

Abstract
Excitotoxic insults such as cerebral ischemia are thought to enhance neuronal autophagy, which is then thought to promote neuronal cell death. Excitotoxic insults indeed increase autophagy markers. Notably, however, autophagy markers can be increased either by autophagy induction (as this enhances their production) or by late-stage autophagy inhibition (as this prevents their degradation during autophagic flux). By comparing each condition with and without protease inhibitors that prevent autophagic degradation of the autophagy marker, the results of this study show that excitotoxic glutamate increases autophagy markers by a late-stage block of autophagy. Initially, this study set out to test if the CaMKII inhibitor tatCN21 mediates its post-insult neuroprotection by regulating autophagy. While tatCN21 partially inhibited basal autophagy in hippocampal neurons, it had no effects on the already blocked autophagy after excitotoxic glutamate insults, indicating that autophagy inhibition is not its neuroprotective mechanism. Additionally, while the autophagy inhibitor chloroquine had no effect, significant neuroprotection was seen instead with two drugs that enhance autophagy induction by different mechanisms, rapamycin (mTOR dependent) and trehalose (mTOR-independent). This suggests that therapeutic approaches should seek to enhance rather than inhibit autophagy, not only in neurodegenerative diseases (where such approach is widely accepted) but also after acute excitotoxic insults. Together, these findings significantly reshape the current view on the mutual cross-regulation of autophagy and excitotoxicity.
Keywords: CaMKII, glutamate, excitotoxicity, autophagy, neuronal cell death
1. Introduction
Cerebral ischemia causes anoxic depolarization of neurons, which triggers excessive release of the excitatory neurotransmitter glutamate. The excessive glutamate then causes excessive Ca2+-influx (largely by activating Ca2+-conducting glutamate receptors such as the NMDA receptor), which then results in excitotoxic neuronal cell death (for review see (Aarts & Tymianski 2004, Hara & Snyder 2007, Doyle et al. 2008, Szydlowska & Tymianski 2010, Coultrap et al. 2011). Indeed, transient ~5 min application of ~100 μM glutamate to cultured neurons triggers massive cell death within 24 h that is largely dependent on NMDA receptors and Ca2+. Among the Ca2+–activated proteins is the Ca2+/calmodulin-dependent protein kinase II (CaMKII), a multifunctional protein kinase that is extremely abundant in the brain and constitutes well over 1% of total protein in the hippocampus, a brain area required for learning and memory that is especially susceptible to neuronal cell death after global cerebral ischemia (for review see (Coultrap & Bayer 2012b, Coultrap et al. 2011). Stimulation of CaMKII activity by Ca2+/calmodulin can also stimulate autophosphorylation at T286, which in turn generates Ca2+-independent “autonomous” CaMKII activity that outlasts the initial stimulus (Miller & Kennedy 1986, Lou et al. 1986, Coultrap et al. 2012). A novel CaMKII inhibitor, tatCN21 (Vest et al. 2007), is neuroprotective even when applied hours after excitotoxic insults in hippocampal or cortical neuron cultures (Vest et al. 2010, Ashpole & Hudmon 2011) or after ischemic insults in vivo (Vest et al. 2010). tatCN21 is a highly selective peptide inhibitor (Vest et al. 2007) that is derived from the natural CaMKII inhibitor protein CaM-KIIN (Chang et al. 1998) and that penetrates cells and the blood-brain-barrier (Vest et al. 2007, Vest et al. 2010, Buard et al. 2010). By contrast, the traditional CaMKII inhibitors KN62 and KN93 also inhibit other CaM kinases as well as PKC and voltage-dependent Ca2+- and K+-channels (Enslen et al. 1994, Brooks & Tavalin 2011, Li et al. 1992, Ledoux et al. 1999). Most importantly, KN62 and KN93 are competitive with Ca2+/calmodulin and block only Ca2+-stimulated but not autonomous CaMKII activity (Tokumitsu et al. 1990, Sumi et al. 1991, Vest et al. 2010), while tatCN21 inhibits both stimulated and autonomous CaMKII activity with equal potency (Buard et al. 2010). As a result, KN62 or KN93 are neuroprotective only when present during excitotoxic insults (a time when they can block the autophosphorylation that generates autonomous activity) but not when added after the insults (a time when autonomous activity has already been generated) (Vest et al. 2010, Ashpole & Hudmon 2011). Thus, tatCN21 but not KN62 or KN93 has therapeutic potential for post-insult neuroprotection after cerebral ischemia.
