

Summary
Programmed necrosis mediated by receptor interacting protein kinase (RIP)3 (also called RIPK3) has emerged as an alternate death pathway triggered by TNF family death receptors, pathogen sensors, interferon receptors, Ag-specific TCR activation and genotoxic stress. Necrosis leads to cell leakage and acts as a ‘trap door’, eliminating cells that cannot die by apoptosis due to the elaboration of pathogen-encoded caspase inhibitors. Necrotic signaling requires RIP3 binding to one of three partners, RIP1, DAI or TRIF, via a common RIP homotypic interaction motif (RHIM). Once activated, RIP3 kinase targets the pseudokinase MLKL to drive cell lysis. Although necrotic or apoptotic death can enhance T cell cross-priming during infection, mice that lack these extrinsic programmed cell death pathways are able to produce antigen-specific T cells and control viral infection. The entwined relationship of apoptosis and necrosis evolved in response to pathogen-encoded suppressors to support host defense and contribute to inflammation.
Synopsis
Regulated cell death is a potent arm of host defense (1–4), involving alternate strategies that evolved with animals to counteract pathogen-encoded cell death suppressors (3, 5). Intrinsic (mitochondrial) apoptosis is necessary for development (6), whereas extrinsic apoptosis and programmed necrosis play out as alternate innate immune countermeasures to control infection (3, 5, 7). Although mechanistically distinct from Casp8-mediated extrinsic apoptosis, RIP3 necrosis similarly eliminates infected cells prior to release of viral progeny, halting infection and triggering an inflammatory response (7). Importantly, extrinsic apoptosis and necrotic cell death machinery is distributed in all somatic cells. These pathways reduce the burden of infection while also producing cell debris to promote Ag cross-presentation by DCs, thereby supporting a robust adaptive immune response that ultimately controls infection. The study of virus-encoded cell death suppressor mutants brought RIP3 necrosis to light, revealing interdependencies fostered by a pathogen-host arms race centered on cell death measures and countermeasures (5). Based on the variety of strategies that have been observed, cell death suppressors are crucial to the pathogenesis of all large DNA viruses (2, 3, 5, 8, 9). Due to the fact that cell death is triggered by pre-existing cellular machinery, dysregulation can inadvertently kill cells and cause inflammatory disease even in the absence of infection (7). The distribution of these pathways in all somatic cells opens possible routes to improve host resistance to natural pathogens as well as to prevent infection of novel biothreat agents. This review will provide a perspective on recent advances in RIP3 necrosis. The intention is to highlight triggers and alternate pathways of extrinsic cell death where therapeutic intervention might improve innate resistance to infection or drive better cross-presentation during vaccination without risking increased inflammatory disease (10). The derivation of viable, fertile and immunocompetent mice with combined deficiency in Casp8 and RIP3 (11) dismisses any key role of Casp8-regulated pathways in development, but certainly raises important questions as to how apoptosis as well as necrosis contribute to the function of the immune system.
Alternate Casp8 apoptosis and RIP3 necrosis pathways
To set the stage for discussing the current understanding of RIP3 kinase in host defense, it is important to consider the crucial role that TNF-mediated signal transduction has played in the elaboration of alternate cytoprotective and cytotoxic pathways (1, 12). Three distinct outcomes of signal transduction via the TNF death receptor, TNFR1, are now recognized: (i) cytokine activation, (ii) extrinsic apoptosis and (iii) programmed necrosis. These converge on death domain (DD) signaling that is orchestrated via the adaptor FAS-associated protein with DD (FADD) in complex with Casp8 and specific inhibitor, FLIP (12, 13), as depicted in Fig 1. Pathogen sensors, interferons, TCRs and genotoxic stress all trigger analogous outcomes. Insights from TNF receptor DD-signaling and identification of virus-encoded cell death suppressors using TNF-based assays (14–16) has brought an appreciation of core cell death machinery operating as an integrated pathogen sensor system (5). In line with the view that extrinsic death came into existence in order to support host defense, both TNF antagonist immunotherapy (17) as well as genetic linkage studies (18) show that TNF signaling contributes as a redundant factor in host defense like many other innate immune mechanisms. A goal of this review is to highlight the growing evidence that TNF opened the awareness to a broadly distributed innate cell death system able to prevent infection.
Figure 1. Regulation of extrinsic apoptosis and RIP3 necrosis by a ‘Necrosome’ or ‘Ripoptosome’ complex.
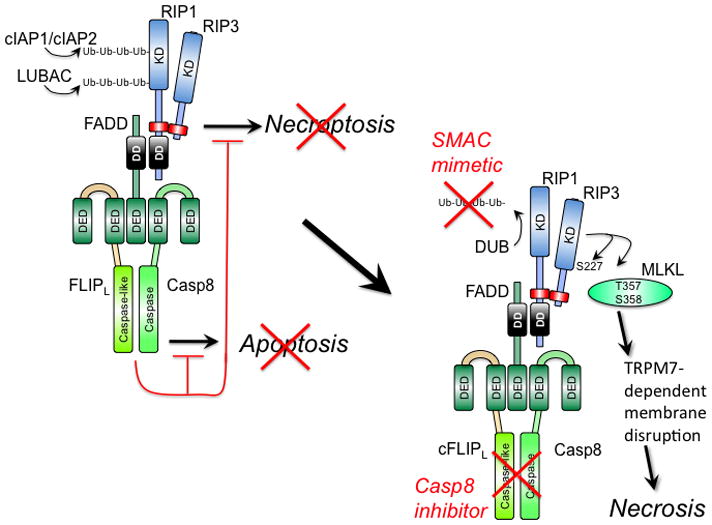
(Left) Cytoprotection. Signal transduction via death receptors (e.g. TNF) (37–39), pathogen sensors (e.g. TLR3 signaling) (27, 51), TCR activation (32, 33) or intracellular genotoxic stress (34) supports FADD association with the FLIP-Casp8 heterodimer via DED as well as RIP1 via DD interaction. RIP1 orchestrates recruitment of RIP3 via a RHIM interaction (red rectangle). The FLIP-Casp8 association prevents self-cleavage activation of Casp8 and maintains sufficient basal protease activity to also prevent necroptosis. E3 ubiquitin ligases cIAP1/cIAP2 and LUBAC also prevent necroptosis by maintaining K63 or linear polyubiquitination (Ub-Ub) of RIP1 and other targets (34, 100, 101). (Right) Activation of necroptosis. When Casp8 activity is blocked by an inhibitor or E3 ubiquitin ligases are compromised (red “X”) by a mimetic of second mitochondria-derived activator of caspases (SMAC), RIP3 kinase autophosphorylates at S277 and targets MLKL (42) for phosphorylation at T357 and S358 (41). These modifications drive trimerization of MLKL and membrane disruption associated with Ca2+ influx via a TRPM7-dependent plasma membrane channel (43). Deubiquitinase (DUB) activity removes polyubiquitin chains in the presence of SMAC mimetic, sensitizing to necrosis when Casp8 activity is compromised.
