

Abstract
Major nuclear envelope abnormalities, such as disruption and/or presence of intranuclear organelles, have rarely been described in cardiomyocytes from dilated cardiomyopathy (DCM) patients. In this study, we screened a series of 25 unrelated DCM patient samples for (a) cardiomyocyte nuclear abnormalities and (b) mutations in LMNA and TMPO as they are two DCM-causing genes that encode proteins involved in maintaining nuclear envelope architecture. Among the 25 heart samples investigated, we identified major cardiomyocyte nuclear abnormalities in 8 patients. Direct sequencing allowed the detection of three heterozygous LMNA mutations (p.D192G, p.Q353K and p.R541S) in three patients. By multiplex ligation-dependant probe amplification (MLPA)/quantitative real-time PCR, we found a heterozygous deletion encompassing exons 3–12 of the LMNA gene in one patient. Immunostaining demonstrated that this deletion led to a decrease in lamin A/C expression in cardiomyocytes from this patient. This LMNA deletion as well as the p.D192G mutation was found in patients displaying major cardiomyocyte nuclear envelope abnormalities, while the p.Q353K and p.R541S mutations were found in patients without specific nuclear envelope abnormalities. None of the DCM patients included in the study carried a mutation in the TMPO gene. Taken together, we found no evidence of a genotype–phenotype relationship between the onset and the severity of DCM, the presence of nuclear abnormalities and the presence or absence of LMNA mutations. We demonstrated that a large deletion in LMNA associated with reduced levels of the protein in the nuclear envelope suggesting a haploinsufficiency mechanism can lead to cardiomyocyte nuclear envelope disruption and thus underlie the pathogenesis of DCM.
Keywords: Lamin A/C, Thymopoietin, Mutation, Dilated cardiomyopathy, Cardiomyocyte, Nucleus ultrastructure
Introduction
Dilated cardiomyopathy (DCM) is characterized by dilatation of cardiac chambers and impaired contraction. Severity of symptoms and age of onset are highly variable. To date, mutations in 28 genes have been associated with autosomal dominant DCM [3, 9–11, 18, 19, MIM#115200]. LMNA, which encodes lamin A/C, is one of the most commonly implicated genes in DCM. Mutations in LMNA are associated with a high risk of dysrhythmia, sudden death and heart failure [4, 35]. Furthermore, symptomatic DCM patients carrying LMNA mutations display a worse prognosis than DCM patients carrying a mutation in another DCM-associated gene [2, 17, 19, 26, 44, 46, 48].
The A-type lamins A and C, alternatively spliced products from LMNA, are type V intermediate filament proteins expressed in terminally differentiated somatic cells. They are components of a thin filamentous mesh-work—the nuclear lamina—underlying the nucleoplasmic side of the inner nuclear membrane, and are also present in the nucleoplasm [8, 20, 28, 30]. Lamins have traditionally been considered key components in providing structural support to the nucleus and anchoring chromatin and nuclear pore complexes to the nuclear envelope [24, 40, 52]. Intranuclear lamins have proposed functions in DNA replication [20, 25, 28, 29], transcription [41] and chromatin organization [38, 50]. In order to carry out their functions, lamins interact with a number of nuclear envelope proteins, chromatin and transcription factors. Among the genes encoding the lamin A/C interacting proteins, TMPO, the gene encoding nucleoplasmic thymopoietin alpha [also called lamina-associated polypeptide 2 (LAP2) alpha] has been shown to harbour a mutation in DCM patients [47]. The identified mutation occurred in the C-terminal domain of thymopoietin alpha, a region known to interact with lamin A/C (residues 319–566) [9]. Three thymopoietin isoforms, alpha, beta and gamma are encoded by TMPO [6]. The three isoforms share an identical amino terminal sequence but have divergent carboxy terminal sequences [9]. Thymopoietin is thought to play an important role in maintaining nuclear architecture through its binding with lamins and other proteins.
