apamin (red) and XE991 (green) application. Apamin significantly reduced the adaptation index compared to control conditions (n = 8, P < 0.05 (*)), while XE991 had little effect (not significant; n = 5, P > 0.05).
We found that SK channel blockade by 100 nm apamin reduced the spike frequency adaptation during the pulse-evoked spike train (Fig. 8A, Overlay). Figure 8B (top panel) summarizes the data for all cells tested (n = 8). In order to compare the overall degree of adaptation during the spike train, we fitted a straight line to both control and apamin plots in Fig. 8B, and compared the slope (termed ‘adaptation index’; Gu et al. 2005) of the linear fits. Apamin significantly reduced the adaptation index to 67.4% of the value under control conditions (Fig. 8C, red; n = 8, P < 0.05). In contrast, 500 μm 1-EBIO produced the opposite effect to apamin (Supporting information Fig. S12D). Thus, the SK opener significantly increased the adaptation index to 172.1% of the control value (Fig. S12E and F; n = 5, P < 0.01).
When we used the same conditions to test the effects of 10 μm XE991, we found that this caused only a slight reduction in the late ISIs (Fig. 8A, green trace) and hence only a slight reduction in the mean adaptation index (to 91.5 ± 20.5% of control, Fig. 8B, bottom). The effect on the adaptation index was variable and not statistically significant (Fig. 8C, green; n = 5, P > 0.05). Thus, for both XE991 and apamin, a similar reduction in the current pulse strength was needed to keep the spike number constant at six, and the first ISI constant (Fig. 8A, right: see overlays of current traces). These observations indicate that although Kv7/M channels strongly dampen the excitability of DGCs, they play at most a minor role in early spike frequency adaptation in this neuron type.
Taken together these results indicate that both SK and Kv7/M channels strongly limit the excitability of DGCs, with about equal reductions in the overall spike frequency. However, only SK channels seem important for specifically reducing the spike frequency towards the end of the 100 ms-long spike train, thus causing early spike frequency adaptation. In contrast, the Kv7/M channels seem to decrease the spike frequency quite uniformly throughout the spike train under the conditions tested here.
SK channels and mAHPs control postsynaptic summation under near-physiological conditions
In order to test whether the mAHP and its underlying SK channel activity can regulate synaptic summation and integration, we injected (into the soma) trains of αEPSCs at different frequencies. To create near-physiological conditions for these tests, the temperature was raised to 36–37°C and the extracellular Ca2+ and K+ concentrations adjusted (to 1.6 and 3.5 mm, respectively; see Methods). We injected trains of ten αEPSCs at frequencies in the theta (10 Hz) and gamma (40 Hz) bands (Fig. 9A, control), which are particularly relevant for hippocampal network activity in vivo (Traub et al. 1996; Buzsaki, 2002). At both frequencies, we adjusted the αEPSC amplitude (which was constant throughout each train) so that the summated αEPSPs reached the AP threshold during the third artificial post synaptic potential (αEPSP), and the overall spiking within the train in control medium was approximately 1–2 APs at 10 Hz, and 3–4 APs at 40 Hz (Fig. 9B).
At 10 Hz, the αEPSPs showed only slight summation (Fig. 9A, 10 Hz). When an AP was triggered, it was followed by a mAHP (seen as an undershoot below baseline; marked by a triangle in Fig. 9C). Hence, the absolute peak voltage of the next αEPSP, occurring during the mAHP, was significantly lower (Fig. 9D, black plot; –51.1 ± 0.8 mV ‘Pre’, –54.6 ± 0.9 mV ‘mAHP’; n = 5, P < 0.05), falling below the spike threshold, presumably due to the mAHP. Furthermore, the αEPSP decay time constant was also significantly reduced during the mAHP (Fig. 9F, black plot; 38.8 ± 3.4 ms ‘Pre’, 31.6 ± 2.4 ms ‘mAHP’; n = 5, P < 0.05), reflecting shunting by the open SK channels during the mAHP. However, after application of apamin, the mAHP was blocked and the αEPSP following the AP usually evoked a second spike (Fig. 9A and C, 10 Hz, red). In these conditions, the αEPSP amplitude, its absolute peak level, and its decay time constant values were not significantly different from those measured before the first AP (Fig. 9D, E and F, red plots; n = 5, P > 0.05). Thus, apamin significantly increased the number of spikes during trains of αEPSPs at 10 Hz (Fig. 9B, 10 Hz, Apamin).
We next applied 40 Hz trains which produced stronger summation (Fig. 9A, 40 Hz, Control). Therefore, we had to reduce the αEPSP amplitude in order to reach the AP threshold within the third αEPSP. Again, under control conditions, mAHPs after single APs shunted subsequent αEPSPs, allowing periods of two or three subthreshold αEPSPs without firing, before the mAHP had declined and αEPSP summation again reached the spike threshold (Fig. 9C, 40 Hz, Control). Apamin dramatically enhanced αEPSP summation after the first AP (Fig. 9A, 40 Hz, red), and thus significantly increased the number of APs during the train (Fig. 9B, 40 Hz). In conclusion, we observed that SK channels opened by a single AP effectively shunted subsequent somatic αEPSP summation under conditions of physiological temperature and extracellular potassium.
Kv7/M channels regulate postsynaptic integration by controlling subthreshold excitability in DGCs
Finally, we aimed to test the effects of Kv7/M channel blockade on postsynaptic integration under the same experimental conditions as Fig. 9. We showed before that Kv7/M channel blockade lowers the AP threshold and increases Rinput at subthreshold potentials, although it had little effect on the mAHPs after single APs. Therefore, we hypothesized that these effects may impact postsynaptic integration and EPSP–spike coupling in DGCs.