TNF family death receptors
TNFR1 as well as the Fas/CD95 and TRAIL death receptors control NF-κB activation, extrinsic apoptosis and programmed necrosis by DD-signal transduction, functioning in collaboration with death effector domain (DED) interactions (6) mediated via a critical complex consisting of Casp8, FLIP, FADD and RIP1 (denoted ‘Complex IIB’ downstream of TNFR1 signal transduction (19), and known as the ‘Necrosome’ or ‘Ripoptosome’ complex (3, 12, 13, 20, 21) shown in Figure 1). This cytosolic complex maintains control over alternate death outcomes downstream of TNF family death receptors (22), while also metering RIP1-enhanced induction of NF-κB (19) and RIP1 kinase-dependent programmed necrosis, also called necroptosis (3, 5, 12, 22, 23). RIP1 kinase-dependent necroptosis is blocked by small molecule drugs, the necrostatins (24). Cell death triggered by death receptors, pathogen sensors (25–28), interferons (29–31), Ag-specific TCR engagement (32, 33) or genotoxic stress (34) is all regulated by heterodimeric Casp8-FLIP within this core complex, preventing Casp8 self-activation and extrinsic apoptosis (12, 13) while allowing sufficient basal protease activity to suppress necrosis (3, 12, 13, 20, 21). The ability of this core Casp8 complex to prevent extrinsic apoptosis as well as necroptosis first emerged in studies of TNFR1 DD-signaling (22, 35, 36). Necroptosis is triggered when Casp8 becomes compromised during death signal transduction. In these settings, RIP1 functions as both a RHIM-dependent adaptor and a protein kinase to phosphorylate RIP3 (37–39), a partnership that results in formation of an amyloid-like complex (40). RIP3 kinase undergoes autophosphorylation and subsequently activates a target protein, mixed lineage kinase domain-like (MLKL), by phosphorylating key amino acids (7, 41, 42). The final steps in this pathway involve the formation of an MLKL homotrimer that translocates to the plasma membrane to mediate Ca2+ influx via a transient receptor potential melastatin related 7 (TRPM7) channel (43). A similar RIP3-MLKL axis (28) is apparently shared by the three pathways leading to RIP3 necrosis, whether RIP1-dependent or RIP1-independent (Fig. 2).
Figure 2. Three distinct RHIM complexes trigger RIP3 necrosis.
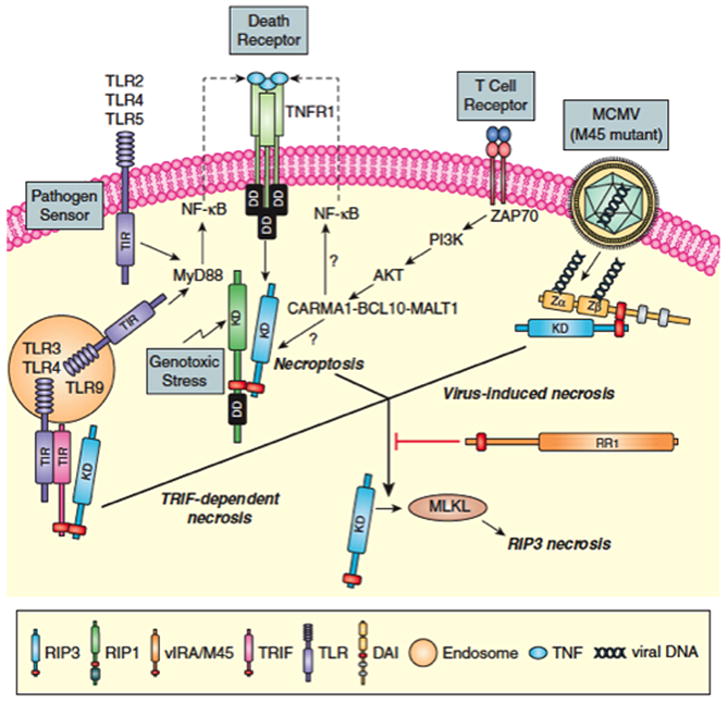
RIP1-RIP3 necroptosis first characterized downstream of death receptor activation via RIP1-RIP3 complex formation (37–39) is also induced by pathogen sensor (e.g. TLR2, TLR4, TLR5 or TLR9 MyD88-dependent signaling) (27, 28), TCR activation (32, 33), intracellular genotoxic stress (34) and vaccinia virus infection (37). Virus-induced DAI-RIP3 necrosis (3, 25, 26) is activated by MCMV M45 mutant virus infection. TRIF-RIP3 necrosis in fibroblasts is activated TLR3 or TRL4 ligands (27, 28). RIP3 complexes with RIP1, DAI or TRIF depend on RHIM interactions that activate RIP3 kinase-dependent modification of MLKL (7, 41, 42) (see Fig. 1). MCMV M45-encoded vIRA functions as a dominant RHIM-inhibitor preventing RIP3 association with RIP1, DAI or TRIF.