Macroautophagy (here referred to as autophagy) is a fundamental cellular process that can be triggered by starvation and various stress factors (for review see (Mizushima et al. 2008, Levine & Kroemer 2008, Gump & Thorburn 2011, Rubinsztein et al. 2012). Autophagy is an alternative pathway for protein degradation, and is especially important for removal of damaged organelles and aggregated protein (Fig. 1). Depending on the circumstance, autophagy can promote either cell survival or cell death (Mizushima et al. 2008, Levine & Kroemer 2008, Gump & Thorburn 2011, Rubinsztein et al. 2012). While the situation in cerebral ischemia is still controversial, with numerous studies describing autophagy either as mediating neuronal death or protection (for review see (Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011), the currently prevailing view appears to be that autophagy contributes to ischemic neuronal cell death, as inhibition of autophagy by brain-specific Atg7 knock-out desensitized newborn mice to hypoxia-induced neuronal death (Koike et al. 2008). It is widely accepted, however, that cerebral ischemia indeed triggers not only apoptotic and necrotic cell death, but also autophagy (for review see (Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011). There is no doubt that ischemic insults increase markers of autophagy, such as autophagosome number and levels of microtubule-associated protein light chain 3 (LC3)-II (Fig. 1). However, it should be noted that these autophagy markers are not only generated during autophagy but are also degraded during autophagic flux (Mizushima & Yoshimori 2007, Klionsky et al. 2008, Klionsky et al. 2012). Thus, the currently available data that show an increase in autophagosomes are actually consistent both with induction of more autophagic flux (i.e. an increase in the whole process of autophagy) and with a late-stage block of autophagic flux (i.e. a decrease in the whole process of autophagy). Indeed, there is one previous study to support that cerebral ischemia causes a late-stage block of autophagic flux rather than autophagy induction (Liu et al. 2010).
Figure 1. Autophagy model.
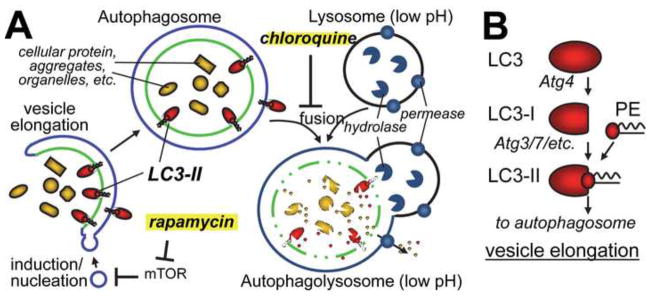
(A) LC3-II dependent vesicle elongation engulfs cellular components into a double-membraned autophagosome. Fusion with the lysosome then leads to degradation in macro-autophagy (while chaperone mediated- and micro-autophagy deliver proteins directly to the lysosome). As indicated, rapamycin induces autophagy while chloroquine causes a late-stage block. The mechanism of autophagy induction by trehalose is unknown, but is independent from mTOR.
(B) Processing of LC3 to LC3-II is required for autophagy induction, and the ratio of LC3-II over LC3-I provides a biochemical marker for changes in autophagy. However, assessing these changes is complex, as LC3-II is then also degraded during autophagic flux. PE: phosphatidyl-ethanolamine.
This study set out to test if neuroprotection by the CaMKII inhibitor tatCN21 (Vest et al. 2010) is mediated by regulation of autophagy. We found that tatCN21 inhibited basal autophagy in hippocampal neurons, but that this was not its neuroprotective effect: tatCN21 did not affect autophagy after excitotoxic insults, and two mechanistically different autophagy enhancers were neuroprotective in our system, while an autophagy inhibitor was not. Most importantly, however, we found that excitotoxic insults caused inhibition of autophagic flux.
2. Results
2.1 tatCN21 inhibits basal autophagy in hippocampal neurons
LC3-II is one of few useful biochemical markers of autophagy (Mizushima & Klionsky 2007, Kabeya et al. 2000). LC3 (called Atg8 in yeast) is constitutively cleaved into LC3-I. As this cleavage only removes one amino acid, these two LC3 form are not distinguishable by Western-blot (and the resulting LC3/LC3-I band is here referred to as LC3-I). However, autophagy requires further modification by phosphatidylethanolamine conjugation to produce LC3-II (see Fig. 1), and this form can be clearly distinguished by Western-analysis (as in Fig. 2). Changes in the LC3-II/I ratio then indicate changes in autophagy. However, the direction of the change is not necessarily clear, as LC3-II is both formed and degraded during autophagy. For instance, an increase in the LC3-II/I ratio indicates either increased autophagy induction or late-stage autophagy inhibition. This problem can be overcome by testing each of the treatment conditions in the absence and in the presence of protease inhibitors that block the autophagic degradation of LC3-II (Mizushima & Yoshimori 2007, Klionsky et al. 2008, Klionsky et al. 2012). Then, the level of increase in LC3-II caused by the protease inhibitors provides an indication for the autophagic flux that occurred during the time of protease inhibition.
Figure 2.