Cardiomyocytes from DCM patients with LMNA mutations exhibit the following characteristics: (a) reduced lamin A/C expression in myocyte nuclei on immunofluorescence/immunohistochemistry studies and (b) two types of ultrastructural changes, minor nuclear envelope damage, such as focal disruptions, blebs and nuclear pore clustering, and major morphologic alterations, including a complete loss of the nuclear envelope and accumulation of mitochondria, glycogen and/or lipofuscin in the nucleoplasm [2, 7, 13, 44, 51]. To date, these major nuclear envelope defects were only found in a DCM patient carrying the p.D192G LMNA mutation [7, 13, 44]. However, in non-genotyped DCM patients as well as in heart failure patients, minor nuclear defects such as irregular nuclear envelope, enlarged and bizarre shaped nuclei, indentations, giant and multiple nucleoli and irregularly distributed chromatin are commonly found [1, 4, 34, 37]. There is considerable heterogeneity in the abnormalities observed from one cardiac sample to the next as well as within one particular sample [1, 37].
The aim of the present study was to ascertain whether: (1) major nuclear envelope abnormalities are a common feature in patients with DCM and (2) LMNA and/or TMPO mutations are associated with nuclear envelope defects in cardiomyocytes from a series of 25 unrelated DCM patients.
Materials and methods
Patient recruitment
Written informed consent was obtained from all patients in accordance with study protocols approved by the hospital ethics committees. Diagnosis of DCM was established based on WHO/ISFC criteria modified by Mestroni and colleagues [27] that included severe LV systolic dysfunction (LV dilatation exceeding 117% of normal value corrected for age and body surface area) and LV ejection fraction (LVEF) ≤45% measured in angiography, without significant coronary artery disease (>50% lumen diameter reduction of one of the main coronary arteries). Exclusion criteria included hypertension, or acquired or congenital heart disease. All patients underwent clinical examination, evaluation of functional status according to NYHA classification, ECG study, two-dimensional echocardiography with Doppler and/or coronary angiography to exclude coronary artery disease. Left ventricular enlargement was calculated according to the method of Henry. A total of 25 index cases recruited in Canada and Poland were enrolled in the study.
Cardiac tissue collection, electron microscopy and immunostaining
Cardiac tissue samples were collected from endomyocardial biopsies performed on clinical indication or from explanted heart tissue. Samples were immediately processed in 1.6–3% gluteraldehyde. Fixed heart tissues were processed into thick and thin sections according to the standard methods. Tissue sections were examined with a Hitachi 7100 or JEM 1200EX electron microscope.
For indirect immunofluorescence examinations, 8 μm cryostat sections were stained with two monoclonal antisera against lamin A/C (NCL-LAM-A/C) (Novocastra Laboratories, Newcastle, UK) and A4, kindly provided by Dr. Hutchison (Department of Biological Science, University of Dundee, UK). A4 antibody detects only lamin A as its recognition site lies after amino acid 572. NCL-LAM-A/C recognizes both lamin A and lamin C. In brief, antibodies diluted 1:10 were applied to tissue sections for 60 min. Sections were rinsed with PBS and incubated for another 60 min with the appropriate Rodamine Red X conjugated goat anti-rabbit IgG secondary antibody diluted 1:30 with PBS. After being washed with PBS, the sections were mounted in gelmount and viewed with an Opton Zeiss standard LAB16 microscope with epifluorescence optics.
Screening of LMNA and TMPO coding sequences
Genomic DNA was isolated from white blood cells (Qiagen Flexigene kit). To screen for somatic mutations, DNA was extracted from cardiac tissues using a Qiamp Mini DNA extraction kit (Qiagen).
Intronic oligonucleotide primers flanking each of the exons were designed based on published sequences (Gen-bank accession number: L123399, L12400 and L12401 for LMNA and UCSC genome bioinformatics database for TMPO). All DNA samples were subjected to PCR and direct sequencing (ABI Prism Big Dye, ABI PRISM 3100 genetic analyzer). If there was any suspicion of genomic variation in a given patient, another sample of DNA collected independently was systematically double-strand sequenced (Table 1). Sequences were compared to two control samples from individuals without known cardiovascular disease. Each sample’s electropherogram was analyzed by two independent investigators.