Pathogen sensors
Pathogen recognition receptors (PRRs) trigger NF-κB and IFN regulatory factor (IRF)3/IRF7 (44–46), activating production of interferon (IFN) and other cytokines (47, 48). These sensors regulate alternate activation of cytokines or cell death in a manner analogous to TNF family death receptor signaling (3, 49), subject to modulation by virus-encoded cell death suppressors (3, 23). RIP1-RIP3 necroptosis (5, 7, 50) occurs downstream of TLR signaling (27, 28, 51) as well as via retinoic acid-inducible gene 1 (RIG-I) or melanoma differentiation-associated protein 5 (MDA5) dsRNA helicase enzymes (52, 53). Similar signaling also lies downstream of seemingly distinct pathogen response categories, including genotoxic stress (34), interferon activation (31), Ag-dependent activation of T cells (32, 33), or infection with viruses such as vaccinia (37, 54), murine CMV (MCMV) (3, 25, 26) and reovirus (55). Two RHIM-containing adaptors in addition to RIP1 are involved in activating RIP3: (i) DNA-dependent activator of IFN regulatory factors (DAI, also called ZBP1 or DLM1) (25) and (ii) TIR-domain-containing adapter-inducing IFNβ TRIF) (28), the key TLR3 and TLR4 signaling adaptor (Fig. 2). RIP3 directly engages the pathogen sensor DAI independent of RIP1 when cells or mice are infected with a mutant MCMV strain that lacks the M45-encoded viral inhibitor of RIP activation (vIRA) (25). Furthermore, RIP3 engages TRIF downstream of TLR3 in both RIP1-dependent and RIP1-independent pathways (27, 28, 51). DAI and TRIF engage in RHIM-dependent interactions that converge on RIP3 kinase (5). The necrotic death mediated by DAI-RIP3 (25, 26) and TRIF-RIP3 (28) may proceed independent of RIP1, but nevertheless follow similar parameters (37–39) and converge on MLKL (28, 41, 42, 56, 57) (Fig. 1). Thus, a RIP3 necrosis ‘trap door’ lies downstream of innate immune signaling, and cell death may be triggered either independent of or dependent on death receptors and RIP1 kinase activity (3, 5, 7), through the various pathogen-related signaling events depicted in Figure 2.
Interferons
Type I (IFNα and IFNβ) or type II (IFNγ) interferons act on receptors (INFαβR or IFNγR, respectively) to trigger JAK-STAT signal transduction and mediate antiviral and immunomodulatory outcomes. Interferons also induce cell death (58) analogous to death receptors and pathogen sensors (3, 49), but is carried out by distinct JAK-STAT signaling cascades. Recent studies implicate both IFNβ (29) and IFNγ (30) in RIP1-dependent cell death with characteristics of RIP3 necrosis. Activation of cell death by IFNγ requires JAK-STAT function as well as RIP1 and RIP3 (31). The impact of either FADD or caspase compromise produces a picture that points to the same core cytosolic Casp8-FADD-FLIP-RIP1 ‘Ripoptosome’ complex (Fig. 1). Beyond death receptor signaling, pathogen sensors as well as related pathogen alarm and control mechanisms as diverse as interferons, genotoxic stress and antigen activation of lymphocytes trigger alternate cell death pathways via the same core Casp8-FADD-FLIP complex (27, 32, 34, 37–39, 51, 52) in position as a mammalian pathogen supersensor (28).
Purpose of RIP3 necrosis
DNA virus-encoded cell death suppressors are crucial to pathogenesis of viral infection and disease progression (1–4). These functions have contributed to dissection of extrinsic death pathways (3, 5, 7). Initially, apoptosis-prone L929 (59) and necrosis-prone L-M variant cell line (60) led to assays (61) that allowed for the identification of adenovirus-encoded cell death suppressors (14). Poxviruses and herpesviruses provided the first examples of DED-containing Casp8 suppressors, so-called viral (v)FLIPs (62, 63), opening the way toward understanding the prosurvival role of NF-κB (64) as well as consequences of cellular FLIP-Casp8 heterodimerization (12). Cowpox caspase and serine protease inhibitor, CrmA, was crucial in characterizing necrosis as a caspase-independent pathway triggered by TNF under conditions that prevent Casp8-dependent extrinsic apoptosis (65). The concept that RIP3 necrosis may be a host countermeasure against viruses encoding caspase inhibitors (3, 9, 33) has been refined with the demonstration that the highly specific CMV-encoded viral inhibitor of Casp8 activation (vICA) predisposes to RIP3 necrosis (11). Consistent with this understanding, TNF-induced necroptosis makes a striking contribution to host defense against the poxvirus and vaccinia in mice (37, 54) where a virus-encoded inhibitor related to CrmA likely unleashes the pathway.
WT MCMV is insensitive to RIP3 necrosis; however, MCMV-encoded M45 mutant viruses that are deficient in vIRA induce necrosis within a few hours of invading cells (3, 25, 26) via a DAI-RIP3 complex. Curiously, mathematical models of MCMV vIRA and vICA function (66) have completely missed the mark (11, 20). vIRA acts as a virion protein (67) to block RHIM-dependent signaling (3, 25, 26) upon delivery to cells during initial penetration (67–70). vIRA-deficient virus fails to gain a foothold in the host as infection halts due to the elimination of virus-exposed cells prior to the production of progeny virus. Two key issues remain to be fully addressed: (i) whether MCMV-encoded vICA suppression of apoptosis is responsible for unleashing DAI-RIP3 necrosis under natural infection conditions in the host animal (3, 28), and (ii) how the core Casp8 complex communicates with RIP3 kinase without the benefit of the adaptor RIP1 (Fig. 1).
Human CMV (HCMV) and MCMV both encode vICA (71) and block Casp8 apoptosis, a viral strategy that is particularly important during infection of macrophages (121). HCMV has a homolog of M45 (72), called UL45, but this fails to suppress cell death (73, 74). The parallels of vICA function aside, most of the immunomodulators encoded by MCMV and HCMV act independently on conserved host defense pathways. HCMV infection blocks necrosis (Omoto and Mocarski, in preparation); however, the nature of HCMV-encoded necrosis inhibitor remains to be determined. Based on early experimental data (3, 9), some vFLIPs may act as suppressors of necrotic death. RIP3 necrosis plays out in humans as well as mice, although humans encode two self-processing caspases (Casp8 and Casp10) where rodents have only Casp8. Nevertheless, primary human peripheral blood cells retain the capacity for necroptosis under experimental conditions that parallel what is known in mice (75).