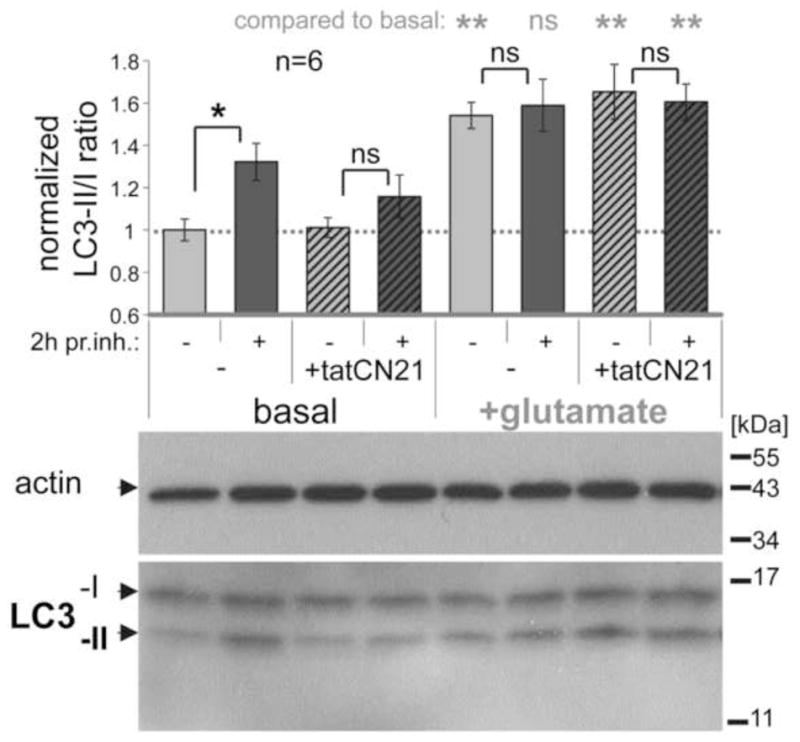
Autophagic flux in hippocampal cultures is inhibited by tatCN21 and by glutamate, as revealed by the LC3-II/I immuno-detection ratio with or without a 2 h treatment with protease inhibitors (pr. inh.). Shown are example Western-blots (lower panel) and their quantification (upper panel; n=6) normalized to the basal LC3-II/I ratio (which was ~0.5). Under basal conditions, protease inhibitor treatment significantly increased the LC3-II/I ratio, due to block of autophagic degradation of LC3-II. This increase was reduced by tatCN21 (5 μM) and completely blocked by excitotoxic glutamate (100 μM for 5 min). In comparison of different conditions without protease inhibitors, glutamate treatment increased the LC3-II/I ratio (indicating a late stage block of autophagic flux) while tatCN21 treatment did not (indicating instead an early stage inhibition of autophagy). **: p<0.01; *: p<0.05 in ANOVA with Newman-Keuls post-hoc analysis; ns: p>0.05 even in one-tailed t-test. Bar graphs indicate mean±SEM.
Here, we first determined the LC3-II/I ratio in primary hippocampal neurons in dissociated cultures with and without protease inhibitors, and with and without tatCN21 (Fig. 2, left side). While several review articles have suggested measuring the LC3-II/actin ratio instead (Mizushima & Yoshimori 2007, Klionsky et al. 2008, Klionsky et al. 2012), the LC3-II/I ratio has been specifically endorsed for the use in central nervous neurons [(Klionsky et al. 2008)]. The LC3-II/I ratio provides the additional advantages of having an internal control detected by the same antibody and of detecting two proteins that are present at similar abundance (at least in the cells examined here; see Fig. 2). Protease inhibitors were applied two hours before harvesting the neurons for the Western-blots. In absence of tatCN21, the protease inhibitors significantly increased the LC3-II/I ratio (Fig. 2, left side), indicating that autophagic flux occurs under basal conditions. When 5 μM tatCN21 was added 20 min prior to the protease inhibitors (in order to ensure complete cellular uptake and CaMKII inhibition), the increase in LC3-II/I ratio was no longer significant (Fig. 2, left side), indicating that tatCN21 inhibited the basal autophagic flux in the neurons. This inhibition of autophagic flux appeared to occur at an early stage, as tatCN21 application by itself did not increase the LC3-II/I ratio compared to untreated control (Fig. 2, left side), an increase that would be expected from a late stage inhibition (which would reduce only degradation but not formation of LC3-II).
