

Abstract
HIV-1 uses its trimeric gp160 envelope (Env) protein consisting of non-covalently associated gp120 and gp41 subunits to mediate entry into human T lymphocytes. A facile virus fusion mechanism compensates for the sparse Env copy number observed on viral particles and includes a 22 amino acid lentivirus-specific adaptation at the gp41 base (amino acid residues 662–683), termed the membrane proximal external region (MPER). We show by NMR and EPR that the MPER consists of a structurally conserved pair of viral lipid-immersed helices separated by a hinge with tandem joints that can be locked by capping residues between helices. This design fosters efficient HIV-1 fusion via inter-converting structures while at the same time affording immune escape. Disruption of both joints by double alanine mutations at Env positions 671 and 674 (AA) results in attenuation of Env-mediated cell-cell fusion and hemifusion as well as viral infectivity mediated by both CD4-dependent and CD4-independent viruses. The potential mechanism of disruption was revealed by structural analysis of MPER conformational changes induced by AA mutation. A deeper acyl chain-buried MPER middle section and the elimination of cross-hinge rigid-body motion almost certainly impede requisite structural rearrangements during the fusion process, explaining the absence of MPER AA variants among all known naturally occurring HIV-1 viral sequences. Furthermore, those broadly neutralization antibodies directed against the HIV-1 MPER exploit the tandem joint architecture involving helix-capping, thereby disrupting hinge function.
Keywords: viral membrane fusion, NMR solution structure, helix-hinge-helix motif, helix capping, broadly neutralizing antibody
Introduction
Lentiviruses such as HIV-1, the causative agent of acquired immunodeficiency syndrome (AIDS), are encapsulated in a membrane derived from the infected host cell as virus buds (reviewed in1; 2). A trimeric Env gp160 spike consisting of three pairs of non-covalent associated gp120 and gp41 subunits is the only viral protein on the HIV-1 membrane. The Env gp120 mediates attachment and entry into human CD4+ T lymphocytes upon binding its primary cellular receptor, CD4, and CCR5 or CXCR4 co-receptor. Viral infectivity is dependent on membrane fusion between HIV-1 and the host cell through formation of a gp41 six-helix bundle complex3; 4; 5. The efficiency of this mechanism is especially critical, given fewer than 12 copies of Env clustered on each viral particle6.
The MPER is a tryptophan rich segment located at the base of the gp41 subunit, and appears to destabilize the viral membrane during the fusion process7; 8. Deletion of the HIV-1 MPER, or concurrent mutation of three conserved tryptophan residues on its N-terminal helix to alanines, abolishes membrane fusion activity9; 10. However, the presence of these conserved tryptophan alone is not sufficient to support the viral fusion activity8. Other MPER residues, even the exposed, primarily hydrophilic ones, may also be involved despite sequence variability therein. Previously, we solved the solution structure of a clade B HxB2 strain MPER peptide in detergent micelle with an unusual helix-hinge-helix motif11. Interestingly, the central hinge region is the target of several broadly neutralizing antibodies (BNAb), including 4E10, 10E8 and Z13e112; 13; 14. Here, we provide detailed structural and functional results on this specialized hinge region relating to its potentially important role during the intermediate stages of the HIV membrane fusion process.
Results and Discussion
MPER sequence conservation and limited variability
Bioinformatics studies show that while considerably conserved within each lentivirus group, the MPER sequence from HIV-1 and its ancestor SIV-CPZ (chimpanzee) are distinctive from those of HIV-2 and their related SIV-MAC (macaque) and SIV-AGM (African green monkey) sequences, and distant from non-primate lentiviruses (Fig. 1). As shown in Figures 1b and 2a, the HIV-1 MPER is highly conserved across different clades. Structurally, the clade B HxB2 MPER peptide in dodecyl-phosphocholine (DPC) detergent micelle shows a helix-hinge-helix motif dictated by the segment's unique amphipathic pattern (Fig. 2b), with the membrane-buried residues mostly conserved and solvent-exposed residues relatively more variable. This pattern is prominent at the central hinge region, where the two key epitope residues for BNAb 4E10 and 10E8, W672 and F673 are essentially invariant and buried in the membrane, while the exposed N671 and N/D674, key epitope residues for Z13e1, manifest significant sequence variability (Figs. 2b and 2c). Using the “Motif-finder” software15 and Los Alamos HIV database, we have identified all 36 amino acid residue combinations at the 671 and 674 positions. This is a surprisingly small number taken from among 21,967 HIV-1 strain sequences across all major clades (Fig. 2c), and the residue types are dominated by asparagine, aspartate, serine, and threonine (NDST set) (Table 1).
Fig. 1.
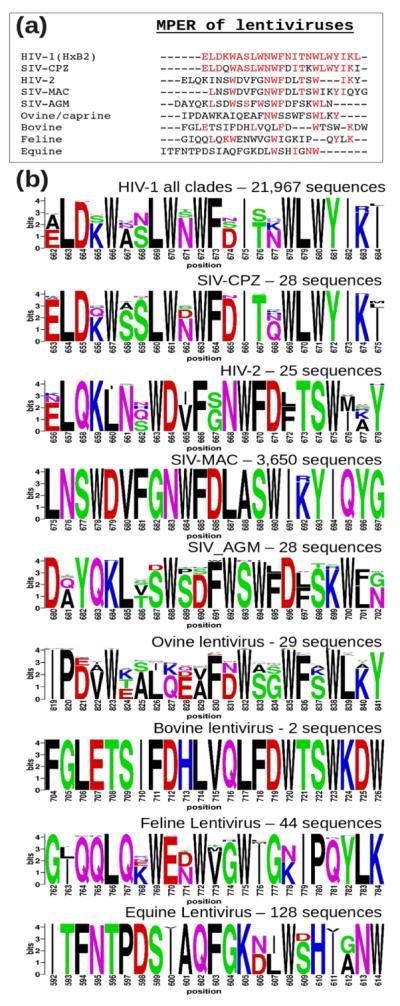
Comparison of MPER segments from 9 groups of lentiviruses. The 23 amino acid long lentiviral segments N-terminal to their respective annotated transmembrane helices were extracted from SwissProt Database. (a) Sequence alignment with colored residues being identical to the reference sequence (HIV-1 HxB2). (b) Logos showing conservation within each of the 9 lentivirus groups. The extremely conserved 3,650 SIV sequences from rhesus macaque (SIV-MAC) is a result of infection in research primate centers.