The increased susceptibility of RIP3-deficient mice to vaccinia infection (37) stands in striking contrast to natural infection with MCMV, where vIRA sustains infection by preventing DAI-RIP3 necrosis (25) (Fig. 2), but where elimination of RIP3 pathways does not alter WT virus pathogenesis or control (26). Thus, RIP3 necrosis is a mechanism of host defense that threatens virus-infected cells, making the dialogue between vIRA and RIP3 crucial in both immunocompetent (26, 69) as well as in immunodeficient mice lacking NK, T and B cell functions (25, 26, 67). The need for vIRA is reversed in RIP3- and DAI-deficient mice (25) where the mutant virus replicates and disseminates. Thus, RIP3 necrosis operates within infected cells, without an apparent contribution to the function of immune cells that respond to and control of infection. RIP3 partner DAI has the capacity to trigger RHIM- and RIP3-dependent IFN activation in mouse and human cells (76, 77), although neither NF-κB nor IFN contribute to virus-induced necrosis (25). DAI-dependent IFN activation can be suppressed by MCMV-encoded vIRA (76, 78); however, the contribution of this one pathogen sensor in dictating levels of NF-κB and IRF3 activation during natural infection in the host has not been gauged with any accuracy. Parenthetically, DAI certainly contributes to HCMV virion-induced IFN response in cell culture (79). With MCMV infection, RIP3 necrosis becomes subdued, enabling virus infection and dissemination to proceed (25, 26, 68). In aggregate, the elaboration of a potent suppressor by MCMV reveals RIP3 necrosis can completely arrest viral infection by killing off infected cells independent of other immune mechanisms as well as the particular inoculation route. It would be a remarkable feat to harness RIP3 by therapeutic intervention to confer pathogen-independent resistance to a wide range of infectious agents such as those that pose a potential biothreat.
Although potentiators of RIP3 necrosis have not been yet investigated, necrotic death is experimentally blocked by recently described small molecule RIP3 kinase inhibitors (28) that act regardless of whether triggered by RIP1-RIP3, DAI-RIP3 or TRIF-RIP3 complex formation. Such inhibitors promise to expand understanding of RIP3 kinase in necrotic death, similar to the powerful impact that necrostatins have had on defining the specific role of RIP1 kinase activity in necroptosis (22).
RIP3 necrosis underlying developmental failure and inflammation
Host defense mechanisms involving immune cells that protect from infection through innate and adaptive mechanisms have the potential to trigger immunopathology (10). Death pathways emanating from the core Casp8 complex are known to undermine development and tissue homeostasis by unleashing RIP3 necrosis and inflammation (3, 5, 12, 49). When germ line Casp8- or FADD-deficiency is rescued by elimination of RIP3 (11, 20) or RIP1 (80), respectively, RIP1-RIP3 necroptosis emerges as a specific risk when the Casp8 complex becomes compromised during development (3, 5, 12). Although RIP3 engages DAI (25) and TRIF (28) as alternatives to RIP3, neither of these RHIM adaptors contributes to midgestational death in mice. Casp8-FLIP association within the core complex (Fig. 1) blocks RIP3 necrosis (20) potentially targeting RIP1, RIP3 or some component of polyubiquitylation/deubiquitylation machinery (Fig. 1); however, the precise target(s) of basal caspase activity that prevents necrosis remain to be clarified. Casp8-deficient humans survive development but exhibit immunodeficiency (81), a phenotype that is remarkably similar to T cell-specific disruption of either Casp8 (32, 82) or FADD (83) in mice where T cells die by necroptosis upon TCR activation (32, 83). The ability of TCR to trigger RIP3 necrosis indicates that the CARMA1-BCL10-MALT1 complex that normally activates NF-κB also influences the core ‘Ripoptosome’ complex or, alternatively, contributes to increased production of TNF followed by TNFR1-induced necroptosis (Fig. 2).
All settings in mice where deficiency of Casp8 (11, 20), FADD, or FLIP (21) has been rescued by eliminating RIP3 produce viable and fertile mice that exhibit lymphoid hyperplasia accompanied by the accumulation of an abnormal B220+ T cell population as they age (84). This phenotype aligns with the importance of Fas-dependent extrinsic apoptosis in the homeostatic turnover of T cells. tCasp8−/−Rip3−/− or tFaddddRip3−/− (83) mice phenocopy this defect, revealing a requirement for Casp8 function to eliminate excess T cells that is independent of RIP3. Curiously, humans with Casp10 deficiency exhibit an autoimmune lymphoproliferative syndrome (ALPS) characteristic of Fas signaling-deficiency (81) that also matches the phenotype of Fas signaling-deficiency in mice.
Other than lymphoid hyperplasia that develops with age, Casp8−/−Rip3−/− mice exhibit none of the severe developmental defects, homeostatic collapse or increased inflammation that result from the disruption of either Casp8 or FADD in specific tissues (49, 52, 85–99). Thus, RIP3 necrotic death and unleashed inflammation are both consequences of compromised Casp8 function. Compromise in E3 ubiquitin ligases cIAP1 and cIAP2, or the SHARPIN component of the linear ubiquitination complex (LUBAC), results in similar inflammatory outcomes (34, 100, 101), and has been the topic of a recent review (102). Thus, the interdependency of Casp8 and RIP3 pathways, which evolved for host defense, leads to serious developmental, homeostatic and inflammatory complications. Disparate observations in the fields of immunology, cell and development biology, as well as in signal transduction center on dysregulation of a core ‘Ripoptosome’ complex that can side track cell cycle progression, NF-κB activation, autophagy, cell adhesion and migration, and inflammation (12, 32, 33, 49). The picture reveals a striking system-wide role for Casp8 in silencing RIP3-dependent pathways to prevent inflammatory damage and disease throughout development and during life (7).