Table 1.
Primer pairs and annealing temperatures used to amplify LMNA and TMPO coding regions
| Primer sequences (5′ to 3′) | PCR annealing temperature (°C) | |||
|---|---|---|---|---|
| LMNA | Exon | Forward | Reverse | |
| 1A | TCTGTCCTTCGACCCGAG | GTAGACCGCCAAGCGATC | 51 | |
| 2A | GGAGGACCTGCAGGAGCT | GCCCTCTCACTCCCTTCC | 50 | |
| 3 | CCTTCAAGTTCTTGTGTTCTGTGAC | CCTAGCCCAGCCCAAGTCTGTC | 58 | |
| 4 | GGCCTCCCAGGAACTAATTCTG | CTCCCTGCCACCATCTGC | 63 | |
| 5 | GCTGTAGCAGTGATGCCCAAC | CCAAAGCCCTGAGAAGTGAAG | 62 | |
| 6 | ATCCTGGAGAGAGTAGCCAG | TCTAGTCAAGGCCAGTTGCC | 60 | |
| 7 | CCCCACTTGGTCTCCCTCTCC | CCCTGATGCAGCTGTATCCCC | 60 | |
| 8 | GAGGCCTCAATTGCAGGCAGGC | GAAAAGGACACTTACCCCAGC | 62 | |
| 9 | GGAGCGCTGGGGTAAGTGTC | CTCGTCCAGCAAGCAGCCAG | 60 | |
| 10 | GTAAGCAGCAGGCCGGACAAAG | GATGCCATGGAATATTCCTGTG | 58 | |
| 11A | GGTCAGTCCCAGACTCGC | ACCAGATTGTCCCCGAAG | 55 | |
| 11B | GTCACTCGCAGCTACCGC | CCACCTCGTCCTACCCCT | 52 | |
| 12 | CTTGTCTGAGCCCCAGACTGGAG | AGGGAAAAGGAAGGGAGGAGAAAT | 65 | |
| TMPO | 1 | GTCTAAGGGGAAGGGTGGAG | CAGACCCACACGTCAAAGAAC | 60 |
| 2 | CCAATTTGGTAGTGAGTTTGCA | TAATTTGGGGTCCTGCTTCA | 60 | |
| 3 | TGAGCTGCATCCTAAATGAAAC | TGAGCTGCATCCTAAATGAAAC | 57 | |
| 4 | GCTTTGTCTACCAGGGCAAATC | GGCAGCCATCTTCACTCATC | 60 | |
| 5 | AAACCAGGGTTCCCGATTA | TGGATTAGTGTTGTCAGGAGGTT | 60 | |
| 6 | CCAGTATGGCCGTTATTAAAGTA | CTCCCTCCCACTCCAAAAA | 60 | |
| 7 | AAGAGAGCCTTGGAAGCATGTG | ACCATTGTACCTGGCTCCAAA | 60 | |
| 8 | TCAGGGAATGTGCTTGGAAT | GCAGTTTTTATTCAGCAGAGAA | 60 | |
Multiplex-ligation dependant probe amplification (MLPA) analysis was performed to screen for deletions and duplications in the LMNA gene coding sequence according to the manufacturer’s instructions (SALSA MLPA KIT P048 LMNA, MRC Holland). The probe mix contained probes for 10 of the 12 coding exons of LMNA, but not for exons 5 and 9 because of the close proximity of exons 4 and 5, and exons 8 and 9. The probe mix also contained 13 probes for other human genes. Two of these probes recognize genes localized on chromosome 1. One of these probes was located 94 kb upstream of LMNA exon 1 and another 100 kb downstream of LMNA exon 12. MLPA amplification products were analyzed on an ABI model 3130XL Genetic Analyzer (Applied Biosystems) with the GeneMapper software V.3.7, using Genescan 500LIZ internal size standard (Applied Biosystems). Each patient’s electropherogram was compared to three controls. We used the Coffalyzer MLPA DAT (MRC-Holland) software to analyze MLPA data. Resulting normalized ratios were ~1.0 for every wild-type peak, 0.5 for heterozygous deletions and 1.5 for heterozygous duplications.