Fig. 2.

Sequence conservation and variation of HIV-1 MPER. (a) Amino acid sequences of MPER peptides used for structural studies, with conserved residues colored in red. (b) HxB2 MPER structure in a DPC detergent micelle with conserved and variable residues colored according to the scale shown. (c) Population of amino acid combinations at the 671 and 674 residue positions at the central hinge region, with BlockLogo sequence conservation diagram of the 671–674 segment shown as an insert at the top with conventional Shannon entropy representation of individual amino acid position variability at the bottom.
Table 1.
Sequence (singly) variability of residues 671 and 674 in all HIV-1 strains. More than 96.5% of the residue pairs (doubly) in the MPER of 21,967 HIV-1 strains are exclusively from the NSTD set (shaded); and all but two pairs had at least one residue from this set.
| Residue 671 | ||||
| Rank | Amino Acid | Frequency | Accumulated frequency | Sequences |
|
| ||||
| 1 | N | 78.42% | 78.42% | 17226 |
| 2 | S | 14.91% | 93.33% | 3276 |
| 3 | T | 5.99% | 99.32% | 1316 |
| 4 | D | 0.64% | 99.96% | 140 |
| 5 | X | 0.02% | 99.98% | 4 |
| 6 | G | 0.01% | 99.99% | 3 |
| 7 | H | 0.00% | 100.00% | 1 |
| 8 | K | 0.00% | 100.00% | 1 |
|
| ||||
| Residue 674 | ||||
| Rank | Amino Acid | Frequency | Accumulated frequency | Sequences |
|
| ||||
| 1 | D | 62.41% | 62.41% | 13709 |
| 2 | S | 21.06% | 83.47% | 4627 |
| 3 | N | 12.05% | 95.52% | 2646 |
| 4 | G | 1.81% | 97.33% | 398 |
| 5 | T | 1.03% | 98.36% | 226 |
| 6 | E | 0.51% | 98.86% | 111 |
| 7 | H | 0.51% | 99.37% | 111 |
| 8 | A | 0.33% | 99.70% | 73 |
| 9 | K | 0.23% | 99.93% | 50 |
| 10 | X | 0.04% | 99.96% | 8 |
| 11 | Q | 0.01% | 99.98% | 3 |
| 12 | R | 0.01% | 99.99% | 3 |
| 13 | Y | 0.00% | 100.00% | 1 |
| 14 | V | 0.00% | 100.00% | 1 |
Hinge mutations impacting HIV-1 viral infectivity
To determine the functional role of the MPER hinge region and investigate the impact of sequence variations at residues 671 and/or 674 on virus entry, viruses pseudotyped with MPER mutants were produced by co-transfection of 293T cells with an Env-deficient HIV-1 (pSG3ΔEnv) backbone and an Env-expressing plasmid. The infectivity of pseudotyped viruses was then determined in TZM-bl (CD4+CXCR4+CCR5+) cells and read out as relative light units (RLU). The mutations of the HIV-1 Con089 clade C strain were generated by changing serine S671 and S674 residues (SS) in the wild-type (WT) sequence to NN, NG, NA and AA respectively. Likewise, T671 and D674 (TD) of the WT Env from the clade B CAAN strain were mutated to DD, ND and SD. All except the AA mutations represent combinations found in naturally occurring HIV strains (Fig. 2c).
Figure 3a shows that changing the residue in the wild-type sequence at these two amino acid positions to commonly represented residues (such as N, D S or T) was well-tolerated by the mutant pseudoviruses. In contrast, viral infectivity was reduced 6 fold for the non-native AA mutation. Serial dilutions of Env-pseudotyped virus stocks with AA mutants from both Con089 and HxB2 strains further confirmed the effect of hinge region AA mutation on virus infectivity in TZM-bl cells (Fig. 3b). The block to virus infectivity was greater with the AA mutation in the CXCR4-dependent strain HxB2 (reduction by more than 10 fold) but nonetheless significant (6–7 fold reduction) in both CCR5-dependent Con089 (Fig. 3b) and PB7 strains (data not shown), indicating that the attenuation of viral infectivity was independent of co-receptor usage. Note the log10 scale used for both X- and Y-axes.
Fig. 3.
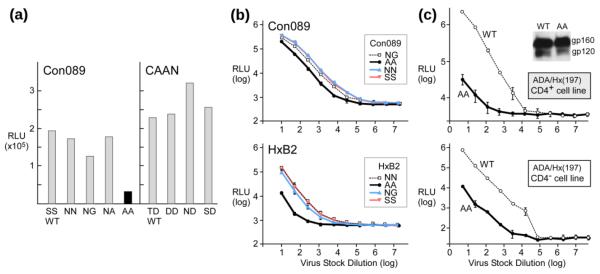
HIV-1 pseudovirus infectivity affected by AA mutations. (a) Infectivity of pseudoviruses harboring the Env mutations at positions 671 and 674. Comparable number of viral particles were used as determined by p24 Elisa. (b) Titration (log10) of Con089 and HxB2 Env WT or mutant pseudoviruses on TZM-bl cells. Mean and standard deviation (sd) of each dilution is shown. (c) Dose dependent infectivity of CD4-independent ADA/Hx(197) pseudoviruses on CD4+ CCR5+ TZM-bl cells and CD4− Cf2Th/ Syn CCR5 cells. The insert shows anti-gp120 mAb Western-blot of ADA/Hx(197) Env from pseudovirus.