RIP3 necrosis contribution to host defense
Despite deficiency in extrinsic apoptosis and RIP3 necrosis, Casp8−/−Rip3−/− mice can control viral infection like WT or RIP3-deficient mice (11), mounting CD8 T cell responses to control acute MCMV infection (Livingston-Rosanoff and Mocarski, in preparation) that compare to matched C57BL/6 mice (103, 104). In addition, tCasp8−/−Rip3−/− (32, 82) and tFaddddRip3−/− (83) mice retain full immune control over the RNA viruses lymphocytic choriomeningitis virus and mouse hepatitis virus. Casp8−/−Rip3−/− mice support MCMV-specific CD8 T cell expansion, contraction and recall like WT controls, including characteristic memory inflation and complete protection from secondary challenge. Thus, extrinsic death pathways are redundant, in a pattern that also characterizes other immune mechanisms (10). Experiments with Casp8−/−Rip3−/− mice have shown that cytotoxicity, turnover of responding cells, memory T cell maintenance and recall, as well as aspects of the cellular immune response to virus infection that interface with other immune and non-immune cell types, can proceed completely independent of extrinsic apoptosis. This comes as a surprise given the range of lymphocytes, macrophages and DCs known to collude in control of viral infection and the repeated implication of death receptors as well as pathogen sensors and other signaling pathways that trigger extrinsic cell death in the overall immune response to infection. Most surprising of all is that elimination of extrinsic cell death does not impact the intensity of the Ag-specific CD8 T cell response, which is dependent on APCs that present viral peptides by cross-presentation (105). In line with other infections (50, 106, 107), the immune response to MCMV is influenced by levels of cross-presentation that depend on dying virus-infected cells for a protective CD8 T cell response (108). This occurs via immunoproteasome-dependent APC function (109, 110) that counters virus-mediated MHC class I downregulation in infected APCs (111). Casp8−/−Rip3−/− mice probably depend on intrinsic, Bcl2 family member Bim-dependent apoptosis (6) for purposes of lymphocyte contraction as well as for turnover in the antiviral CD8 T cell response because there is no other known pathway to support cross-presentation (108). Casp8−/−Rip3−/− mice are able to support memory inflation that accompanies latent infection, a pathway that depends on direct Ag presentation (112) as well as CD4 T cell function (113). The ability of Casp8−/−Rip3−/− mice to mount diverse innate and adaptive immune responses in the absence of extrinsic death machinery indicates that Fas, other death receptors, or any innate signaling via the ‘Ripoptosome’ complex is dispensable for a cellular immune response that controls infection (84).
Immunogenicity of pro-apoptotic and pro-necrotic viruses
The remarkable ability of mice lacking Casp8 and RIP3 pathways to mount a protective CD8 T cell immune response raises a very important question about the contribution of extrinsic cell death to the immune response in the WT host. This question is important on several levels: (i) the basis of vaccine immunogenicity rests on empirical comparisons to natural virus infection (114), (ii) little is known about the independent contribution of innate immune cell death independent of innate immune induction of cytokine production, and, (iii) experimental studies have long suggested a correlation between induction of cell death and immunogenicity (50, 106, 107), although this has not yet been addressed in hosts that are deficient in major cell death pathways or with pathogens that are specifically susceptible to apoptotic or necrotic death. Extrinsic apoptosis and programmed necrosis certainly influence immune response parameters towards virus-infected cells in infection models (50, 106, 107). In the setting of systemic MCMV infection, where cross-presentation dominates CD8 T cell priming (108) and relies on the CD8α subset of DCs (115), the impact of particular cell death pathways on CD8 T cell immunity remains to be established. This area has relevance because CMVs have potential as vaccine vectors to protect against pathogens such as HIV (116–119). In natural infection, viral load is a major driver of adaptive immunity. Attenuated or replication-defective viral vectors typically drive a weaker T cell response that may exhibit different qualitative parameters than the original viral pathogen (114). This has triggered a growing literature on the topic of rational vaccine vector design (120) as well as the search for vectors that have the potential to deliver supernatural immunogenicity, such as observed with rhesus macaque CMV (119). Contributions of cross-presentation to CD8 T cell priming can be addressed by disrupting either MCMV (108) or mouse (115) genetic determinants such as virus-encoded cell death suppressors or the extrinsic apoptosis and programmed necrosis pathways that they target (16).
Pro-apoptotic vICA (121) and pro-necrotic vIRA (26) mutant MCMV induce premature Casp8 apoptosis (121, 122) and RIP3-dependent necrosis (25, 26), cutting short infection (16). Replication levels of vICA- and vIRA-deficient viruses become normalized in Casp8−/−Rip3−/− (DKO) mice due to the absence of the pathways that these cell death suppressors target. Systemic inoculation (123) delivers sufficient virus to trigger a cross-presentation-mediated response even when a replication defective virus is employed, although peak antiviral responses are lower (108). WT MCMV produces sizeable virus loads in spleens, livers and lungs of WT and DKO mice and virus disseminates to salivary glands where persistent infection occurs for at least a month. In contrast, virus strain-matched pro-apoptotic vICA (ΔM36) (121, 122, 124) and pro-necrotic vIRA (M45mutRHIM) (25, 26) mutant virus are attenuated and fail to produce significant virus loads in any tissue. The differences between replication competent and replication deficient MCMV significantly impact the size of virus-specific CD8 T cell response (108). Despite replication compromise, pro-apoptotic and pro-necrotic mutants induce a MCMV-specific CD8 T cell response that is as robust as WT virus when assessed by cytoplasmic IFNγ in virus specific T cells. In this setting, the increased cross-presentation from either enhanced apoptosis (121, 122) or enhanced necrosis (25, 26) may compensate for reduced Ag load (Fig. 3) a relationship that contrasts observations with the replication-deficient viral mutant that does not enhance cross-presentation, leaving peak MCMV-specific IFNγ+ CD8 T cell levels that are many fold lower than WT (108). It appears that pro-necrotic or pro-apoptotic viruses promote Ag cross-presentation (Livingston-Rosanoff and Mocarski, in preparation), leading to a model that relates Ag load and CD8 T cell response (Fig. 3). The recent success of rhesus macaque CMV as a model SIV vaccine vector (119, 125) may certainly stem from predicted pro-apoptotic nature of this vector due to the disruption of vICA activity (71). It will be of interest to determine whether matched pro-apoptotic or pro-necrotic rhesus macaque CMV mutants show enhanced cross-presentation properties comparable to murine CMV mutants in mice.
Figure 3. Model: Viral Ag load and cell death pathways collaborate in cross-presentation to drive CD8 T cell immunity during infection.
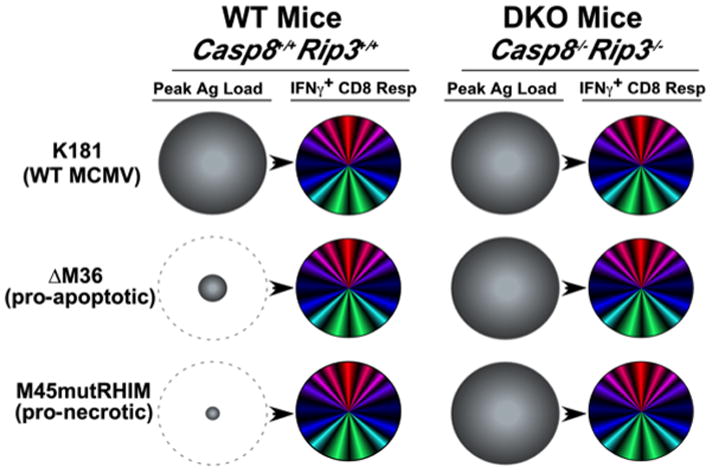
Model derived from studies on the impact of apoptotic and necrotic cell death pathways on cross-presentation in the CD8 T cell response (50, 106, 107) as well as developing understanding of MCMV immune response parameters. Relative peak viral load (shaded grey circles) at day 3 pi and peak CD8 T cell response at day 7–10 pi (multicolored circles) with WT MCMV (K181 strain), pro-apoptotic mutant ΔM36 or pro-necrotic mutant M45mutRHIM. The benefit of enhanced cross-presentation from either pro-apoptotic or pro-necrotic viruses is depicted by the dashed grey circles.