Again, we injected αEPSC trains in the DGC soma at 10 and 40 Hz (Fig. 10A). XE991 strongly increased the number of suprathreshold αEPSPs (Fig. 10A, green), thus significantly increasing the number of spikes at both frequencies (Fig. 10B, upper panel 10 Hz, 1.86 ± 0.3 APs Control, 8 ± 0.7 APs XE991, n = 7, P < 0.001; Fig. 10B, lower panel 40 Hz, 2.28 ± 0.3 APs Control, 5.14 ± 0.7 APs XE991, n = 7, P < 0.01). Higher magnification of the αEPSP trains (Fig. 10C) showed that XE991 lowered the AP threshold (Fig. 10C, green), consistent with the results shown in Fig. 5.
In order to further clarify whether the increased summation/spiking arises from a combination of different factors and not only from the lowered AP threshold after Kv7/M channel blockade, we applied αEPSPs of smaller amplitude to avoid spiking (Fig. 10D). We found that XE991 significantly increased the amplitude of the first
αEPSP from 4.39 ± 0.3 mV (control) to 6.01 ± 0.6 mV in XE991 (Fig. 10E; n = 6, P < 0.05), as expected from the subthreshold increase in Rinput (Fig. 10B and E). In 2011, Shah et al. reported that Kv7/M channel blockade increased both the αEPSP amplitude and decay time constant in CA1 pyramidal cells. In contrast, we found a small (non-significant) decrease in the decay time of the first αEPSP in the 10 Hz train (Fig. 10E, right, 40.3 ± 8 ms Control, 31.3 ± 5.2 ms XE991; n = 6, P > 0.05). Therefore, the larger summed αEPSPs in XE991 (Fig. 10D) must be mainly due to the increase in αEPSP amplitude that, along with the lowered AP threshold, favours the EPSP–spike coupling. However, the unexpected lack of increase, and even slight reduction, in the αEPSP decay time constant, following Kv7/M channel blockade, suggests that indirect effects on other subthreshold currents affect the αEPSP decay.
Discussion
Complementary roles of SK and Kv7/M potassium channels in DG granule cells
Our main conclusion from this in vitro study is that SK and Kv7/M potassium channels seem to play important and complementary roles in excitability control, postsynaptic integration, and spike frequency adaptation in rat DGCs.
Thus, we found that apamin-sensitive SK channels in DGCs mediate strong negative feedback control of neuronal excitability and signalling, including intracellular [Ca2+]. The SK channels exert this control by generating mAHPs (Figs 1, 2, 4 and 5) regulating the number, frequency and pattern of APs (Figs 7 and 8), and hence the Ca2+ influx into these cells (as inferred from the effect of apamin on the sAHP; Supporting information Fig. S2). These conclusions are based on somatic recordings and tests with the selective SK channel blockers apamin and scyllatoxin, and the SK channel opener 1-EBIO. Thus, SK channels appear to be the main cause of both the mAHP and early spike frequency adaptation in this neuron type, in contrast to other hippocampal principal neurons (see below). SK channels underlie both the mAHPs following single APs during low frequency firing evoked by sustained depolarizing input (Fig. 5), and the mAHP following a high frequency spike train triggered by a single depolarizing pulse (Figs 1, 2 and 4), or by a series of brief depolarizing pulses (Supporting information Fig. S1). However, despite their strong effect on excitability, we found no effect of SK channels on the basal cell Rinput or the spike threshold.
The functions of Kv7/M channels seem to be complementary to those of SK channels in several respects. Thus, unlike SK, the Kv7/M channels are activated already before the first AP and thus regulate both the somatic Rinput and the spike threshold, i.e. a feed-forward type of excitability control. But, again unlike SK channels, the Kv7/M channels seem to be only minimally activated by each AP, and are thus unable to contribute noticeably to the single-spike mAHP. Accordingly, the Kv7/M channels seem to contribute little to the feedback type of excitability control that tipically underlies spike frequency adaptation, and contribute also far less to the post-burst mAHP than the SK channels do.
The DGCs also have a slow, calcium-dependent afterhyperpolarization (sAHP) current that contributes to late spike frequency adaptation (Staley et al. 1992). Indirectly, both SK and Kv7/M channels can also regulate the sAHP through control of spike frequency and thus Ca2+ influx (e.g. Supporting information Fig. S2). However, we found no evidence that SK channels contribute directly to the sAHP and its underlying current (Fig. 1A and C), and it is unclear whether Kv7/M channels may do so (Tzingounis & Nicoll, 2008; Andrade et al. 2012; see below). Our compartmental model seemed unable to reproduce the effects of XE991 on the sAHP in a straightforward, robust manner, that is, unless we supposed the existence of a second XE991-sensitive but Ca2+-activated conductance (Supporting information, section S3 and Fig. S9C), i.e. an XE991-sensitive but retigabine-resistant (Fig. 3A) component of gsAHP, which seems unorthodox (but see Andrade et al. 2012). A characterization of the sAHP is beyond the scope of the present study and will be addressed in a separate project.
Furthermore, our experiments with αEPSCs under near-physiological conditions indicate that the SK and Kv7/M channels also play complementary roles in postsynaptic integration and control of spike latency, spike timing and spike number in these cells. Again, the Kv7/M channels act in a feed-forward manner, decreasing even subthreshold EPSP amplitudes and summation, and raising the spike threshold (Fig. 10). In contrast, the SK channels mediate strictly suprathreshold, feedback control via the mAHP, modulating EPSPs and summation only in the wake of spiking (Fig. 9).