One possible explanation for the reduced viral infectivity might be linked to structural changes within the gp120/gp41 trimer induced by the AA mutation in the MPER impacting receptor binding. This possibility was excluded by examining an AA mutant of the CD4-independent chimeric strain, ADA/Hx(197)16. ADA/Hx-AA mutant resulted in a significantly decreased capacity to infect both CD4+ TZM-bl (Fig. 3c, top) and CD4− Cf2Th/Syn CCR5 (Fig. 3c, bottom) cells (~70 fold reduction from WT). Thus, it appears that the AA mutation effect is significantly more pronounced in a CD4 independent strain. In addition, no effects of this AA mutation on the efficiency of gp160 envelope protein precursor processing to gp120, or expression level were observed on these pseudoviruses (Fig. 3c insert).
Double alanine MPER mutant affect viral membrane fusion
We next examined the effect of AA mutation on gp41-mediated fusion by a fluorescence cell-cell fusion assay. Env-expressing 293T effector cells stained with cytoplasmic dye calcein-AM (green) were co-cultured with 3T3.CD4.CCR5 target cells stained with CellTracker orange CMTMR (red), and the exchange of cytoplasmic dyes between effector and target cells was monitored by fluorescence microscopy. The JRFL-AA mutant exhibited both reduced numbers and sizes of syncytia (appearing orange/yellow) at 4-hour co-culture compared to wild-type (Fig. 4a, top row), at levels comparable to the JRFL cleavage (−) negative control. A similar observation was made with ADA-AA compared to ADA-WT at 4 hour. However, using bright field illumination at 24 hour, while syncytia in JRFL-AA were significantly smaller than JFRL-WT syncytia (Fig. 4a, bottom row), the ADA-WT and ADA-AA syncytia were more comparable (data not shown), arguing that MPER function may be critical for early fusion events. This possibility was confirmed in subsequent lipid mixing/hemifusion assays below.
Fig. 4.

Env-mediated fusion impaired by AA mutations. (a) Qualitative microscopy analysis of cell-cell fusion. Content mixing of Env-expressing 293T effector cells with 3T3.CD4.CCR5 target cells. The top row shows the overlay of fluorescence images after co-incubation of 293T cells (Green, Calcein) with 3T3.CD4.CCR5 cells (Red, CMTMR) at 37°C for 2 h. The bottom row shows representative bright-field images collected 24 h after co-incubation at 37°C. (b) Fusion kinetics of JR-FL (top) or ADA (bottom) Env-expressing 293T cells with TZM-bl cells containing a Tat-driven luciferase reporter. (c) Hemifusion identified by lipid mixing between 293T cells (green) and DiI-labeled 3T3.CD4.CCR5 cells (red). The top row shows the overlay of fluorescence images collected 2 h after co-incubation at 37°C. The bottom row shows the corresponding bright field images. (d) Quantitative lipid mixing efficiency of JRFL-AA relative to WT. Flow-cytometric analysis of lipid-dye transfer between DiO labeled 293T cells and DiD labeled TZM-bl cells were conducted. The percentage lipid mixing activities were determined following the subtraction of background dye redistribution between empty vector-transfected effector and target cells, normalized to that of WT (100%) in three independent experiments.
To quantitatively assess fusion efficiency, TZM-bl cells expressing luciferase under the control of the HIV long terminal repeat promoter responsive to HIV Tat were utilized as targets. The results confirmed impairment of cell-cell fusion by AA mutation in the MPER of JRFL as well as ADA strains. Fusion efficiency was reduced by ~50% in the first 6 hours in both strains (Fig. 4b). The reduced fusion efficiency was independent of surface expression level and envelope protein precursor processing. To further study the fusion block at the early lipid mixing/hemifusion phase, membrane probes were used in an experiment where DiO-labeled Env-293T cells (green) were co-cultured with DiI-labeled 3T3.CD4.CCR5 (red) target cells for 2 hours. Figure 4c shows DiI-labeled individual cells associated with DiO-labeled effector cells, and also transfected with a cleavage site defective Env as a negative control. Whereas extensive dye redistribution with numerous co-localization events (spotting on cell surfaces) was observed for JRFL-WT, reduced co-localization for JRFL-AA was observed which indicated blocking of lipid mixing, similar to the JRFL cleavage (−) negative control. Using flow-cytometry analysis to quantity the double fluorescence cells, hemifusion was observed to be reduced by ~50% for the JRFL-AA compared to JRFL-WT (Fig. 4d). Collectively, these findings document a selective detrimental effect of MPER AA mutation on viral fusion and infection.
MPER helix-hinge-helix motif is a common feature in both clade B and C HIV-1 strains
To understand the impact of hinge residue variation at the 671 and 674 positions on MPER conformation, we carried out NMR and EPR spectroscopy studies on several native and mutant MPER peptides (Fig. 2a). The solution structures of three clade C peptides, Con089, Du151.2, and ZM197M.PB7 in DPC micelles (Fig. 5a and Table 2) adopt the same helix-hinge-helix motif as the previously solved clade B HxB2 peptide11. These new peptides used in NMR studies all contain five additional native N-terminal residues from the gp41 C-terminal heptad repeat (CHR), EQELL for clade B and EKDLL for clade C strains. As represented by the clade C Con089 peptide in DPC detergent micelles shown in Figure 5b, the N-terminal extension forms a fishhook-like turn stabilized by two conserved leucine residues L660 and L661 inserted into the membrane phase, as well as a hydrogen bond between the side-chain amide of W666 and the backbone carbonyl of L661. This N-terminal extension from CHR is highly mobile and apparently has little effect on the rest of the peptide structure.
Fig. 5.

Solution structures of MPER peptides. (a) NMR structure ensembles of Con089, Du151.2, ZM197M.PB7, and HxB2-AA MPER peptides in DPC detergent micelles, superimposed by their N-terminal helices (666–672). (b) The additional CHR residues at the N-terminal end of MPER, as represented here by the Con089 peptide in DPC micelles, all adopt a conserved beta-turn stabilized by L661/L662 and W666. (c) Ribbon diagram of a representative HxB2-AA peptide with the side-chains of alanine substituted residues colored in pink. (d) The left panels show JNH RDC values with good correlation between Con089 and Du151.2, but weaker correlation between HxB2 and HxB2-AA. The right panels show difference in JNH RDC values (normalized according to fitted linear correlation parameters) between Con089 and Du151.2, and between HxB2 and HxB2-AA. The errors are derived from NMR amide peak position estimates.