Additionally, we examined a second autophagy marker, p62 (Fig. 3), in this case relative to actin (as p62 does not allow for an internal control with the same antibody). Again, p62 accumulated in neurons during protease inhibition in basal conditions while tatCN21 blocked further accumulation during protease inhibition (Fig. 3, left side). This further corroborated inhibition of basal autophagic flux by tatCN21. However, it should be noted that analysis of p62 (in contrast to LC3-II) cannot distinguish between early and late stage inhibition of autophagic flux, as inhibition at either stage causes accumulation of p62, because early stage autophagic flux does not require generation of new p62 (in contrast to LC3-II) (Mizushima & Yoshimori 2007, Klionsky et al. 2008, Klionsky et al. 2012). It should also be noted that the analysis of the p62/actin ratio showed more variability compared to the LC3-II/I ratio (consistent with additional error introduced by lack of an internal standard provided by use of a single antibody), and that the accumulation of p62 with protease inhibitor treatment that indicated basal autophagy was significant only by t-test but not by ANOVA (in contrast to the analysis of the LC3-II/I ratio).
Figure 3.
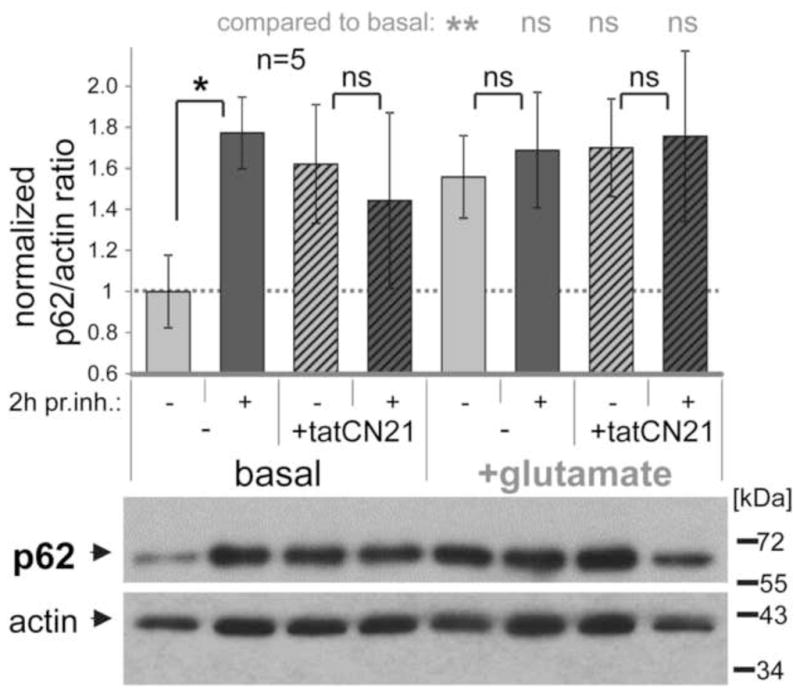
Autophagic flux in hippocampal cultures is inhibited by tatCN21 and by glutamate, as revealed by the p62/actin immuno-detection ratio with or without a 2 h treatment with protease inhibitors (pr. inh.). Shown are example Western-blots (lower panel) and their quantification (upper panel; n=5) normalized to the basal p62/actin ratio. Under basal conditions, protease inhibitor treatment significantly increased the p62/actin ratio, due to block of autophagic degradation of p62. This increase was reduced by tatCN21 (5 μM) and by excitotoxic glutamate (100 μM for 5 min). In comparsion of different conditions without protease inhibitors, an increase in p62/actin ratio is consistent with inhibition of autophagic flux at either early or late stages. **: p<0.01; *: p<0.05; ns: p>0.3 in one-tailed t-test. Bar graphs indicate mean±SEM.
2.2 Excitotoxic glutamate inhibits autophagic flux
tatCN21 is neuroprotective even when applied after excitotoxic insults (Vest et al. 2010, Ashpole & Hudmon 2011). Thus, we also tested how tatCN21 affects the changes in autophagy caused by such insults (Figs. 2+3; right sides). In contrast to its inhibition of basal autophagy, tatCN21 had no effect on the autophagy in hippocampal neurons after excitotoxic glutamate insults (Figs. 2+3). Most notably, however, these glutamate insults themselves resulted in a complete block of autophagic flux, as protease inhibition did not cause any further increase in the LC3-II/I (Fig. 2) or p62/actin (Fig. 3) ratio.
While treatments that enhance autophagy and treatments that block autophagy at a late stage can both increase the LC3-II/I ratio compared to basal conditions without protease inhibitors, only a late stage block of autophagy prevents additional further LC3-II accumulation during protease inhibition. Indeed, the increase in LC3-II/I ratio after glutamate treatment compared to basal conditions was statistically significant only in the absence but not in the presence of protease inhibitors (Fig. 2). These results demonstrate that excitotoxic glutamate causes a late-stage block of autophagy.
Treatments that enhance autophagic flux generally reduce p62 levels and treatments that reduce autophagic flux generally increase p62 levels, due to enhanced or reduced autophagic degradation of p62. A potential caveat can be introduced by possible additional regulatory effects of the treatments on p62 transcription/translation, which may confound the results. Nevertheless, the increase in p62 levels after excitotoxic glutamate treatment corroborates (Fig. 3) the block of autophagic flux shown by the analysis of LC3-II/I.