This differential effect on AP threshold is probably related both to the fact that SK channels are Ca2+ activated whereas Kv7/M channels are voltage gated, and to different subcellular localizations of these channels. Thus, SK channel activation in DGCs seems normally to depend on spike-triggered Ca2+ influx causing a sufficient rise in [Ca2+]i near the SK channels for < ∼200 ms (the duration of the SK-dependent mAHP). Therefore they cannot affect the threshold of the first AP after a longer silent period, unlike the Kv7/M channels, which are activated by subthreshold depolarizations. In contrast, SK channels in globus pallidus neurons have been shown to influence the spike threshold (Deister et al. 2009). However, these neurons fire rhythmically at 15–20 Hz even in the absence of synaptic input, and thus receive a regular supply of Ca2+ that keeps their SK channels open, unlike the DGCs, which normally fire at a low rate, below 0.5 Hz, in vivo (Buzsaki & Czeh, 1992; Jung & McNaughton, 1993; Wiebe & Staubli, 1999; Leutgeb et al. 2007). In addition, whereas the Kv7/M channels are concentrated in the AIS, thus being ideally situated to regulate the spike threshold through shunting and polarization (Cooper et al. 2001; Hu et al. 2007; Shah et al. 2008), the SK channels have a largely dendritic or somato-dendritic distribution in many central neurons (Sailer et al. 2002, 2004; Adelman et al. 2012) and are thus less able to affect the spike threshold.
Kv7/M channel blockade by XE991 changed the spike latency during αEPSP trains (Fig. 10), apparently via two mechanisms. By increasing Rinput and by lowering the spike threshold. These effects can be compared to the interplay between D-current and M-current in CA1 pyramidal cells, where IM causes a slow, polarizing sag that counteracts the ID ramp, thus tending to reduce the maximal AP latency in response to minimal (rheobase) stimulation (see Fig. 6A and B in Storm, 1990). However, IM blockade will of course always tend to reduce the AP latency in response to constant excitatory input, even in CA1, although it may still increase the maximal spike latency when the input current is reduced to rheobase, allowing other delaying processes (slow ID ramp or EPSP summation) to increase the delay (Storm, 1990).
The mAHP in DG granule cells is mainly generated by SK channels; comparison to other cell types
SK channels are expressed in many parts of the vertebrate nervous system, and have been found to underlie mAHPs, spike frequency adaptation, and excitability control in a variety of neuron types (for review, see: Vogalis et al. 2003; Fakler & Adelman, 2008; Faber, 2009; Adelman et al. 2012). SK channels also play such roles in neurons of the medial temporal lobe memory system, including some hippocampal interneurons (Lawrence et al. 2006) and subicular pyramidal cells (Gu et al. 2005). In hippocampal CA1 pyramidal cells, however, convergent evidence indicates that the mAHP and the related excitability control are generated primarily by Kv7/M channels, with little or no SK contribution (Storm, 1989; Williamson & Alger, 1990; Gu et al. 2005, 2008; Peters et al. 2005), although some reports concluded differently (Stocker et al. 1999).
Thus, curiously, the relative importance of SK and Kv7/M channels in feedback spike frequency adaptation and mAHPs seem to be almost exactly opposite in CA1 pyramidal cells and DGCs: while Kv7/M channels dominate these forms of feedback regulation in CA1 pyramids, SK channels dominate in DGCs. In this respect, the DGCs also differ from some other ‘prototype’ vertebrate neurons, including frog sympathetic ganglion cells, where both Kv7/M and SK channels are jointly important for spike frequency adaptation, in a highly synergistic manner (Adams et al. 1986; Storm, 1989; Gu et al. 2005; Peters et al. 2005).
The main root of these differences in SK and Kv7/M channel functions between neuron types seems to be differences in how efficiently each channel type is activated by an action potential. Thus, in CA1 pyramidal cells, our group previously found that M-current is substantially activated by normal Na+ spikes and therefore generates a spike-dependent mAHP and feedback spike frequency adaptation (Storm, 1989; Gu et al. 2008). This is in contrast to both SK channels, which are activated by Ca2+ spikes but not by Na+ spikes in CA1 pyramidal cells, and the h/HCN channel-dependent mAHP component that is mainly due to subthreshold depolarizations (Storm, 1989; Gu et al. 2008). In frog sympathetic ganglion cells, two-electrode voltage clamp experiments showed activation of both SK and BK but not Kv7/M channels by brief (2 ms long), large, spike-like depolarizations (Lancaster & Pennefather, 1987).
Why, then, are Kv7/M channels apparently not substantially activated by spikes in DGCs, whereas SK channels are? Probably, the duration of the Na+ spikes in this cell type is too brief to allow the Kv7/M channels to activate appreciably (Fig. 6D and Supporting information Fig. S8B) given the comparatively slow gM kinetics suggested by our modelling studies (Supporting information, section S4 and Fig. S6). Thus, the AP duration (measured at half-amplitude) is only 0.6 ms (Geiger & Jonas, 2000) in rat DGCs (34°C), compared to 0.95 ms (Staff et al. 2000) in rat CA1 pyramidal cells (35°C) and ∼2.5 ms in bullfrog sympathetic ganglion neurons (22°C; Adams et al. 1982). That SK channels are nevertheless activated sufficiently by a single DGC spike to generate a robust mAHP (Fig. 5), must presumably be due to a more efficient Ca2+ channel activation or coupling to SK channels in these cells than in CA1 pyramidal cells.
Furthermore, the absence of an SK contribution to the mAHP in CA1 pyramidal cells may perhaps in part reflect their larger dendritic arbor and a greater soma-to-dendrite voltage attenuation compared with DGCs (Supporting information, section S4 and Fig. S10). Simulations using a CA1 pyramidal cell model (based on that of Hu et al. 2009; Supporting information, section S5) suggest that these cells produce a Kv7/M-generated mAHP by having a larger effective somatic M-conductance, faster gM kinetics and broader APs than DGCs (Supporting information, section S4 and Figs S6C and S11).