Quantitative real-time PCR was used to confirm the deletion found in LMNA (Roche LightCycler 2.0) [36]. Primers were designed for LMNA exon 9 (forward: 5′-ggagcgctggggtaagtgtc-3′; reverse: 5′-ctcgtccagcaagca gcccag-3′). PCR was set up in capillaries in a total volume of 20 μl; each PCR mixture contained 2.5 mM MgCl2 (2.5 mM), 2 μl of sybr green mix, 0.6 μM of mixture of forward and reverse primers and 20 ng of DNA. Standard curves for both the housekeeping gene (β2-microglobulin) and target gene were generated. Two different types of sample DNA (i.e., one sample from the DCM patient carrying the deletion and one sample from a DCM patient without the deletion) were utilized to ensure that the gene dosage ratio of the target to the housekeeping gene was disrupted only when a deletion was present. Using the slope and y-intercept of the standard curve, the LightCycler system calculated the concentrations (M) of sample and control DNA for both the reference gene (β2 microglobulin) and the target gene (Lamin A/C). These concentrations were then used to calculate the gene dosage ratio using the following equation:
where R = 0.5 (0.4–0.7) indicates deletion, R = 1 (0.8–1.2) indicates normal copy number and R = 1.5 (1.3–1.7) indicates duplication
Statistical analysis
LVEF and LVEDD in percentage values were compared between patients with major cardiomyocyte nuclear abnormality and patients with non-specific cardiomyocyte nuclear abnormality using a two-tailed Student’s t test (level of significance 5%).
Results
Ultrastructural characteristics of endomyocardial samples
Ultrastructural analysis of endomyocardial samples enabled us to distinguish two groups of DCM patients. One group consisted of eight individuals exhibiting major nuclear envelope abnormalities on electron micrographs (Fig. 1) and the second consisted of 17 patients with minor and non-DCM specific abnormalities.
Fig. 1.

Electron micrographs of cardiomyocytes from dilated cardiomyopathy patients with or without LMNA mutation. a, b, c and d Major nuclear abnormalities in, respectively, patient 1, 3, 4 and 5 (Table 2). Patient 1 carried the LMNA deletion exons 3–12, while patients 3, 4 and 5 had no LMNA nor TMPO mutation; a accumulation of mitochondria around the nuclear envelope; b blebbing of the nuclear envelope and separation of the inner and the outer nuclear membrane; c extrusion of nucleoplasm from the cardiomyocyte nucleus into the cytoplasm; d accumulation of cytoplasmic organelles in the nucleoplasm; e non-specific nuclear alteration in patient 10 (Table 2) carrying the p.Q353K LMNA mutation; f non-specific alterations in dilated cardiomyopathy patient 14 (Table 2) with wild-type LMNA and TMPO (original magnifications: a, 20,000×; b, 18,000×; c, 12,000×; d, 10,000×; e, 4,000×; and f, 4,000×)
Among the patients with nuclear envelope defects, the proportion of defective cells ranged from a small percentage to as much as 30% (patients 2 and 6). The most obvious abnormality, occurring in four of eight patients, was intrusion and accumulation of mitochondria and other cytoplasmic organelles into the nuclear matrix (patients 1, 2, 5 and 7) (Fig. 1a, d). The membranes of these organelles appeared discontinuous from the nuclear envelope (Fig. 1a, d). The nuclear envelope was usually irregular (Fig. 1a–c) and partially disrupted or totally missing in six of eight patients (patients 1, 2, 4, 5, 6 and 7) (Fig. 1a, c, d). Chromatin disorganization was also observed (patients 2 and 6). In contrast, samples from the 17 patients in the second group displayed modest and non-specific nuclear membrane alterations, such as nuclear irregularity, that are commonly found in DCM patients regardless of the presence of LMNA mutations (Fig. 1e, f).
Clinical characteristics of the patient cohort
Clinical characteristics of the 25 DCM patients (19 males and 6 females) included in our study are shown in Table 2. There was considerable heterogeneity in terms of the clinical characteristics and severity of DCM across the cohort of 25 patients.