Table 2.
NMR statistics.
| Con089* | Du151.2 | ZM197M.PB7 | HxB2-AA† | |
|---|---|---|---|---|
| Total NOE restraints | 440 | 383 | 430 | 335 |
| Intra-residue | 145 | 138 | 175 | 159 |
| Medium range (<=4) | 286 | 233 | 251 | 170 |
| Long range(>4) | 9 | 12 | 4 | 6 |
|
| ||||
| Dihedral angle restraints | 45 | 47 | 46 | 47 |
|
| ||||
| RDC restraints | 60 | 58 | 55 | 50 |
|
| ||||
| Membrane depth restraints | 14 | 15 | 15 | 16 |
|
| ||||
| Backbone <RMSD> to mean | ||||
| (666–682) | 0.78 Å | 0.94 Å | 0.32 Å | 0.59 Å |
| N-helix (666–672) | 0.04 Å | 0.05 Å | 0.05 Å | 0.09 ņ |
| C-helix (675–682) | 0.06 Å | 0.32 Å | 0.06 Å | 0.08 ņ |
|
| ||||
| Ramachandran statistics | ||||
| Most favored regions | 81.2 % | 76.8 % | 83.6 %% | 78.8 % |
| Additionally allowed regions | 18.4 % | 21.6 % | 16.4 % % | 21.2 % |
| Generously allowed regions | 0.4 % | 1.6 % | 0 % % | 0 % |
| Disallowed regions | 0 % | 0 % | 0 % | 0 % |
|
| ||||
| Alignment tensor | ||||
| Da (N-helix) | 9.32+/−0.11* | 9.32+/−0.04 | 4.51+/−0.02 | 7.19+/−0.06† |
| R (N-helix) | 0.13+/−0.01* | 0.18+/−0.01 | 0.38 +/−0.01 | 0.43+/−0.01† |
| Da (C-helix) | 4.74+/−0.04* | 4.12+/−0.09 | 2.72+/−0.03 | 4.98+/−0.04† |
| R (C-helix) | 0.53+/−0.02* | 0.41+/−0.04 | 0.13+/−0.01 | 0.31+/−0.01† |
The alignment tensor parameters for Con089 shown above were obtained with D-JNH, D-JCAHA RDCs only. RDCs of D-JNCO, D-JCOCA were acquired with a second DNA sample that yield Da=9.71+/−0.04, R=0.24+/−0.01 for N-helix, Da=4.24+/−0.03, R=0.48+/−0.01 for C-helix.
The HxB2-AA ensemble has 10 accepted conformers selected from 30 calculated structures (20 for other peptides). The N-and C-helix regions for HxB2-AA are defined as 666–673, and 678–682 respectively.
The statistical results of NMR structural calculations are summarized in Table 2, including residual dipolar coupling (RDC) constants measured using a DNA nanotube detergent-resistant alignment medium17. The RDC values obtained from multiple experiments including D-JNH, D-JNCO, D-JCOCA and D-JCAHA are incompatible with a single alignment tensor for the entire MPER peptide. However, the N-terminal and C-terminal helical segments of these MPER peptides can be fitted with two independent alignment tensors, consistent with the flexibility afforded by the central hinge (Fig. 5a). The highly mobile MPER N-terminal region (657–665) and the C-terminal residue (683), as well as the central hinge region (672–673) are excluded from RDC data analysis. The similarity of RDC alignment tensors between Con089 and Du151.2 confirmed that the two peptides have very similar orientations as they differ only by two residues at 671 and 674 positions (Figs. 2a and 5a). RDC values for the clade C peptide ZM197M.PB7 are rather different due to its distinctive charged residue distribution (Fig. 2a), resulting in different alignment tensor in the DNA nanotube alignment medium.
Distinctive conformation of MPER AA mutant peptide
The structure of HxB2-AA peptide does not have the generally L-shaped bend as observed with the other MPER peptides (Fig. 5a). In addition, the N-terminal helix is extended by one residue to F673, while the C-terminal helix is shortened by two residues at the N-terminal end (Fig. 5c). This distorts the original central hinge region, resulting in an elongated and partially unstructured linker region between residues 674 and 677. The alanine residues at 671 and 674 positions are not exposed to the solvent as is typical for other MPER peptides (Figs. 5b and 5c).
Measured JNH RDC values between HxB2 WT and HxB2-AA mutant peptides in DPC micelles show a weaker correlation with a reduced R2 value compared to that between Con089 and Du151.2 (Fig. 5d, left panels), which have similar orientations. This is despite the fact that each pair differs only at residue positions 671 and 674 by the change of asparagine to serine or alanine, and maintains the same charged residue distribution (Fig. 2a). Thus, the RDC values from HxB2 and HxB2-AA mutant MPER peptides must diverge as a result of structural differences on the membrane surface, most noticeably near the distorted hinge regions (Fig. 5d, right panels). The same patterns are observed for other RDC types, but JNH RDC values shown here are more accurate and easiest to measure experimentally.
Atypical behavior of MPER AA mutant on membrane surface
EPR immersion depth measurements for the three clade C MPER peptides, Con089, Du151.2 and ZM197M.PB7, also showed comparable overall structure and membrane immersion in POPC:POPG (4:1 by weight) liposomes (Fig. 6a). Similar to HxB211, the acyl chain-facing and aqueous-facing side-chains judging by their immersion depths alternate every third or fourth sequence position from 666 to 673 and from 675 to 681, consistent with helical conformation. However, the immersion depth data are out of phase at residue position 674, supporting the helix-hinge-helix motif. Taken together, the helix-hinge-helix motif in MPER is likely a conserved feature in all HIV-1 clades and potentially the ancestral SIV from chimpanzee (Fig. 1).
Fig. 6.