Comparison with previous recordings from DG granule cells
Previously, a small apamin-sensitive SK current, ISK, was recorded by somatic whole-cell voltage clamp in rat (Sailer et al. 2002) and human DGCs (Beck et al. 1997), apparently in reasonable agreement with the small size of these cells, and the low or moderate levels of SK1–3 mRNA (Stocker & Pedarzani, 2000) and SK1–3 protein (Sailer et al. 2002, 2004) detected in the DGC layer. Furthermore, a recent study using KCNQ3–/– and KCNQ2+/– knock-out mice concluded that Kv7.2/Kv7.3-dependent M channels (KCNQ2/3) generate more than half of the so-called ‘mAHP current’ (ImAHP) in mouse DGCs, suggesting that IM is an important generator of the mAHP in this cell type (Tzingounis & Nicoll, 2008). However, because of the small size and high Rinput (mean ∼200 MΩ; Fig. 7B) of the DGCs, a small SK current of about 10–50 pA is sufficient to generate the observed somatic mAHP amplitudes of ∼3–5 mV (∼4 mV/200 MΩ = ∼20 pA). Furthermore, it should be recalled that somatic whole-cell voltage clamp recordings from cells with extensive dendritic and axonal processes are hard to interpret because of severe space clamp limitations (Williams & Mitchell, 2008), in particular when the channels studied are likely to be primarily non-somatic, such as SK channels (found at high density in dendritic spines of hippocampal and neocortical pyramidal cells; Ngo-Anh et al. 2005; Ballesteros-Merino et al. 2012) and Kv7/M channels (concentrated in the axon/AIS of DGCs and cortical pyramidal cells; Cooper et al. 2001; Devaux et al. 2004; Klinger et al. 2011). Voltage clamp recordings of Ca2+-activated currents are particularly problematic, because of unclamped Ca2+ spikes and uncontrolled feedback (Pedarzani & Storm, 1993). To avoid these complications, we relied mainly on current clamp recordings in this study (Figs 10).
Although characterization of the sAHP is beyond the scope of the present study, the apparently paradoxical effects of XE991 and retigabine on the sAHP shown in Fig. 3 deserve some comments. Why did both XE991 and retigabine seem to reduce the sAHP, although these drugs have opposite effects on Kv7/M channels? The sAHP reduction by XE991 may seem to support the idea that the sAHP in DGCs is partly generated by Kv7/M channels (Tzingounis & Nicoll, 2008; Andrade et al. 2012). However, this hypothesis predicts that the sAHP should be enhanced by the Kv7/M channel opener retigabine, whereas we found a reduction. It might be suggested that retigabine perhaps opened the Kv7/M channels so much already before the spike train that it partly occluded further opening after the spikes. Our modelling supports this interpretation and suggests that the effect of retigabine on the mAHP depends on the size of the increase in gM during the spike train. The effect of retigabine on the sAHP may also be (partly) indirect, via an effect on the Ca2+ influx that triggers the sAHP. Thus, Kv7/M channels opened by retigabine may reduce the opening of voltage-gated Ca2+ channels (Cav) through local shunting and polarization, which may not be visible in the soma. Our modelling is basically consistent with this, although a small reduction in AP amplitude can be seen at the soma. Unless gsAHP is partly blocked by XE991 (Andrade et al. 2012; Supporting information, section S3), even the curious, apparent reduction of the sAHP by XE991 (Fig. 3A and C) might perhaps be indirect. For example, by blocking IM, XE991 might possibly cause depolarization in non-somatic parts of the cell, causing inactivation of low-threshold Cav channels that trigger gsAHP (Blaxter et al. 1989; Vervaeke et al. 2006).
Functional implications
In vivo recordings show that DGCs fire at low frequencies during a variety of behavioural tasks, e.g. with mean rates of 0.01–0.5 Hz in a spatial memory task (Jung & McNaughton, 1993; Wiebe & Staubli, 1999; Leutgeb et al. 2007). When the animal traverses a place field of a DGC, it can fire at frequencies up to ∼30 Hz (Leutgeb et al. 2007), phase-locked to theta oscillations (Skaggs et al. 1996). The spike transmission probability from a DGC to a CA3 pyramidal cell varies from single APs to spike bursts, in a frequency-dependent fashion (Henze et al. 2002). Recordings from connected pairs of DGC–CA3 pyramidal cells in hippocampal slice cultures showed that different network mechanisms can be recruited depending on the granule cell firing pattern, which strongly regulates the information flow from DG to CA3 (Mori et al. 2004, 2007). This suggests that the intrinsic mechanisms controlling excitability and spike frequency adaptation may be important for regulating the network mechanisms triggered by the DGC activity.
If the SK and Kv7/M channels of DGCs are similarly activated in vivo as we have found in this in vitro study, they may contribute to the regulation of functionally important parameters. Thus, by modulating both EPSP summation and spike threshold in response to trains of synaptic input, Kv7/M channels in DGCs regulate the initial discharge latency (delay) and thus spike timing (Fig. 10), which is crucial for Hebbian spike-timing-dependent plasticity (STDP) such as LTP and LTD (Caporale & Dan, 2008). In particular, this may regulate LTP at the perforant path (PP) synapses between MEC layer 2 stellate cells and DGC dendrites (Bliss & Lømo, 1973), which carry EC grid cell information to the hippocampus (Moser et al. 2008; Treves et al. 2008). Also SK channels can regulate spike timing, albeit only for spikes with a frequency of at least ∼5 Hz (corresponding to a mAHP duration of ∼200 ms), but not for the first spike in a burst (Fig. 9). The regulation of spike delay by K+ channels may also be relevant for how information in single DGCs is converted into a ‘temporal delay code’ in target CA3 pyramidal cells and interneurons (Henze et al. 2002).