Conclusion
The capacity of the host to switch-hit between apoptosis and necrosis pathways very likely facilitates innate clearance of many intracellular pathogens, despite the fact that adenoviruses, poxviruses and herpesviruses all encode potent cell death suppressors that have limited the impact of cell death in host defense (3, 14, 15, 25, 26, 37, 68). The balance of cell death contributes to inflammation, cross-presentation and control of viral infection as well as life-long adaptive immune memory that prevents reinfection. Cell death pathways that evolved to support host defense are completely dispensable for development. It is attractive to consider how these pathways may be harnessed to enhance innate host resistance to infection and improve immunogenicity of vaccines.
Acknowledgments
We thank colleagues at the Emory Vaccine Center for the environment where the concepts presented here were developed and discussed.
Footnotes
This work was supported by National Institutes of Health Grants RO1 AI030363 and AI020211 (to E.S.M.), T32GM008169 and an ARCS Fellowship (to D.L.R.), OD012198 (to WJK) and start-up funds from the University of Texas at Austin and the Cancer Prevention Research Institute of Texas (CPRIT) (to JWU).
Literature Cited
- 1.Lamkanfi M, V, Dixit M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008;4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. 2011;12:79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castanier C, Arnoult D. Mitochondrial localization of viral proteins as a means to subvert host defense. Biochim Biophys Acta. 2011;1813:575–583. doi: 10.1016/j.bbamcr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Kaiser WJ, Upton JW, Mocarski ES. Viral modulation of programmed necrosis. Curr Opin Virol. 2013;3:296–306. doi: 10.1016/j.coviro.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green DR. Means to and End: Apoptosis and Other Cell Death Mechanisms. Cold Spring Harbor Press; Cold Spring Harbor, NY: 2010. [Google Scholar]
- 7.Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27:1640–1649. doi: 10.1101/gad.223321.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mocarski ES, Grakoui A. Persistent and Latent Infection. In: Mahy B, van Regenmortel M, editors. Encyclopedia of Virology. 3. Elsevier; Oxford, UK: 2007. pp. 108–116. [Google Scholar]
- 9.Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci. 2010;35:434–441. doi: 10.1016/j.tibs.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnaba V, Paroli M, Piconese S. The ambiguity in immunology. Front Immunol. 2012;3:18. doi: 10.3389/fimmu.2012.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS. RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell. 2011;44:9–16. doi: 10.1016/j.molcel.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 14.Mahr JA, Gooding LR. Immune evasion by adenoviruses. Immunol Rev. 1999;168:121–130. doi: 10.1111/j.1600-065x.1999.tb01287.x. [DOI] [PubMed] [Google Scholar]
- 15.Rahman MM, McFadden G. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2006;2:e4. doi: 10.1371/journal.ppat.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCormick AL, Mocarski ES. Cell death pathways controlled by cytomegaloviruses. In: Reddehase MJ, editor. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Caister Scientific Press; Norfolk, United Kingdom: 2013. pp. 263–276. [Google Scholar]
- 17.Domm S, Cinatl J, Mrowietz U. The impact of treatment with tumour necrosis factor-alpha antagonists on the course of chronic viral infections: a review of the literature. Br J Dermatol. 2008;159:1217–1228. doi: 10.1111/j.1365-2133.2008.08851.x. [DOI] [PubMed] [Google Scholar]
- 18.Bousfiha A, Picard C, Boisson-Dupuis S, Zhang SY, Bustamante J, Puel A, Jouanguy E, Ailal F, El-Baghdadi J, Abel L, Casanova JL. Primary immunodeficiencies of protective immunity to primary infections. Clin Immunol. 2010;135:204–209. doi: 10.1016/j.clim.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 20.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-Caspase-8-cFLIP(L) complex. Cell Rep. 2012;1:401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benedict CA, Banks TA, Ware CF. Death and survival: viral regulation of TNF signaling pathways. Curr Opin Immunol. 2003;15:59–65. doi: 10.1016/s0952-7915(02)00018-3. [DOI] [PubMed] [Google Scholar]
- 24.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 25.Upton JW, Kaiser WJ, Mocarski ES. DAI (ZBP1/DLM-1) complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like Receptor 3-mediated necrosis via TRIF, RIP3 and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thapa RJ, Basagoudanavar SH, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ, Beg AA, Madesh M, Balachandran S. NF-kappaB Protects Cells from Gamma Interferon-Induced RIP1-Dependent Necroptosis. Mol Cell Biol. 2011;31:2934–2946. doi: 10.1128/MCB.05445-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110:3109–3118. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hedrick SM, I, Ch’en L, Alves BN. Intertwined pathways of programmed cell death in immunity. Immunol Rev. 2010;236:41–53. doi: 10.1111/j.1600-065X.2010.00918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, Macfarlane M, Cain K, Meier P. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci. 2011;36:541–552. doi: 10.1016/j.tibs.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 37.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 40.Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 42.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109:5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2013 doi: 10.1038/ncb2883. (Epub Dec 8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoshi T, Koyama S, Kobiyama K, Akira S, Ishii KJ. Innate and adaptive immune responses to viral infection and vaccination. Curr Opin Virol. 2011;1:226–232. doi: 10.1016/j.coviro.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 45.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 46.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 47.Verma S, Benedict CA. Sources and signals regulating type I interferon production: lessons learned from cytomegalovirus. J Interferon Cytokine Res. 2011;31:211–218. doi: 10.1089/jir.2010.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 49.Wallach D, Kovalenko A, Kang TB. ‘Necrosome’-induced inflammation: must cells die for it? Trends Immunol. 2011;32:505–509. doi: 10.1016/j.it.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, Macfarlane M, Hacker G, Leverkus M. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajput A, Kovalenko A, Bogdanov K, Yang SH, Kang TB, Kim JC, Du J, Wallach D. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34:340–351. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 53.Zou J, Kawai T, Tsuchida T, Kozaki T, Tanaka H, Shin KS, Kumar H, Akira S. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity. 2013;38:717–728. doi: 10.1016/j.immuni.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 54.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 55.Berger AK, Danthi P. Reovirus activates a caspase-independent cell death pathway. mBio. 2013;4:e00178–00113. doi: 10.1128/mBio.00178-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 57.Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural Insights into RIP3-Mediated Necroptotic Signaling. Cell Rep. 2013;5:70–78. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 58.Kotredes KP, Gamero AM. Interferons as inducers of apoptosis in malignant cells. J Interferon Cytokine Res. 2013;33:162–170. doi: 10.1089/jir.2012.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kull FC, Jr, Cuatrecasas P. Preliminary characterization of the tumor cell cytotoxin in tumor necrosis serum. J Immunol. 1981;126:1279–1283. [PubMed] [Google Scholar]
- 61.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141:2629–2634. [PubMed] [Google Scholar]
- 62.Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci U S A. 1997;94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 64.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 65.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 66.Philippi N, Walter D, Schlatter R, Ferreira K, Ederer M, Sawodny O, Timmer J, Borner C, Dandekar T. Modeling system states in liver cells: survival, apoptosis and their modifications in response to viral infection. BMC Systems Biol. 2009;3:97. doi: 10.1186/1752-0509-3-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lembo D, Donalisio M, Hofer A, Cornaglia M, Brune W, Koszinowski U, Thelander L, Landolfo S. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol. 2004;78:4278–4288. doi: 10.1128/JVI.78.8.4278-4288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 2008;283:16966–16970. doi: 10.1074/jbc.C800051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brune W, Menard C, Heesemann J, Koszinowski UH. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. 2001;291:303–305. doi: 10.1126/science.291.5502.303. [DOI] [PubMed] [Google Scholar]
- 70.Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci U S A. 2008;105:3094–3099. doi: 10.1073/pnas.0800168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology. 2003;316:221–233. doi: 10.1016/j.virol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 72.Lembo D, Brune W. Tinkering with a viral ribonucleotide reductase. Trends Biochem Sci. 2009;34:25–32. doi: 10.1016/j.tibs.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 73.Hahn G, Khan H, Baldanti F, Koszinowski UH, Revello MG, Gerna G. The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J Virol. 2002;76:9551–9555. doi: 10.1128/JVI.76.18.9551-9555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patrone M, Percivalle E, Secchi M, Fiorina L, Pedrali-Noy G, Zoppe M, Baldanti F, Hahn G, Koszinowski UH, Milanesi G, Gallina A. The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J Gen Virol. 2003;84:3359–3370. doi: 10.1099/vir.0.19452-0. [DOI] [PubMed] [Google Scholar]
- 75.Muller-Sienerth N, Dietz L, Holtz P, Kapp M, Grigoleit GU, Schmuck C, Wajant H, Siegmund D. SMAC mimetic BV6 induces cell death in monocytes and maturation of monocyte-derived dendritic cells. PLoS One. 2011;6:e21556. doi: 10.1371/journal.pone.0021556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181:6427–6434. doi: 10.4049/jimmunol.181.9.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 78.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10:916–922. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DeFilippis VR, Alvarado D, Sali T, Rothenburg S, Fruh K. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol. 2010;84:585–598. doi: 10.1128/JVI.01748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Snow AL, Pandiyan P, Zheng L, Krummey SM, Lenardo MJ. The power and the promise of restimulation-induced cell death in human immune diseases. Immunol Rev. 2010;236:68–82. doi: 10.1111/j.1600-065X.2010.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ch’en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, Hedrick SM. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci U S A. 2008;105:17463–17468. doi: 10.1073/pnas.0808043105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu JV, Weist BM, van Raam BJ, Marro BS, Srinivas P, Bell BD, Luhrs KA, Lane TE, Salvesen GS, Walsh CM. Complementary roles of FADD and RIPK3 in T cell homeostasis and antiviral immunity. Proc Natl Acad Sci U S A. 2011;108:15312–15317. doi: 10.1073/pnas.1102779108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ben Moshe T, Barash H, Kang TB, Kim JC, Kovalenko A, Gross E, Schuchmann M, Abramovitch R, Galun E, Wallach D. Role of caspase-8 in hepatocyte response to infection and injury in mice. Hepatology. 2007;45:1014–1024. doi: 10.1002/hep.21495. [DOI] [PubMed] [Google Scholar]
- 86.Maelfait J, Beyaert R. Non-apoptotic functions of caspase-8. Biochem Pharmacol. 2008;76:1365–1373. doi: 10.1016/j.bcp.2008.07.034. [DOI] [PubMed] [Google Scholar]
- 87.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase- 8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, Dittrich-Breiholz O, Kracht M, Brenner O, Wallach D. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med. 2009;206:2161–2177. doi: 10.1084/jem.20090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee P, Lee DJ, Chan C, Chen SW, Ch’en I, Jamora C. Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature. 2009;458:519–523. doi: 10.1038/nature07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P, Syrbe U, Sieper J, Qin Z. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122:4094–4104. doi: 10.1172/JCI64115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 92.Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 94.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity. 2012;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 95.Scharner D, Rossig L, Carmona G, Chavakis E, Urbich C, Fischer A, Kang TB, Wallach D, Chiang YJ, Deribe YL, Dikic I, Zeiher AM, Dimmeler S. Caspase-8 is involved in neovascularization-promoting progenitor cell functions. Arterioscler Thromb Vasc Biol. 2009;29:571–578. doi: 10.1161/ATVBAHA.108.182006. [DOI] [PubMed] [Google Scholar]
- 96.Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, Wallach D. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol. 2008;181:2522–2532. doi: 10.4049/jimmunol.181.4.2522. [DOI] [PubMed] [Google Scholar]