Structural comparison with MPER AA mutant. (a) EPR membrane immersion depths of Con089, Du151.2, ZM197M.PB7, and HxB2-AA in liposomes. Depth values between −5 Å to 0 Å and larger than 0 Å correspond to lipid head-group region and acyl-chain region, respectively. The white-colored bars indicate complete exposure to aqueous phase (depth < −5 Å). (b) NMR structure models of representative Con089A, Du151.2, ZM197M.PB7, and HxB2-AA peptides on the membrane surface. The lipid head-group and acyl-chain regions are shown in light blue and yellow, respectively. Residues 671 and 674 are colored in pink. The N-terminal extended regions (657–661) are omitted for simplicity. (c) EPR DEER spectra of singly spin-labeled HxB2-AA MPER peptides at residue positions 670 (top), 678 (middle) and 681 (bottom) showing spin-spin correlation consistent with peptide dimerization. (d) NMR 15N-HSQC spectra of HxB2-AA MPER in DPC micelles, showing broadening and disappearance of some backbone amide peaks at higher peptide concentrations consistent with aggregation.
In contrast, EPR immersion depth measurements show an altered membrane immersion pattern for HxB2-AA mutant as compared to other MPER peptides (Fig. 6a). There are more residues buried in the aliphatic region, and fewer residues with complete solvent exposure in the case of the HxB2-AA mutant. The depth pattern of Con089-AA mutant peptide was also an outlier (data not shown). Figure 6b shows the side-views of the MPER peptides embedded in the membrane by incorporating EPR immersion depth restraints in the NMR structure calculations. The HxB2-AA mutant peptide has a deeply buried middle section, bending in a different orientation with respect to other native MPER peptides (Figs. 5a and 6b).
EPR double electron-electron resonance (DEER) spectra of singly spin-labeled HxB2-AA MPER peptides at residue position W670, or W678, Y681 show detectable spin-spin distances (< 30 Å) consistent with peptide dimerization (Fig. 6c). NMR results also suggest that the HxB2-AA MPER mutant tends to aggregate at high concentrations, as the amide backbone peaks become broadened and some of them disappeared (Fig. 6d) even in DPC micelles. These changes were not observed for other native MPER peptides and can be attributed to the double substitution of asparagine at residue positions 671 and 674 by alanines and the associated increase in hydrophobicity.
Alternative N-capping of the MPER C-terminal helix
How does the double alanine substitution of MPER 671/674 hinge residues functionally impact HIV-1 infection during the early membrane fusion stages (Fig. 7a)? First, MPER hinge distortion and aggregation might alter gp41 trimer interface and indirectly induce structural change around the CD4 binding site in the pre-fusion state. However, this possibility cannot explain AA mutation effects on CD4-independent virus infectivity (Fig. 3c). Alternatively, the AA mutation might destabilize gp41 6-helix bundle-mediated membrane pore formation manifest in the post-fusion state. This is also unlikely given only minor contacts involving residues 671 and 674 in a 6-helix bundle structure including MPER segment18. Third, the AA mutant MPER could impede the assembling of the gp41 six-helix bundle during the fusion intermediate stages, hindering the kinetics of the early membrane fusion process (Fig. 7a) in agreement with our experimental findings and the structural implications discussed below.
Fig. 7.

MPER conformation change during HIV membrane fusion. (a) Illustrations of the HIV fusion process following CD4 binding. Chemokine receptors are omitted at the pre-fusion stage and only one gp41 monomer is drawn in the intermediate stages for simplicity. MPER is highlighted in red. (b) Hinge conformation revealed by backbone dihedral angles for free and BNAb-bound HxB2 wild-type MPER derived from observed NMR chemical shifts, except for 10E8 where the angles are extracted from a crystal structure (PDB: 4G6F). The backbone dihedral angles extracted from unbound Con089 and HxB2-AA peptide NMR structures are shown for comparison. (c) Side-chain oxygen to backbone amide hydrogen bonds observed in crystal structures of MPER bound to BNAbs: (top) between N671 to D674 in 10E8, (middle) between N671 to F673 in 4E10 (PDB: 2FX7), and (bottom) between D674 to T676 in Z13e1 (PDB: 3FN0). (d) Orientation change of W672/F673 with respect to the membrane with different N-capping of the C-terminal helix in 10E8-bound and unbound MPER.
Native MPER 671 and 674 position residues overwhelmingly belong to the NDST set (Table 1), a prominent feature for all HIV-1, HIV-2 and SIV lentiviruses (Fig. 1). The asparagine, aspartate, serine and threonine residues are favored as helix N-cap residues by forming side-chain to main-chain hydrogen bonds to stabilize the N-terminal end of alpha helices19; 20. This is consistent with the breaking of N- and C-terminal helices of the MPER to adopt a helix-hinge-helix motif. Figure 7b show backbone dihedral angles predictions based on observed NMR chemical shifts that reveal the breakage of helices in the 2F521; 22-bound MPER23 and the 4E10-bound MPER24 at residue 671, and at residue 674 in the Z13e1-bound MPER24. Helix breakage at residue 671 is also observed in the X-ray crystal structure of the 10E8-bound MPER13. Indeed, the side-chains of MPER residues N671/D674 form hydrogen bonds to the backbone amides of the C-terminal helix residues in all of the aforementioned MPER-neutralizing antibody complexes (Fig. 7c). Similar helix breakage at residue 674 can also be observed for unbound Con089 MPER peptides in DPC micelles, but not for HxB2-AA (Fig. 7b).
Helix N-capping has been shown to regulate kinetics of light-induced conformational changes in a PYP PAS domain25, correlate with allosteric activation in a cAMP receptor via helix breakage26, as well as maintain continuous curvature on the surface of a retroviral capsid via an inter-domain hydrogen bond27. The first structural implication of our results is that the gp41 ectodomain could pivot in a rigid body motion via the MPER central hinge region. The helix-hinge-helix motif, strengthened by the N-capped MPER C-terminal helix, could exert force on the lipid bilayer while at the same time adapting to membrane curvature during viral membrane fusion. Secondly, as shown in Figure 7d, the orientations of the two central residues, W672 and F673, relative to the membrane in relation to the helical break at either 671 (left) or 674 (right) joint positions are strikingly different. It appears that the formation of a “sliding” hinge region, facilitated by helix N-capping at a pair of tandem joint positions of the MPER (Fig. 7b) could potentially promote lipid mixing by allowing such “rotary” motion of bulky aromatic residues with respect to the viral membrane.