Since both SK and Kv7/M channels regulate spike number and frequency of DGCs in response to αEPSC trains (Figs 9 and 10), these channels may regulate NMDA-dependent Hebbian STDP/LTP at their PP input synapses, as well as transmission and plasticity at their output synapses from DGCs to CA3 neurons via MF synapses. In vivo recordings in anaesthetized rats indicate that the MF synapses function as ‘conditional detonators’, dependent on the DGC firing pattern. Thus, single spikes generally failed to discharge CA3 pyramidal neurons and interneurons, whereas spike trains effectively discharged both cell types (Henze et al. 2002). The NMDA receptor (-R)-independent presynaptic MF-LTP is highly spike frequency dependent (Nicoll & Schmitz, 2005) and may therefore also be regulated by SK and Kv7/M channels (Figs 9 and 10). Finally, in addition to the SK channel functions that we have studied here by somatic recordings, it is possible that SK channels may be coupled to postsynaptic NMDA-Rs in DGC dendrites, and thereby regulate synaptic plasticity also in a local manner, as previously reported for hippocampal and amygdala pyramidal cells (Faber et al. 2005; Ngo-Anh et al. 2005).
The impact of SK and Kv7/M channels and other intrinsic DGC mechanisms in vivo will probably depend on the behavioural state of the animal, and thus the functional state of the hippocampal system. In particular, Kv7/M channels are highly regulated by acetylcholine and several other transmitters that vary with the arousal level, attention, and other behavioural states (Cobb & Lawrence, 2010). Other synaptic bombardment will also alter the impact of intrinsic mechanisms (Destexhe, 2010). This will have to be determined by future in vivo studies. However, available results from in vivo manipulations of SK and Kv7/M channels suggest that both channel types do in fact regulate hippocampal functions. In particular, several studies have shown that systemic administration of apamin or knock-out of SK channel genes facilitates different forms of hippocampus-dependent learning and memory (Messier et al. 1991; Deschaux et al. 1997; Ikonen & Riekkinen, 1999; Stackman et al. 2002; Deschaux & Bizot, 2005; Hammond et al. 2006; Brennan et al. 2008), and enhances synaptic plasticity (Behnisch & Reymann, 1998). Although the effects on memory have often been attributed to SK channels and AHPs in CA1 neurons (Norris et al. 1998; Stackman et al. 2002), and particularly to NMDA-R-coupled SK channels in CA1 dendritic spines (Ngo-Anh et al. 2005; Hammond et al. 2006; Lin et al. 2008), some of these effects may actually be mediated by SK channels and SK-dependent mAHPs and excitability control of the DGCs. Thus, apamin and SK channel knock-out would increase the excitability, spike number and frequency of the DGCs, and SK channels in dendritic spines of DGCs may also contribute through NMDA-R-dependent activation mechanisms, as reported for CA1 pyramidal neurons (Ngo-Anh et al. 2005; Hammond et al. 2006; Lin et al. 2008).
Suppression or enhancement of Kv7/M channel activity by genetic manipulations (Peters et al. 2005) have shown that this channel type has a strong impact on hippocampus-dependent memory. Since Kv7/M channels are involved in multiple processes in several cell types, it is likely that the mechanisms underlying the observed behavioural effects are complex and varied. This complexity may explain the seemingly opposite effects of Kv7/M channel suppression in different experiments, where both improved (‘cognitive enhancement’) and impaired memory have been reported (see e.g. Peters et al. 2005; Fontán-Lozano et al. 2011). The Kv7/M channel functions that we found in DGCs may contribute to this complexity. Thus, the increased discharge of DGCs after Kv7/M channel blockade would by itself be expected to enhance Hebbian plasticity (‘cognitive enhancement’), whereas disruption of proper spike timing (Fig. 10) might reduce coincidence detection and STDP.
Key points
Previous studies showed that different firing patterns of hippocampal dentate granule cells (DGCs) can trigger different network responses. However, the intrinsic DGC mechanisms controlling their excitability, spike patterns and synaptic integration in DGCs, remain poorly understood.
SK and Kv7/M channels play important roles controlling neuronal integration and excitability, but their specific roles vary between cell types. Both channel types are expressed in DGCs, but their roles are unclear.
We found that SK channels are the main generators of medium afterhyperpolarizations in DGCs, thus causing negative feedback regulation of spiking (spike frequency adaptation) and calcium influx.
In contrast, Kv7/M perform subthreshold and ‘feed-forward’ control of input resistance, postsynaptic integration, action potential threshold and excitability, thus weakening EPSP–spike coupling.
Thus, in DGCs, the SK and Kv7/M channels seem to perform complementary functions in postsynaptic integration and excitability control. This may have important consequences for dentate network physiology.
Acknowledgments
We thank members of the Storm laboratory for helpful comments and discussions on the manuscript.
Glossary
- αEPSCs
artificial excitatory postsynaptic currents
- ADP
afterdepolarization
- AHP
afterhyperpolarization
- AIS
axon initial segment
- AP
action potential
- DG
dentate gyrus
- DGC
dentate granule cell
- fAHP
fast afterhyperpolarization
- gM
M-conductance
- gSK
SK-conductance
- IaAHP
apamin-sensitive afterhyperpolarization current
- Ih
H-current
- IM
M-current
- ImAHP
mAHP-current
- IsAHP
sAHP-current
- ISI
interspike interval
- ISK
SK-current
- LTP
long-term potentiation
- mAHP
medium afterhyperpolarization
- MEC
medial entorhinal cortex
- MF
mossy fibre
- NMDA-R
NMDA receptor
- PP
perforant path
- Rinput
input resistance
- RMP
resting membrane potential
- sADP
slow afterdepolarization
- sAHP
slow afterhyperpolarization
- STDP
spike timing-dependent plasticity
- Vhalf
voltage for half-maximal conductance, τ, time constant.