Table 2.
Patients’ clinical characteristics at the time of the heart sampling, LMNA mutation status and nuclear envelope defect status
| Patient | LMNA mutation | Family history | Nuclear envelope defect | Sex/age at onset (years) | NYHA class | Echocardiography | Weight (kg)/height (cm)/LVEDD (%) | Dysrhythmias | Clinical status |
|---|---|---|---|---|---|---|---|---|---|
| Major nuclear envelope defects and LMNA mutation | |||||||||
| 1 | Deletion exons 3–12 | Yes | Irregular and broken nuclear envelope; accumulation of mitochondria within and around the nuclei | F/39 | II | LVEDD 54 mm, LVEF 50% | 57/164/120.0 | nsVT, couplets, frequent VE, ICD | Mild progressive HF No MD |
| 2* | p.D192G (c.575A > G) | Yes | Complete loss of nuclear envelope, accumulation of mitochondria, glycogen and/ or lipofuscin in the nucleoplasm, chromatin disorganization | M/26 | IV | LVEDD 60 mm, LVEF 20% | 67/175/127.2 | 1 AVB, LAFB | Died at 27 while awaiting for a heart transplant No MD |
| Major nuclear envelope defects and no LMNA mutation | |||||||||
| 3 | No | Yes | Blebbing of nuclear envelope, separation of inner and outer nuclear membrane | M/63 | III | LVEDD 64 mm, LVEF 26% | 71/180/136.5 | SVT, VT | Died of progressive HF, 4 years after diagnosis No MD |
| 4 | No | Yes | Extrusion of nucleoplasm from the cardiomyocyte nucleus into the cytoplasm | M/21 | III | LVEDD 75 mm, LVEF 25% | 80/176/153.7 | nsVT | HTx at 24 No MD |
| 5 | No | No | Irregular nucleus border; lack of nuclear membrane, presence of mitochondria within nuclear matrix | M/29 | III | LVEDD 59 mm, LVEF 30% | 84/176/120.5 | RBBB, LPFB, nsVT, Pacemaker at 32 | Progressive HF, HTx at 40 No MD |
| 6 | No | No | Local disruption of nuclear envelope, chromatin disorganization | F/14 | IV | LVEDD 64 mm, LVEF < 10% | 55/152/144.7 | Recurrent VT/ VF | HTx at 14 No MD |
| 7 | No | No | Misshapen nuclei, local disruption of nuclear envelope, penetration of mitochondria into nucleus | M/14 | III | LVEDD 71 mm, LVEF 20% | 57/168/155.6 | nsVT, couplets, frequent VE, ICD at 20 | Progressive HF No MD |
| 8 | No | No | Irregular shape of nuclei | M/18 | III | LVEDD 68 mm, LVEF 20% | 47/160/156.0 | LBBB, nsVT | Progressive HF No MD |
| Non-specific nuclear envelope defects and LMNA mutation | |||||||||
| 9* | p.R541S (c. 1621C >A) | Yes | No | M/12 | IV | LVEDD 64 mm, LVEF 25% | 49/152/147.3 | NA | HTx at 13 No MD |
| 10 | p.Q353K (c.1057C > A) | Yes | No | M/38 | IV | LVEDD 62 mm, LVEF 22%, | 59/163/138.1 | ICD | HTx at 38 Skeletal myopathy |
| Non-specific nuclear envelope defects and no LMNA mutation | |||||||||
| 11 | No | Yes | No | M/27 | II | LVEDD 60 mm, LVEF 38% | 100/186/117.2 | Permanent AF, single VE | Improved after AF ablation, LVEF 45% No MD |
| 12 | No | Yes | No | M/26 | III | LVEDD 75 mm, LVEF 20% | 73/183/154.8 | Single VE | Stable HF No MD |