Taken together, analysis of LC3-II/I and p62/actin ratio showed that excitotoxic glutamate causes a block of autophagic flux in hippocampals neurons, and the LC3-II/I analysis showed that this block occurs at a late stage.
2.3 Neuroprotection by autophagy enhancers, not by an inhibitor
The neuroprotective effects of tatCN21 (Vest et al. 2010, Ashpole & Hudmon 2011) are likely not due to inhibition of autophagy, as tatCN21 inhibited only basal autophagy but had no effect on the already blocked autophagy after excitotoxic glutamate insults (see Fig. 2). Nevertheless, we decided to determine the effect of other autophagy modulators on excitotoxic cell death. The late-stage autophagy inhibitor chloroquine had no effect on the number of neurons surviving 24 h after a 5 min treatment with 100 μM glutamate (Fig. 4). By contrast, significant neuroprotection was seen for two different drugs that enhance autophagy by different mechanisms, rapamycin (mTOR-dependent (Ravikumar et al. 2004)) and trehalose (mTOR-independent (Sarkar et al. 2007)). Notably, while rapamycin and trehalose showed the same extent of neuroprotection, trehalose had to be added earlier before the glutamate insult in order to achieve this neuroprotection (Fig. 4). This is consistent with a faster induction of autophagy by rapamycin compared to trehalose.
Figure 4.
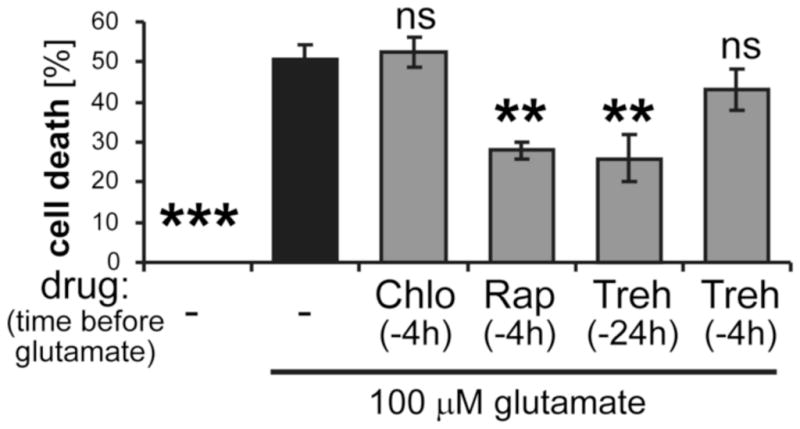
Neuroprotection by autophagy enhancers in hippocampal cultures. Neuronal cell death 24 h after a transient 5 min excitotoxic treatment with 100 μM glutamate was not affected by the autophagy inhibitor chloroquine (Chlo; 5 μM), but significantly reduced by rapamycin (Rap; 1 μM) or trehalose (Treh; 100 μM), which enhance autophagy by fundamentally different mechanisms. Compared to rapamycin, neuroprotection by trehalose required application at an earlier timepoint before the glutamate insult, consistent with a more delayed induction of autophagy by trehalose. The fraction of dying neurons was identified and quantified (n=4 coverslips) by ethidium homo-dimer staining. The statistical analysis indicates comparison to glutamate treatment without drug treatment (by ANOVA with Newman-Keuls post-hoc analysis). Bar graphs indicate mean±SEM.
Taken together, these data indicate that enhancing autophagy provides neuroprotection from excitotoxic neuronal cell death, and that tatCN21 is neuroprotective not because of but despite its inhibition of autophagy.
3. Discussion
The currently prevailing view is that autophagy contributes to neuronal cell death during ischemia, although this is still controversial (for review see Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011, Koike et al. 2008). By contrast, it is currently universally accepted that cerebral ischemia indeed induces neuronal autophagy (for review see Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011). The results of this study instead indicate that excitotoxic insults cause a late-stage block of autophagy and that enhancing autophagy protects from excitotoxic cell death. Additionally, tatCN21 inhibited only basal but not glutamate-regulated autophagy, further demonstrating that the post-insult neuroprotection provided by tatCN21 is not linked to its effects on autophagy.