- 98.Ben Moshe T, Kang TB, Kovalenko A, Barash H, Abramovitch R, Galun E, Wallach D. Cell-autonomous and non-cell-autonomous functions of caspase-8. Cytokine Growth Factor Rev. 2008;19:209–217. doi: 10.1016/j.cytogfr.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 99.Kang TB, Ben-Moshe T, Varfolomeev EE, Pewzner-Jung Y, Yogev N, Jurewicz A, Waisman A, Brenner O, Haffner R, Gustafsson E, Ramakrishnan P, Lapidot T, Wallach D. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173:2976–2984. doi: 10.4049/jimmunol.173.5.2976. [DOI] [PubMed] [Google Scholar]
- 100.Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J, Vaux DL. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012;31:1679–1691. doi: 10.1038/emboj.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Hacker G, Silke J, Tschopp J. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 102.Silke J, Hartland EL. Masters, marionettes and modulators: intersection of pathogen virulence factors and mammalian death receptor signaling. Curr Opin Immunol. 2013;25:436–440. doi: 10.1016/j.coi.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 103.Andrews DM, Estcourt MJ, Andoniou CE, Wikstrom ME, Khong A, Voigt V, Fleming P, Tabarias H, Hill GR, van der Most RG, Scalzo AA, Smyth MJ, Degli-Esposti MA. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J Exp Med. 2010;207:1333–1343. doi: 10.1084/jem.20091193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sumaria N, van Dommelen SL, Andoniou CE, Smyth MJ, Scalzo AA, Degli-Esposti MA. The roles of interferon-gamma and perforin in antiviral immunity in mice that differ in genetically determined NK-cell-mediated antiviral activity. Immunol Cell Biol. 2009;87:559–566. doi: 10.1038/icb.2009.41. [DOI] [PubMed] [Google Scholar]
- 105.Lin ML, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol. 2008;86:353–362. doi: 10.1038/icb.2008.3. [DOI] [PubMed] [Google Scholar]
- 106.Albert ML. Death-defying immunity: do apoptotic cells influence antigen processing and presentation? Nat Rev Immunol. 2004;4:223–231. doi: 10.1038/nri11308. [DOI] [PubMed] [Google Scholar]
- 107.Ferguson TA, Choi J, Green DR. Armed response: how dying cells influence T-cell functions. Immunol Rev. 2011;241:77–88. doi: 10.1111/j.1600-065X.2011.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Snyder CM, Allan JE, Bonnett EL, Doom CM, Hill AB. Cross-presentation of a spread-defective MCMV is sufficient to prime the majority of virus-specific CD8+ T cells. PLoS One. 2010;5:e9681. doi: 10.1371/journal.pone.0009681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Seckert CK, Schader SI, Ebert S, Thomas D, Freitag K, Renzaho A, Podlech J, Reddehase MJ, Holtappels R. Antigen-presenting cells of haematopoietic origin prime cytomegalovirus-specific CD8 T-cells but are not sufficient for driving memory inflation during viral latency. J Gen Virol. 2011;92:1994–2005. doi: 10.1099/vir.0.031815-0. [DOI] [PubMed] [Google Scholar]
- 110.Hutchinson S, Sims S, O’Hara G, Silk J, Gileadi U, Cerundolo V, Klenerman P. A dominant role for the immunoproteasome in CD8+ T cell responses to murine cytomegalovirus. PLoS One. 2011;6:e14646. doi: 10.1371/journal.pone.0014646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Doom CM, Hill AB. MHC class I immune evasion in MCMV infection. Med Microbiol Immunol. 2008;197:191–204. doi: 10.1007/s00430-008-0089-y. [DOI] [PubMed] [Google Scholar]
- 112.Snyder CM, Cho KS, Bonnett EL, Allan JE, Hill AB. Sustained CD8+ T cell memory inflation after infection with a single-cycle cytomegalovirus. PLoS Pathog. 2011;7:e1002295. doi: 10.1371/journal.ppat.1002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Walton SM, Torti N, Mandaric S, Oxenius A. T-cell help permits memory CD8(+) T-cell inflation during cytomegalovirus latency. Eur J Immunol. 2011;41:2248–2259. doi: 10.1002/eji.201141575. [DOI] [PubMed] [Google Scholar]
- 114.Pulendran B, Ahmed R. Immunological mechanisms of vaccination. Nat Immunol. 2011;12:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Torti N, Walton SM, Murphy KM, Oxenius A. Batf3 transcription factor-dependent DC subsets in murine CMV infection: differential impact on T-cell priming and memory inflation. Eur J Immunol. 2011;41:2612–2618. doi: 10.1002/eji.201041075. [DOI] [PubMed] [Google Scholar]
- 116.Dudek T, Knipe DM. Replication-defective viruses as vaccines and vaccine vectors. Virology. 2006;344:230–239. doi: 10.1016/j.virol.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 117.Paris RM, Kim JH, Robb ML, Michael NL. Prime-boost immunization with poxvirus or adenovirus vectors as a strategy to develop a protective vaccine for HIV-1. Expert Rev Vaccines. 2010;9:1055–1069. doi: 10.1586/erv.10.106. [DOI] [PubMed] [Google Scholar]
- 118.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AV. Viral vectors as vaccine platforms: deployment in sight. Curr Opin Immunol. 2011;23:377–382. doi: 10.1016/j.coi.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 119.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473:523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Arens R. Rational design of vaccines: learning from immune evasion mechanisms of persistent viruses and tumors. Adv Immunol. 2012;114:217–243. doi: 10.1016/B978-0-12-396548-6.00009-3. [DOI] [PubMed] [Google Scholar]
- 121.Cicin-Sain L, Ruzsics Z, Podlech J, Bubic I, Menard C, Jonjic S, Reddehase MJ, Koszinowski UH. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol. 2008;82:2056–2064. doi: 10.1128/JVI.01803-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ebermann L, Ruzsics Z, Guzman CA, van Rooijen N, Casalegno-Garduno R, Koszinowski U, Cicin-Sain L. Block of death-receptor apoptosis protects mouse cytomegalovirus from macrophages and is a determinant of virulence in immunodeficient hosts. PLoS Pathog. 2012;8:e1003062. doi: 10.1371/journal.ppat.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hsu KM, Pratt JR, Akers WJ, Achilefu SI, Yokoyama WM. Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J Gen Virol. 2009;90:33–43. doi: 10.1099/vir.0.006668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Daley-Bauer LP, Mocarski ES. Myeloid cell recruitment and function in cytomegalovirus immunity and pathogenesis. In: Reddehase MJ, editor. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. Caister Scientific Press; Norfolk, United Kingdom: 2013. pp. 363–373. [Google Scholar]
- 125.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Fruh K, Picker LJ. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science. 2013;340:1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]