| 13 | No | Yes | No | M/40 | NA | LVEDD 77 mm, LVEF 19% | 73/168/164.3 | ICD, pacemaker | Died at 59, poor candidate denied for HTx No MD |
| 14 | No | Yes | No | F/14 | IV | LVEDD 58 mm, LVEF 14% | 37/155/139.4 | No | HTx at 14 No MD |
| 15 | No | Yes | No | M/36 | II | LVEDD 78 mm, LVEF 25% | 94/185/154.9 | Single Vex | Stable HF No MD |
| 16 | No | Yes | No | M/27 | III | LVEDD 78 mm, LVEF 10% | 72/186/160.7 | Sinus tachycardia | HTx within several months No MD |
| 17 | No | Yes | No | M/34 | IV | LVEDD 78 mm, LVEF 20% | 73/178/163.1 | I-degree AVB | Fulminant HF leading to HTx within several months No MD |
| 18 | No | Yes | No | M/35 | III | LVEDD 58 mm, LVEF 15% | 80/170/121.1 | Frequent sVT | Atrial septal aneurysm; HTx at 39 No MD CPK value up to 600 U/I |
| 19 | No | No | No | F/13 | NA | LVEDD 69 mm, LVEF 10% | 41/164/160.3 | NA | HTx at 13 No MD |
| 20 | No | No | No | F/61 | NA | LVEDD 69 mm, LVEF 12% | 88/165/145.3 | ICD | Pericarditis, HTx at 62 No MD |
| 21 | No | No | No | M/54 | III | LVEDD 88 mm, LVEF 20% | 109/175/175.1 | Pacemaker | Died at 60, waiting for HTx No MD |
| 22 | No | No | No | M/40 | III–IV | LVEDD 68 mm, LVEF 27% | 71/160/147.8 | No | HTx at 62 No MD |
| 23 | No | No | No | M/27 | NA | LVEDD 96 mm, LVEF 13% | 70/170/203.8 | ICD | HTx at 57 No MD |
| 24 | No | No | No | F/12 | NA | LVEF 13% | 42/153/NA | NA | HTx at 12 No MD |
| 25 | No | No | No | M/20 | NA | LVEF 13% | 86.5/197/NA | NA | HTx at 22 No MD |
AF atrial fibrillation, AVB atrioventricular block, HF heart failure, HTx heart transplantation, ICD implantable cardioverter defibrillator, LAFB left anterior fascicular block, LPFB left posterior fascicular block, LVEDD left ventricle end diastolic diameter, LVEF left ventricle ejection fraction, MD muscular disease, NA not available, RBBB right bundle branch block, VEX ventricular extrasystole, VE ventricular ectopy, VT ventricular tachycardia, nsVT nonsustained ventricular tachycardia, SVT supraventricular tachycardia, VT/VF ventricular tachycardia/ventricular fibrillation
In the group of eight patients who exhibited nuclear envelope abnormalities, age at disease onset ranged from 14 to 63 years, four patients had a family history of DCM (Table 2, Fig. 2, [44]), while none had a suspicion of muscular dystrophy. Disease severity ranged from mild DCM to progressive heart failure and LVEF values ranged from <10 to 50%. Various arrhythmias were present in all eight patients; two patients required ICD (patients 1 and 7) and one required pacemaker (patient 5). Three patients had received a heart transplant (patients 4, 5 and 6) and two patients had died of heart failure (patients 2 and 3). In addition, patient 5 presented with hypothyroidism and patient 7 with mild mental retardation. No other phenotypic abnormality was identified within this population.
Fig. 2.