Inhibition of autophagy (by brain-specific knock-out of Atg5 or Atg7) leads to massive neurodegeneration around week 3 after birth (Koike et al. 2008, Komatsu et al. 2006, Hara et al. 2006). Thus, in neurodegenerative diseases that involve protein-aggregation (and/or mitochondria damage), enhancing the autophagy-mediated removal of these aggregates (and/or damaged mitochondria) is generally thought to be a promising therapeutic strategy (Rubinsztein et al. 2012, Hara et al. 2006, Komatsu et al. 2006, Lee et al. 2010, Hartman 2012, Ravikumar et al. 2004, Tanaka et al. 2004, Sarkar et al. 2007). By contrast, it is much less clear if therapy of acute neuronal cell death after ischemia should also seek to enhance autophagy, or instead to inhibit it (for review see Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011, Koike et al. 2008). The currently prevailing view that autophagy should be inhibited in the treatments of acute conditions is largely based on the observation that inhibition of autophagy by brain-specific Atg7 knock-out desensitized newborn mice to hypoxia-induced neuron loss death (Koike et al. 2008). However, many of the proteins required for autophagy also have other cellular functions, and Atg7 depletion can also reduce apoptosis (Gump & Thorburn 2011, Grishchuk et al. 2011, Walls et al. 2010), which would be expected to alter cell survival. Furthermore, as described above, the Atg7 knock-outs show massive neuronal cell death starting around postnatal week 3 (Koike et al. 2008, Komatsu et al. 2006), and milder earlier effects could conceivably lead to partial neuroprotection by mechanisms akin to the well described ischemic pre-conditioning (where mild insults lead to partial protection from subsequent stronger insults) (Liu et al. 2009, Morris et al. 2011). Indeed, the results of this study instead showed that autophagy enhancing drugs protected neurons from excitotoxic death, while an autophagy inhibitor had no effect. This suggests that therapeutic approaches should enhance rather than inhibit autophagy, not only in neurodegenerative diseases, but also in acute excitotoxic conditions such as cerebral ischemia. However, given the numerous previous studies with conflicting conclusions (for review see Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011, Koike et al. 2008), it appears unlikely that our study will end this ongoing debate.
In general, a therapy of excitotoxic conditions would require its effectiveness when applied after the insults. However, trehalose is currently used as a dietary supplement, and could therefore also be used effectively as a preventative treatment in at-risk patients. Pharmacological treatments always raise questions about target specificity. However, trehalose and rapamycin enhance autophagy by fundamentally different mechanism (Ravikumar et al. 2004, Sarkar et al. 2007), but showed the same degree of neuroprotection. This provides further rationale for other autophagy-enhancing regimens, such as physical exercise and caloric restriction (Hartman 2012), which would also reduce other risk factors in at-risk patient. In contrast to trehalose, our CaMKII inhibitor tatCN21 provides neuroprotection even when applied post-insult (Vest et al. 2010, Ashpole & Hudmon 2011). As this study showed that the neuroprotection by tatCN21 is not mediated through effects on autophagy, combination therapies with autophagy enhancers could provide additional benefits, with autophagy enhancing regimens as preventative treatments and tatCN21 as additional post-insult therapy.
The previous conclusion that autophagy promotes neuronal cell death after acute insults is clearly conflicting with the results from this study. However, this previous conclusion depends on the notion that such insults indeed enhance autophagy. While there is no doubt that ischemic insults increase autophagy markers such as autophagosome number and LC3-II levels (Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011), such results are also consistent with a late-stage block of autophagy (which prevents the autophagic degradation of these markers during autphagic flux) (Mizushima & Yoshimori 2007, Klionsky et al. 2008, Klionsky et al. 2012). Importantly, the results of our study showed that excitotoxic glutamate insults cause a late-stage block of autophagy. While such an autophagy-block is in contrast to current perception (for review see Gabryel et al. 2012, Uchiyama et al. 2008, Smith et al. 2011), there is one previous study to support a similar conclusion (Liu et al. 2010). Specifically, this study utilized the late-stage autophagy blocker chloroquine in vivo, and found that cerebral ischemia did not cause any additional increase in LC3-II levels in the hippocampus (Liu et al. 2010). Like our results with lysosomal protease inhibitors, this finding supports late-stage autophagy block rather than autophagy induction.
It should be noted that block of late-stage autophagic flux by ischemic insults does not necessarily preclude that the same insults might also promote autophagy induction at an earlier stage. Exclusive late-stage block of autophagy (without any additional autophagy induction) by glutamate should increase LC3-II levels compared to basal conditions only in the absence of protease inhibitors but not in their presence. By contrast, simultaneous late-stage block and early stage induction by glutamate should result in higher LC3-II levels compared to basal conditions also in presence of the protease inhibitor. Indeed, glutamate appeared to increase the LC3-II levels further even when comparing the protease inhibitor conditions, although this was not statistically significant (p=0.112 in a two-tailed t-test). Thus, while our results clearly demonstrate inhibition of autophagic flux by glutamate, they do not conclusively rule out a possible additional increase in autophagy induction.