Additional information
Competing interests
The authors declare no conflicts of interests.
Author contributions
P.M.-A. and J.F.S. conceived and designed the experiments; P.M.-A. performed the experiments and data analysis; R.M. constructed the computational model and performed the simulations. P.M.-A., R.M. and J.F.S. wrote the manuscript. The authors approved the manuscript for publication.
Funding
This study was supported by the Norwegian Research Council and the University of Oslo.
References
- Adams PR, Constanti A, Brown DA, Clark RB. Intracellular Ca2+ activates a fast voltage-sensitive K+ current in vertebrate sympathetic neurones. Nature. 1982;296:746–749. doi: 10.1038/296746a0. [DOI] [PubMed] [Google Scholar]
- Adams PR, Jones SW, Pennefather P, Brown DA, Koch C, Lancaster B. Slow synaptic transmission in frog sympathetic ganglia. J Exp Biol. 1986;124:259–285. doi: 10.1242/jeb.124.1.259. [DOI] [PubMed] [Google Scholar]
- Adelman JP, Maylie J, Sah P. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245–269. doi: 10.1146/annurev-physiol-020911-153336. [DOI] [PubMed] [Google Scholar]
- Alle H, Ostroumov K, Geiger J, Storm JF. Neuroscience Meeting Planner. Chicago, IL, USA: Society for Neuroscience; 2009. M-current, persistent Na+ current, and subthreshold resonance recorded in mossy fiber boutons (MFBs) in rat hippocampus. Program No. 42.2/D10. 2009 Online. [Google Scholar]
- Andrade R, Foehring RC, Tzingounis AV. The calcium-activated slow AHP: cutting through the Gordian knot. Front Cell Neurosci. 2012;6:47. doi: 10.3389/fncel.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aradi I, Holmes WR. Role of multiple calcium and calcium-dependent conductances in regulation of hippocampal dentate granule cell excitability. J Comput Neurosci. 1999;6:215–235. doi: 10.1023/a:1008801821784. [DOI] [PubMed] [Google Scholar]
- Auguste P, Hugues M, Mourre C, Moinier D, Tartar A, Lazdunski M. Scyllatoxin, a blocker of Ca2+-activated K+ channels: structure-function relationships and brain localization of the binding sites. Biochemistry. 1992;31:648–654. doi: 10.1021/bi00118a003. [DOI] [PubMed] [Google Scholar]
- Ballesteros-Merino C, Lin M, Wu WW, Ferrandiz-Huertas C, Cabanero MJ, Watanabe M, Fukazawa Y, Shigemoto R, Maylie J, Adelman JP, Lujan R. Developmental profile of SK2 channel expression and function in CA1 neurons. Hippocampus. 2012;22:1467–1480. doi: 10.1002/hipo.20986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SJ, Opitz T, Merkens M, Kelly T, von der Brelie C, Krueppel R, Beck H. Stable mossy fiber long-term potentiation requires calcium influx at the granule cell soma, protein synthesis, and microtubule-dependent axonal transport. J Neurosci. 2010;30:12996–13004. doi: 10.1523/JNEUROSCI.1847-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Beck H, Clusmann H, Kral T, Schramm J, Heinemann U, Elger CE. Potassium currents in acutely isolated human hippocampal dentate granule cells. J Physiol. 1997;498:73–85. doi: 10.1113/jphysiol.1997.sp021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnisch T, Reymann KG. Inhibition of apamin-sensitive calcium dependent potassium channels facilitate the induction of long-term potentiation in the CA1 region of rat hippocampus in vitro. Neurosci Lett. 1998;253:91–94. doi: 10.1016/s0304-3940(98)00612-0. [DOI] [PubMed] [Google Scholar]
- Blaxter TJ, Carlen PL, Niesen C. Pharmacological and anatomical separation of calcium currents in rat dentate granule neurones in vitro. J Physiol. 1989;412:93–112. doi: 10.1113/jphysiol.1989.sp017605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann N Y Acad Sci. 1999;868:370–378. doi: 10.1111/j.1749-6632.1999.tb11298.x. [DOI] [PubMed] [Google Scholar]
- Brennan AR, Dolinsky B, Vu MA, Stanley M, Yeckel MF, Arnsten AF. Blockade of IP3-mediated SK channel signaling in the rat medial prefrontal cortex improves spatial working memory. Learn Mem. 2008;15:93–96. doi: 10.1101/lm.767408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Czéh G. Physiological function of granule cells: a hypothesis. Epilepsy Res Suppl. 1992;7:281–290. [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Carnevale NT, Hines ML. The NEURON Book. Cambridge, NY: Cambridge UP; 2006. [Google Scholar]
- Cobb S, Lawrence JJ. Neuromodulation of hippocampal cells and circuits. In: Cutsuridis V, Graham B, Cobb S, Vida I, editors. Hippocampal Microcircuits. New York: Springer; 2010. pp. 187–246. [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deister CA, Chan CS, Surmeier DJ, Wilson CJ. Calcium-activated SK channels influence voltage-gated ion channels to determine the precision of firing in globus pallidus neurons. J Neurosci. 2009;29:8452–8461. doi: 10.1523/JNEUROSCI.0576-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschaux O, Bizot JC. Apamin produces selective improvements of learning in rats. Neurosci Lett. 2005;386:5–8. doi: 10.1016/j.neulet.2005.05.050. [DOI] [PubMed] [Google Scholar]
- Deschaux O, Bizot JC, Goyffon M. Apamin improves learning in an object recognition task in rats. Neurosci Lett. 1997;222:159–162. doi: 10.1016/s0304-3940(97)13367-5. [DOI] [PubMed] [Google Scholar]