Since alanines have the highest helical propensity28 but are unsuitable for capping a helix, double alanine mutations of MPER would disrupt the conserved helix-hinge-helix motif. Hence, in MPER-AA mutant, the extension of the N-terminal helix, deeper embedding in the membrane and inefficient C-terminal helix breaking, all contribute to reduced fusion activity and viral entry. In essence, the HIV-1 apparently incorporates a flexible and “fail-safe” tandem-joint region, rationalizing why the double AA mutation in the MPER cannot exist in nature. However, by immobilizing the central hinge region, MPER-targeted BNAbs hinder HIV-1 fusion and infectivity.
Materials and Methods
Materials
Lipids, liposome preparation, synthetic peptide production with and without spin labels as well as procedures for GB1-MPER fusion protein production in E. coli were previously described11; 24. NMR stable isotope labels and d38-dodecyl-phosphocholine (DPC) detergent was purchased from Cambridge Isotope Laboratories (Andover, MA). Env-expressing plasmids JR-FL delCT(+) (cytoplasmic tail deletion with wild-type cleavage site), JR-FL delCT(−) (mutated cleavage site) and Tat-expressing plasmid pcTAT were kindly provided by Dr. Richard T. Wyatt (The Scripps Research Institute). Env-expressing plasmids to make CD4-independent pseudoviruses ADA/Hx(197) were kindly provided by Dr. Joseph G. Sodroski (Dana-Farber Cancer Institute). Con089 Env plasmid was kindly provided by Drs. Bart Haynes (Duke University) and Ronald Swanstrom (University of North Carolina at Chapel Hill). 293T cells were purchased from ATCC. TZM-bl cell, 3T3.CD4.CCR5 cell and CD4− Cf2Th/Syn CCR5 were obtained from the AIDS Research and Reference Reagent Program, NIH.
Preparation of pseudoviruses
Single-round, recombinant HIV-1 viruses (Con089, CAAN, HxB2 and ADA/Hx(197)) were generated by transfection of 293T cells using an Env-deficient HIV-1 (pSG3ΔEnv) backbone and Env-expressing plasmid. Briefly, cells were seeded in 10-cm dish (approximately 3×106 cells per dish) and transfected the next day with pSG3ΔEnv and Env-expressing plasmid. 72 hours after the transfection, virus-containing supernatants were collected, cleared of cell debris by low-speed centrifugation and filtered through 0.45-mm filters. To produce pseudoviruses that contain the luciferase gene to infect Cf2 Th/Syn CCR5 cell, 293T cells were transfected with the HIV-1 packaging plasmid pCMVDP1DenvpA, the firefly luciferase-expressing plasmid pHIvec2.luc and the plasmid expressing the HIV-1 Rev protein and the envelope protein. The amount of virus particles produced was determined using Alliance HIV-1 p24 antigen ELISA Kit (Perkin Elmer, Waltham, MA) per product manual. To prepare viruses pseudotyped with mutant Env protein, mutations were created by site-directed mutagenesis with QuikChange™ Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA).
Virus infection
Target cells (10,000 cells per well) were seeded into all wells of a 96-well flat-bottom culture plate. Serial 5-fold dilutions for a total of 11 dilutions of stock pseudoviruses with comparable level of p24 were added into quadruplicate wells. 20 μg/ml DEAE-dextran were added to enhance virus infection. Target cells were then incubated at 37°C for 48 hours before the measurement of luminescence using Steady-Glo Luciferase assay system (Promega, Madison,WI).
293T cell transfection
Cell-cell fusion was monitored by cell-cell content mixing or cell-cell lipid mixing after co-incubation of effector cells (Env-transfected 293T cells) with target cells (3T3.CD4.CCR5). To express WT or AA mutant Env protein, 293T cells were transfected with Env-expression plasmids using Fugene HD (Roche Diagnostics) at 3:1 ratio (v/w). 36 hours after the transfection, 293T cells were detached and stained with gp120-specific anti-V3 loop antibody 1A3 to determine the expression level. The amounts of Env-expressing plasmids were adjusted to yield comparable expression levels of wild-type Env protein and AA mutant Env protein on the surface of 293T cells.
Luciferase reporter assay of cell-to-cell fusion
To quantitatively analyze Env-mediated cell-cell fusion process, TZM-bl cells, which contain Tat-responsive reporter genes for firefly luciferase, were used as target cells. 293T cells transfected with Env-expressing plasmids together with Tat-expressing plasmids were mixed with TZM-bl cells at 1:1 ratio. Cell mixtures were cultured in 96-well plates in triplicates with 20,000 cells in each well. At various time points cultured cells were taken out and stored at −80°C. After the final collection, cells were lysed and the luciferase activity was measured using Steady-Glo Luciferase assay system (Promega).
Cytoplasmic dye transfer assays
Effector cells were loaded with cytoplasmic dye Calcein-AM at a concentration of 0.5 μM and target cells were loaded with cytoplasmic dye CMTMR (Invitrogen) at a concentration of 10 μM for 30 minutes at 37°C. After washing three times with phosphate-buffered saline, the two cell populations were mixed at 1:1 ratio and 300,000 cells were cultured in 24-well plate. Images of cells were taken using Nikon eclipse Ti fluorescence microscope after co-incubation for 4 hours or overnight.
Lipid mixing assays
The cell-cell lipid mixing was done similar to the cell-cell content mixing, except that effector cells and target cells were stained with lipophilic dyes DiO (Invitrogen) and DiI (Invitrogen) respectively and images were acquired 2 hours after co-incubation. Fusion defective Env-transfected 293T effector cells were compared as a negative control. All experiments were repeated three times and representative images were shown.