But how can enhancing autophagy be neuroprotective, if the insults actually shut down the autophagic flux? Autophagy enhancers would further increase accumulation of autophagosomes, while the insults prevent degradation of the engulfed material and thus their recycling to provide new building blocks in the energy-depleted neurons. However, completed autophagy would not directly provide energy to neurons anyway, as the brain can utilize only glucose, ketone bodies, or lactate for energy production (for review see (Simpson et al. 2007, Belanger et al. 2011)). Thus, the main neuroprotective function of autophagy may be sequestration of damaged mitochondria (which may prevent promotion of apoptosis by cytochrome C release), an effect readily achieved by enhanced autophagy induction even when further progression of autophagy is blocked.
Together, the results of this study lend support to therapeutic strategies that seek to enhance rather than inhibit autophagy. Most importantly, they show that we understand the vice versa effect of these insults on autophagy not nearly as well as generally thought.
4. Experimental Procedures
4.1 Material
Culture media were obtained from Life Technologies, chemicals were obtained from Life Technologies or Sigma (St. Louis, MO), unless indicated otherwise. The CaMKII inhibitor peptide tatCN21 was described previously (Vest et al. 2007, Vest et al. 2010, Buard et al. 2010).
4.2 Neuronal cell culture
Medium density dissociated hippocampal cultures were prepared from newborn rats and cultured on 24-well plates in Neurobasal A medium supplemented with B27 and glutamine as previously described (Vest et al. 2007, Vest et al. 2010). Animal procedures were approved by the University of Colorado Denver IACUC, and followed NIH guidelines. Glial cell growth was suppressed with FDU on day 3. Experiments were done after 14 days in culture. All incubations were done in culture medium at 37°C and 5% C02 in humidified incubators.
4.3 Assessment of autophagy
Antibodies that recognize the autophagy markers LC3-II (Novus Biologicals, Littleton, CO) and p62 (Abnova, Walnut, CA) were used to detect the respective protein levels by Western-blot. LC3-II was distinguished from LC3-I by size. Western-blots were done as previously described (Vest et al. 2007, Buard et al. 2010, Coultrap et al. 2010). Neurons were harvested in 100 μl of 1% SDS-buffer (without dyes) per well. Extracts were then diluted in SDS-loading buffer to equal protein concentrations (as determined by BCA assay; Pierce, Rockford, IL). 6 μg protein was loaded, and immuo-staining of 3-actin (Sigma, St. Louis, MO) was used as a loading control. Bands were visualized with Immobilon Western Chemiluminescent HRP substrate (Millipore, Billerica, MA) on X-ray film. Immuno-detection of LC3-II/LC3-I, actin, and/or p62 was quantified by densitometry (Coultrap & Bayer 2012a). In order to indicate changes caused by different treatments, the LC3-II/I ratio was normalized to the untreated basal condition.
Autophagic flux was assessed by comparing LC3-II/I or p62 for each tested condition after a 2 h incubation with or without the lysosomal protease inhibitors pepstatin A and E64d (Mizushima & Yoshimori 2007, Klionsky et al. 2008, Klionsky et al. 2012). Protease inhibitors were added immediately after excitotoxic insults (5 min, 100 μM glutamate) or mock treatment (for basal conditions). tatCN21 (5 μM; ~125fold IC50 (Buard et al. 2010)) was added 20 min before the excitotoxic or mock treatment. Note that the culture medium contains sufficient glycine for effective NMDA receptor stimulation by glutamate.
4.4 Assessment of neuronal cell death
Neuronal cultures were treated for 5 min with 100 μM glutamate. 24 h later, neuronal cell death was assessed by ethidium homo-dimer 2 (EtDH2) staining, as previously described (Vest et al. 2010). Neurons were identified by morphology and immunostaining with an anti-MAP2 antibody (Pharmingen, San Diego, CA) (Vest et al. 2010). Total and EtDH2 stained neurons were counted in order to determine the percentage of dead/dying neurons.
4.5 Statistical analysis
All data are represented as mean ± SEM. Statistical analysis was done by t-test or by ANOVA with Newman-Keuls post-hoc analysis, as indicated. Differences between the means were considered to be statistically significant if the p-value was <0.05.
Highlights.
Excitotoxic glutamate insults cause a late-stage block of autophagy in hippocampal neurons.
Autophagy enhancers provide neuroprotection from excitotoxic insults.
An autophagy inhibitor had no effect on excitotoxic neuronal cell death.
CaMKII inhibition reduces autophagy basally, but not after excitotoxic insults.
Acknowledgments
This work was supported by elope, Inc., St. Baldrick’s Foundation Scholar Award (to J.M.L.) and by National Institute of Health grants K12HD068372 (to J.M.L.), R01CA150925 (to A.T.), R01CA111421 (to A.T.), P30NS04154 (University of Colorado Center grant), and R01NS052644 (to K.U.B.).