- Destexhe A. Inhibitory “noise. Front Cell Neurosci. 2010;4:9. doi: 10.3389/fncel.2010.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES. Functions and modulation of neuronal SK channels. Cell Biochem Biophys. 2009;55:127–139. doi: 10.1007/s12013-009-9062-7. [DOI] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- Fakler B, Adelman JP. Control of KCa channels by calcium nano/microdomains. Neuron. 2008;59:873–881. doi: 10.1016/j.neuron.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Fontan-Lozano A, Suarez-Pereira I, Delgado-Garcia JM, Carrion AM. The M-current inhibitor XE991 decreases the stimulation threshold for long-term synaptic plasticity in healthy mice and in models of cognitive disease. Hippocampus. 2011;21:22–32. doi: 10.1002/hipo.20717. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Gu N, Hu H, Vervaeke K, Storm JF. SK (KCa2) channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J Neurophysiol. 2008;100:2589–2604. doi: 10.1152/jn.90433.2008. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Graham LJ, Storm JF. Complementary theta resonance filtering by two spatially segregated mechanisms in CA1 hippocampal pyramidal neurons. J Neurosci. 2009;29:14472–14483. doi: 10.1523/JNEUROSCI.0187-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol. 2002;545:783–805. doi: 10.1113/jphysiol.2002.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen S, Riekkinen P., Jr Effects of apamin on memory processing of hippocampal-lesioned mice. Eur J Pharmacol. 1999;382:151–156. doi: 10.1016/s0014-2999(99)00616-0. [DOI] [PubMed] [Google Scholar]
- Jung MW, McNaughton BL. Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus. 1993;3:165–182. doi: 10.1002/hipo.450030209. [DOI] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft J, Spruston N. Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. J Physiol. 2007;578:799–818. doi: 10.1113/jphysiol.2006.124586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernell D. The repetitive impulse discharge of a simple neurone model compared to that of spinal motoneurones. Brain Res. 1968;11:685–687. doi: 10.1016/0006-8993(68)90157-1. [DOI] [PubMed] [Google Scholar]
- Klinger F, Gould G, Boehm S, Shapiro MS. Distribution of M-channel subunits KCNQ2 and KCNQ3 in rat hippocampus. Neuroimage. 2011;58:761–769. doi: 10.1016/j.neuroimage.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Adams PR. Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J Neurophysiol. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Pennefather P. Potassium currents evoked by brief depolarizations in bull-frog sympathetic ganglion cells. J Physiol. 1987;387:519–548. doi: 10.1113/jphysiol.1987.sp016587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GM, Storm JF. Interaction between synaptic excitation and slow afterhyperpolarization current in rat hippocampal pyramidal cells. J Physiol. 2001;536:809–823. doi: 10.1111/j.1469-7793.2001.00809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanthorn T, Storm J, Andersen P. Current-to-frequency transduction in CA1 hippocampal pyramidal cells: slow prepotentials dominate the primary range firing. Exp Brain Res. 1984;53:431–443. doi: 10.1007/BF00238173. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Saraga F, Churchill JF, Statland JM, Travis KE, Skinner FK, McBain CJ. Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. J Neurosci. 2006;26:12325–12338. doi: 10.1523/JNEUROSCI.3521-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI. Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science. 2007;315:961–966. doi: 10.1126/science.1135801. [DOI] [PubMed] [Google Scholar]
- Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J. SK2 channel plasticity contributes to LTP at Schaffer collateral–CA1 synapses. Nat Neurosci. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tilwalli S, Ye G, Lio PA, Pasternak JF, Trommer BL. Morphologic and electrophysiologic maturation in developing dentate gyrus granule cells. Brain Res. 2000;856:202–212. doi: 10.1016/s0006-8993(99)02421-x. [DOI] [PubMed] [Google Scholar]
- Lømo T. Potentiation of monosynaptic EPSPs in cortical cells by single and repetitive afferent volleys. J Physiol. 1968;194:84–85P. [PubMed] [Google Scholar]
- Lubke J, Frotscher M, Spruston N. Specialized electrophysiological properties of anatomically identified neurons in the hilar region of the rat fascia dentata. J Neurophysiol. 1998;79:1518–1534. doi: 10.1152/jn.1998.79.3.1518. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature. 1982;299:636–638. doi: 10.1038/299636a0. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA1 pyramidal neurones in vitro. J Physiol. 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol. 1986;372:221–244. doi: 10.1113/jphysiol.1986.sp016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- Mateos-Aparicio P, Tuvnes FA, Storm JF. SK channels underlying medium afterhyperpolarizations and excitability control in dentate gyrus granule cells. FENS Abstr. 2010a;5:013.26. [Google Scholar]
- Mateos-Aparicio P, Tuvnes FA, Storm JF. SK channels underlying medium afterhyperpolarizations and excitability control in dentate gyrus granule cells. San Diego, CA, USA: Society for Neuroscience; 2010b. Program No. 341.20/G5. 2010 Neuroscience Meeting Planner. Online. [Google Scholar]
- Messier C, Mourre C, Bontempi B, Sif J, Lazdunski M, Destrade C. Effect of apamin, a toxin that inhibits Ca2+-dependent K+ channels, on learning and memory processes. Brain Res. 1991;551:322–326. doi: 10.1016/0006-8993(91)90950-z. [DOI] [PubMed] [Google Scholar]
- Mori M, Abegg MH, Gahwiler BH, Gerber U. A frequency-dependent switch from inhibition to excitation in a hippocampal unitary circuit. Nature. 2004;431:453–456. doi: 10.1038/nature02854. [DOI] [PubMed] [Google Scholar]