Flow cytometric analysis of efficiency of membrane lipid transfer during fusion
293T cells transfected either with Env WT or AA mutant-expressing plasmids or empty plasmids were stained with lipophilic dye DiO while TZM-bl cells were stained with lipophilic dye DiD. 300,000 of mixed cells at 1:1 ratio were cultured in 24-well plate for 2 h at 37°C, dissociated with PBS and 25mM EDTA, washed and immediately subjected to flow cytometric analysis. DiO and DiD double positive population comprises cells with fused membrane. The percentage of fused double positive cells were calculated to quantity hemifusion efficiency of WT and AA mutant following the subtraction of background dye redistribution between empty vector-transfected effector and target cells, and the lipid mixing activity for AA mutant was normalized to that of WT in three independent experiments, averaged and plotted.
EPR measurements
EPR power saturation measurements were performed on a Bruker EMX spectrometer using a loop-gap resonator. The immersion-depths values were calculated by the ratio of accessibility value of O2 to 50 mM nickel (II) ethylenediaminediacetic acid (NiEDDA). Samples were purged by either a stream of air or nitrogen gas. EPR distance measurements were performed with a Bruker ELEXSYS E680 spectrometer using two EPR techniques: conventional EPR (sensitive distance range 8–25 Å) and pulsed EPR (sensitive distance range 15–80 Å). Pulsed EPR DEER experiments, were carried out using a dead-time-free pulse sequence as described previously29. Conventional EPR and DEER spectra were analyzed with a Monte-Carlo/Simplex Gaussian convolution method to extract spin-spin distance30.
NMR structure determination
NMR experiments were performed using several Bruker spectrometers equiped with cryogenic probes operating at 1H frequency between 900 and 600 MHz at 35°C, using typically 1mM isotopically labeled MPER samples in 90%H2O/10%D2O, pH adjusted to 6.6 with 100mM d38-DPC. NMR structural determination of HxB2-AA mutant was carried out at a lower peptide concentration (500uM) to prevent aggregation. 2D and 15N or13C edited 3D NOESY data sets are collected with 60 ms mixing time. RDC data, D-JNH by TROSY experiment, and D-J-CAHA31, D-JNCO32, D-JCOCA33 by quantitative J experiments were collected using aligned samples containing 1mM MPER and 20 mg/ml DNA nanotube material. Weakly oriented HDO with ~20 mg/ml DNA yielded 2H quadrupolar splitting of 7–8 Hz in 500MHz magnet field.
NMR Data were processed by nmrPipe34. NMR resonance and NOE assignments were completed using CARA35. NOE distances were calibrated using CYANA36. Backbone dihedral angles restraints are derived from HNHA experiment and chemical shift based TALOS+ program37 that are consistent with local NOE restraints. EPR membrane immersion data were adopted after taking into account the maximum length (7Å) and dynamics of the nitroxide spin-label used in the EPR experiments. Structure models were calculated using XPLOR-NIH38 software with a TENSO module39 incorporating RDC restraints during high temperature torsion angle dynamics stage and planeDisPot module40 incorporating EPR depth restraints during subsequent low temperature Cartesian coordinate dynamics stage.
DNA nanotubes production
The modified p7308-bases-long M13mp18 phage DNA was generated at the nanomole scale as described previously41; 42. DNA staple oligonucleotide strands were prepared by solid-phase chemical synthesis (Invitrogen, Grand Island, NY) on the 200 nanomole scale in salt-free purification grade and dried format. Each monomer folding mixture was prepared by combining 120 nM phage DNA and 720 nM DNA staple in a pH 8.0 buffer (5 mM Tris Cl, 1 mM EDTA, pH 8.0, 20 mM MgCl2), aliquoted into four 96-well plates with 150 μl per well (36 ml each monomer). The folding ramp was as follows: 80°C for 5 min, decrease by 1 degree / 5 min to 65°C, then decrease by 1 degree / 40 min to 20°C. Each folded monomer sample was pooled and purified separately from excess staple strands via gravity-flow ion-exchange chromatography (Qiagen-Tip 10000 Column), as described previously17. Nanotube heterodimers were self-assembled by combining purified front and rear monomer mixtures together and incubated at 37°C for 2 hours, and then precipitated by addition of 0.25 volumes of 20% PEG8000 followed by incubation at room temperature for 15 minutes. The nanotubes were recovered by centrifugation at 15000 g for 30 minutes at 4°C, resuspended in 2.5 mM Tris, 0.5 mM EDTA, pH 8.0, 10 mM MgCl2, then concentrated to 30 mg/ml and buffer-exchanged into desired protein buffer (90%H2O/10%D2O, 5 mM MgCl2, 5mM NaPO4, pH 6.6) with Centricon-100. Finally, 260 μl of 20 mg/ml nanotubes were mixed with 260 μl of 1 mM 15N/13C labeled MPER sample containing 100mM d38-DPC and 50mM NaCl before concentrated down to 260 μl using a Centricon-3 concentrator.
Highlights
Helix-hinge-helix motif is conserved in gp41 MPER, target for several HIV-1 BNAbs.
Double Ala mutations at MPER hinge region impair membrane fusion, viral infectivity.
AA mutations cause structural changes and prevent helix-capping at the hinge region.
MPER hinge structure is critical despite apparent variability of exposed residues.
Acknowledgments
This work is funded by NIH Grants AI084785, AI091693 to E.L.R., GM047467 to G.W., 1DP2OD004641 and 1U54GM094608 to W.S, Gates Foundation Grants to E.L.R. and M.S.S. Y. C. is affiliated with the PhD Program in Biological Sciences in Public Health at Harvard School of Public Health, Boston, MA, 02115. L.S. acknowledges the support of NHMFL UCGP grant No. 5080. The NHMFL is funded by NSF Cooperative Agreement No. DMR1157490, DOE and the State of Florida. We thank Drs. J.G. Sodroski, B. Haynes, R. Swanstrom and R.T. Wyatt for reagents, Drs. J. Chou and M. Allan for advice with RDC data, J-H. Wang for comments, and G. Heffron for assistance with NMR instruments. The structure coordinates and NMR restraints have been deposited with Protein Data Bank (PDB ID: 2ME1, 2ME2, 2ME3, 2ME4) and Biological Magnetic Resonance Bank (BMRB ID: 19512, 19513, 19514, 19515) databases.