Footnotes
Conflict of interest disclosure
The University of Colorado is currently seeking patent protection for tatCN21, its derivatives, and its uses (PCT/US08/077934 “Compositions and methods for improved CaMKII inhibitors and uses thereof”).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts MM, Tymianski M. Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr Mol Med. 2004;4:137–147. doi: 10.2174/1566524043479202. [DOI] [PubMed] [Google Scholar]
- Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol Cell Neurosci. 2011;46:720–730. doi: 10.1016/j.mcn.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Brooks IM, Tavalin SJ. CaMKII inhibitors disrupt AKAP79-dependent PKC signaling to GluA1 AMPA receptors. Journal of Biological Chemistry. 2011;286:6697. doi: 10.1074/jbc.M110.183558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard I, Coultrap SJ, Freund RK, Lee YS, Dell’Acqua ML, Silva AJ, Bayer KU. CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J Neurosci. 2010;30:8214–8220. doi: 10.1523/JNEUROSCI.1469-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci U S A. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Barcomb K, Bayer KU. A significant but rather mild contribution of T286 autophosphorylation to Ca2+/CaM-stimulated CaMKII activity. PLoS One. 2012;7:e37176. doi: 10.1371/journal.pone.0037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU. Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) In: Mukai H, editor. Neuromethods: Protein Kinase Technologies. Springer; 2012a. pp. 49–72. [Google Scholar]
- Coultrap SJ, Bayer KU. CaMKII regulation in information processing and storage. Trends Neurosci. 2012b;35:607–618. doi: 10.1016/j.tins.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Buard I, Kulbe JR, Dell’Acqua ML, Bayer KU. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J Biol Chem. 2010;285:17930–17937. doi: 10.1074/jbc.M109.069351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Vest RS, Ashpole NM, Hudmon A, Bayer KU. CaMKII in cerebral ischemia. Acta Pharmacol Sin. 2011;32:861–872. doi: 10.1038/aps.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. 2008;55:310–318. doi: 10.1016/j.neuropharm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem. 1994;269:15520–15527. [PubMed] [Google Scholar]
- Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia--a potential target for neuroprotective strategies? Pharmacol Rep. 2012;64:1–15. doi: 10.1016/s1734-1140(12)70725-9. [DOI] [PubMed] [Google Scholar]
- Grishchuk Y, Ginet V, Truttmann AC, Clarke PG, Puyal J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy. 2011;7:1115–1131. doi: 10.4161/auto.7.10.16608. [DOI] [PubMed] [Google Scholar]
- Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 2011;21:387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Snyder SH. Cell signaling and neuronal death. Annu Rev Pharmacol Toxicol. 2007;47:117–141. doi: 10.1146/annurev.pharmtox.47.120505.105311. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hartman AL. Neuroprotection in metabolism-based therapy. Epilepsy Res. 2012;100:286–294. doi: 10.1016/j.eplepsyres.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Tadakoshi M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Ledoux J, Chartier D, Leblanc N. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J Pharmacol Exp Ther. 1999;290:1165–1174. [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Hidaka H, Wollheim CB. Inhibition of voltage-gated Ca2+ channels and insulin secretion in HIT cells by the Ca2+/calmodulin-dependent protein kinase II inhibitor KN-62: comparison with antagonists of calmodulin and L-type Ca2+ channels. Mol Pharmacol. 1992;42:489–488. [PubMed] [Google Scholar]
- Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68–78. doi: 10.1111/j.1471-4159.2010.06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XQ, Sheng R, Qin ZH. The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol Sin. 2009;30:1071–1080. doi: 10.1038/aps.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou LL, Lloyd SJ, Schulman H. Activation of the multifunctional Ca2+/calmodulin-dependent protein kinase by autophosphorylation: ATP modulates production of an autonomous enzyme. Proc Natl Acad Sci U S A. 1986;83:9497–9501. doi: 10.1073/pnas.83.24.9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Morris KC, Lin HW, Thompson JW, Perez-Pinzon MA. Pathways for ischemic cytoprotection: role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J Cereb Blood Flow Metab. 2011;31:1003–1019. doi: 10.1038/jcbfm.2010.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007;27:1766–1791. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RS. Autophagy in acute brain injury: feast, famine, or folly? Neurobiol Dis. 2011;43:52–59. doi: 10.1016/j.nbd.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- Uchiyama Y, Koike M, Shibata M. Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy. 2008;4:404–408. doi: 10.4161/auto.5598. [DOI] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O’Leary H, Port JD, Bayer KU. Dual Mechanism of a Natural CaMKII Inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective post-insult neuroprotection by a novel Ca(2+)/calmodulin-dependent protein kinase II (CaMKII) inhibitor. J Biol Chem. 2010;285:20675–20682. doi: 10.1074/jbc.M109.088617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, Shacka JJ, Zhang J, Roth KA. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem. 2010;285:10497–10507. doi: 10.1074/jbc.M110.103747. [DOI] [PMC free article] [PubMed] [Google Scholar]