- Mori M, Gahwiler BH, Gerber U. Recruitment of an inhibitory hippocampal network after bursting in a single granule cell. Proc Natl Acad Sci U S A. 2007;104:7640–7645. doi: 10.1073/pnas.0702164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser EI, Kropff E, Moser MB. Place cells, grid cells, and the brain's spatial representation system. Annu Rev Neurosci. 2008;31:69–89. doi: 10.1146/annurev.neuro.31.061307.090723. [DOI] [PubMed] [Google Scholar]
- Navratilova Z, Giocomo LM, Fellous JM, Hasselmo ME, McNaughton BL. Phase precession and variable spatial scaling in a periodic attractor map model of medial entorhinal grid cells with realistic after-spike dynamics. Hippocampus. 2012;22:772–789. doi: 10.1002/hipo.20939. [DOI] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci. 2005;8:642–649. doi: 10.1038/nn1449. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Krause M, Haug T, Storm JF, Stuhmer W. Modulation of the Ca2+-activated K+ current sIAHP by a phosphatase-kinase balance under basal conditions in rat CA1 pyramidal neurons. J Neurophysiol. 1998;79:3252–3256. doi: 10.1152/jn.1998.79.6.3252. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Kulik A, Muller M, Ballanyi K, Stocker M. Molecular determinants of Ca2+-dependent K+ channel function in rat dorsal vagal neurones. J Physiol. 2000;527:283–290. doi: 10.1111/j.1469-7793.2000.t01-1-00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, McCutcheon JE, Rogge G, Jensen BS, Christophersen P, Hougaard C, Strobaek D, Stocker M. Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current IAHP and modulates the firing properties of hippocampal pyramidal neurons. J Biol Chem. 2005;280:41404–41411. doi: 10.1074/jbc.M509610200. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, Stocker M, Adelman JP, Fakler B. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J Biol Chem. 2001;276:9762–9769. doi: 10.1074/jbc.M010001200. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Pennefather P, Lancaster B, Adams PR, Nicoll RA. Two distinct Ca-dependent K currents in bullfrog sympathetic ganglion cells. Proc Natl Acad Sci U S A. 1985;82:3040–3044. doi: 10.1073/pnas.82.9.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Sailer CA, Hu H, Kaufmann WA, Trieb M, Schwarzer C, Storm JF, Knaus HG. Regional differences in distribution and functional expression of small-conductance Ca2+-activated K+ channels in rat brain. J Neurosci. 2002;22:9698–9707. doi: 10.1523/JNEUROSCI.22-22-09698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer CA, Kaufmann WA, Marksteiner J, Knaus HG. Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol Cell Neurosci. 2004;26:458–469. doi: 10.1016/j.mcn.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Scharfman H. The dentate gyrus: a comprehensive guide to structure, function, and clinical implications. Prog Brain Res. 2007;163:1–840. [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429:184–187. doi: 10.1038/nature02553. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Brown DA. Differential effects of Kv7 (M-) channels on synaptic integration in distinct subcellular compartments of rat hippocampal pyramidal neurons. J Physiol. 2011;589:6029–6038. doi: 10.1113/jphysiol.2011.220913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaggs WE, McNaughton BL, Wilson MA, Barnes CA. Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus. 1996;6:149–172. doi: 10.1002/(SICI)1098-1063(1996)6:2<149::AID-HIPO6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff NP, Jung HY, Thiagarajan T, Yao M, Spruston N. Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol. 2000;84:2398–2408. doi: 10.1152/jn.2000.84.5.2398. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Otis TS, Mody I. Membrane properties of dentate gyrus granule cells: comparison of sharp microelectrode and whole-cell recordings. J Neurophysiol. 1992;67:1346–1358. doi: 10.1152/jn.1992.67.5.1346. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Pedarzani P. Differential distribution of three Ca2+-activated K+ channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci. 2000;15:476–493. doi: 10.1006/mcne.2000.0842. [DOI] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J Physiol. 1989;409:171–190. doi: 10.1113/jphysiol.1989.sp017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Traub RD, Whittington MA, Colling SB, Buzsáki G, Jefferys JG. Analysis of gamma rhythms in the rat hippocampus in vitro and in vivo. J Physiol. 1996;493:471–484. doi: 10.1113/jphysiol.1996.sp021397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves A, Tashiro A, Witter MP, Moser EI. What is the mammalian dentate gyrus good for? Neuroscience. 2008;154:1155–1172. doi: 10.1016/j.neuroscience.2008.04.073. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogalis F, Storm JF, Lancaster B. SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur J Neurosci. 2003;18:3155–3166. doi: 10.1111/j.1460-9568.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- Wiebe SP, Staubli UV. Dynamic filtering of recognition memory codes in the hippocampus. J Neurosci. 1999;19:10562–10574. doi: 10.1523/JNEUROSCI.19-23-10562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Williamson A, Alger BE. Characterization of an early afterhyperpolarization after a brief train of action potentials in rat hippocampal neurons in vitro. J Neurophysiol. 1990;63:72–81. doi: 10.1152/jn.1990.63.1.72. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Yaari Y. Axo-somatic and apical dendritic Kv7/M channels differentially regulate the intrinsic excitability of adult rat CA1 pyramidal cells. J Neurophysiol. 2006;95:3480–3495. doi: 10.1152/jn.01333.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.