Abbreviations
- AA
double alanine mutation
- BNAb
broadly neutralizing antibody
- Env
envelope protein
- HIV
human immunodeficiency virus
- MPER
membrane proximal external region
- RDC
residual dipolar coupling
- RLU
relative luminescence units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roux KH, Taylor KA. AIDS virus envelope spike structure. Curr Opin Struct Biol. 2007;17:244–252. doi: 10.1016/j.sbi.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 4.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 5.Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chojnacki J, Staudt T, Glass B, Bingen P, Engelhardt J, Anders M, Schneider J, Müller B, Hell SW, Kräusslich HG. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338:524–528. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- 7.Ivankin A, Apellániz B, Gidalevitz D, Nieva JL. Mechanism of membrane perturbation by the HIV-1 gp41 membrane-proximal external region and its modulation by cholesterol. Biochim Biophys Acta. 2012;1818:2521–2528. doi: 10.1016/j.bbamem.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vishwanathan SA, Hunter E. Importance of the membrane-perturbing properties of the membrane-proximal external region of human immunodeficiency virus type 1 gp41 to viral fusion. J Virol. 2008;82:5118–5126. doi: 10.1128/JVI.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muñoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol. 1999;73:6089–6092. doi: 10.1128/jvi.73.7.6089-6092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 12.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22:163–173. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson J, Brunel FM, Jensen R, Crooks ET, Cardoso RM, Wang M, Hessell A, Wilson IA, Binley JM, Dawson PE, Burton DR, Zwick MB. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J Virol. 2007;81:4033–4043. doi: 10.1128/JVI.02588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kudahl U, Simon C, Zhang G, Reinherz EL, Brusic V. Motiffinder. 2012 http://research4.dfci.harvard.edu/cvc/motiffinder/
- 16.Haim H, Strack B, Kassa A, Madani N, Wang L, Courter JR, Princiotto A, McGee K, Pacheco B, Seaman MS, Smith A. B. r., Sodroski J. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS pathogens. 2011;6:e1002101. doi: 10.1371/journal.ppat.1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douglas SM, Chou JJ, Shih WM. DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc Natl Acad Sci USA. 2007;104:6644–6648. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010;6:e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doig AJ, Baldwin RL. N- and C-capping preferences for all 20 amino acids in alpha-helical peptides. Protein Sci. 1995;4:1325–1336. doi: 10.1002/pro.5560040708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richardson JS, Richardson DC. Amino acid preferences for specific locations at the ends of alpha helices. Science. 1988;240:1648–1652. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]
- 21.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, R. W, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–10737. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Julien JP, Bryson S, Nieva JL, Pai EF. Structural details of HIV-1 recognition by the broadly neutralizing monoclonal antibody 2F5: epitope conformation, antigen-recognition loop mobility, and anion-binding site. J Mol Biol. 2008;384:377–392. doi: 10.1016/j.jmb.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 23.Kim M, Sun ZY, Rand KD, Shi X, Song L, Cheng Y, Fahmy AF, Majumdar S, Ofek G, Yang Y, Kwong PD, Wang JH, Engen JR, Wagner G, Reinherz EL. Antibody mechanics on a membrane-bound HIV segment essential for GP41-targeted viral neutralization. Nat Struct Mol Biol. 2011;18:1235–1243. doi: 10.1038/nsmb.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song L, Sun ZY, Coleman KE, Zwick MB, Gach JS, Wang JH, Reinherz EL, Wagner G, Kim M. Broadly neutralizing anti-HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc Natl Acad Sci USA. 2009;106:9057–9062. doi: 10.1073/pnas.0901474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumauch IM, Kaledhonkar S, Philip AF, Wycoff J, Hara M, Li Y, Xie A, Hoff WD. A conserved helical capping hydrogen bond in PAS domains controls signaling kinetics in the superfamily prototype photoactive yellow protein. J Am Chem Soc. 2010;132:15820–15830. doi: 10.1021/ja107716r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu S, Maillard RA, Gribenko AV, Lee JC. The N-terminal capping propensities of the D-helix modulate the allosteric activation of the Escherichia coli cAMP receptor protein. J Biol Chem. 2012;287:39402–11. doi: 10.1074/jbc.M112.404806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey GD, Hyun JK, Mitra AK, Kingston RL. A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J Mol Biol. 2012;417:212–223. doi: 10.1016/j.jmb.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 28.Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson. 2000;213:316–325. doi: 10.1016/j.jmr.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 30.Sale K, Song L, Li Y, Perozo E, Fajer PG. Improvement of EPR distance measurements using molecular modeling of spin probes. J Am Chem Soc. 2005;127:9334–9335. doi: 10.1021/ja051652w. [DOI] [PubMed] [Google Scholar]
- 31.Call ME, Schnell JR, Xu C, Lutz RA, Chou JJ, Wucherpfennig KW. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell. 2006;127:355–368. doi: 10.1016/j.cell.2006.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou JJ, Delaglio F, Bax A. Measurement of one-bond 15N-13C' dipolar couplings in medium sized proteins. J Biomol NMR. 2000;18:101–105. doi: 10.1023/a:1008358318863. [DOI] [PubMed] [Google Scholar]
- 33.Jaroniec CP, Ulmer TS, Bax A. Quantitative J correlation methods for the accurate measurement of 13C'-13Calpha dipolar couplings in proteins. J Biomol NMR. 2004;30:181–194. doi: 10.1023/b:jnmr.0000048946.71249.2f. [DOI] [PubMed] [Google Scholar]
- 34.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 35.Keller RLJ. Computer aided resonance assignment tutorial. Cantina Verlag; Goldau, Switzerland: 2004. [Google Scholar]
- 36.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 37.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 39.Sass HJ, Musco G, Stahl SJ, Wingfield PT, Grzesiek S. An easy way to include weak alignment constraints into NMR structure calculations. J Biomol NMR. 2001;21:275–280. doi: 10.1023/a:1012998006281. [DOI] [PubMed] [Google Scholar]
- 40.Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, Wucherpfennig KW. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berardi MJ, Shih WM, Harrison SC, Chou JJ. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Douglas SM, Dietz H, Liedl T, Högberg B, Graf F, Shih WM. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]