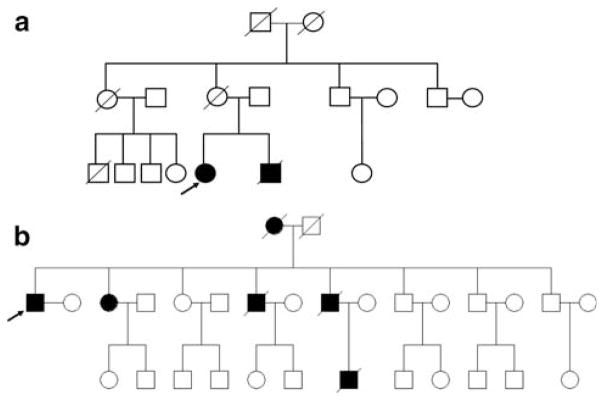
Pedigree of the families with LMNA mutations. Arrow indicates proband; black filled symbol dilated cardiomyopathy patient; open symbol asymptomatic individual. a Deletion of exons 3–12; b p.Q353K
A similar degree of heterogeneity was also observed among the 17 patients with no major nuclear envelope abnormalities: age at disease onset ranged from 12 to 61 years, 10 patients had a family history of DCM (Table 2, Fig. 2, [44]), 1 patient had documented skeletal myopathy (patient 10) and 1 had elevated CPK values [up to 600 U/I (patient 18)]. LVEF values ranged from 10 to 38%; conduction defects were present in all but two patients (data were not available for four patients), four patients required ICD and two had pacemakers. Twelve patients had received heart transplants and two patients had died of heart failure (patients 13 and 21).
We compared mean LVEF and LVEDD in percentage values between patients exhibiting major nuclear envelope abnormalities and patients with minor and non-DCM-specific abnormalities. No significant correlation between the severity of the phenotype and the presence of nuclear envelope defect was detected (p >0.05).
Screening of LMNA and TMPO coding sequences
We screened the complete coding sequence as well as the intron–exon boundaries of LMNA (12 exons) and TMPO (8 exons) for mutations using direct sequencing of DNA extracted from white blood cells of all 25 patients. We identified three patients with LMNA mutations, one in the group with major nuclear envelope defects (patient 2, p.D192G) and two in the group with non-specific nuclear envelope abnormalities (patient 9, p.R541S and patient 10, p.Q353K). Patient 2 and 9 and their LMNA mutations have been described previously [7, 44]. All these variations were absent in DNA from more than 100 controls. No mutation was found in TMPO. Previously reported polymorphisms in both LMNA (synonymous polymorphisms rs538089, rs505058, rs4641) and TMPO (non-synonymous polymorphism rs17459334) were detected. We also found a c.C1341A synonymous polymorphism in LMNA exon 6, which has not been previously reported, in two control individuals. Mutations in the corresponding codon have been previously reported in patients with limb-girdle muscular dystrophy 1B (p.R377H, [31]; p.R377L, [21]).
Sequencing analysis cannot detect large heterozygous deletions or duplications. In an attempt to ascertain whether the defects resulted from deletions or duplications in LMNA, we performed multiplex ligation-dependant probe amplification (MLPA) on the LMNA coding sequence in all 25 DCM patients. One DCM patient (Table 2, patient 1) with major nuclear abnormality displayed a MLPA normalized ratio peak area of ~0.5 for exons 3–12 (Fig. 3). This indicated the presence of a heterozygous deletion encompassing exons 3 to 12 (>4,704 bp). The remaining probes, including those for exons 1 and 2, displayed a normalized ratio of ~1.0 indicating normal copy number (Fig. 3). We confirmed the presence of the deletion using qPCR (R value of 0.5 for the patient with the deletion and 1 for the patient without the deletion) (Fig. 4).
Fig. 3.

MLPA analysis graph showing the heterozygous LMNA deletion of exons 3–12 in patient 1. Exons 3–12 display a normalized ratio of ~0.5; indicating a loss of genetic materiel. Exons 1, 2 and the remaining control probes display a normalized ration of ~1.0; indicating normal copy number. The control probes are targeted within the same chromosome and to different chromosomes as indicated by their locus
Fig. 4.

Confirmation of the deletion of exons 3–12 in LMNA in patient 1 using qPCR. R values were calculated as described in the “Materials and methods”. Patient 14 without the deletion had a R value of 0.955, which indicates a normal copy number of the exon. The R value for patient 1 is 0.476, which indicates the presence of only one copy of the exon (n = 3)
Lastly, to assess whether the nuclear envelope defects in the remaining patients could result from somatic mutations, we screened for LMNA and TMPO mutations in the heart from sporadic cases with nuclear envelope defect for which cardiac tissue was available (patients 5, 6) as well as in heart from patients without defective nuclear envelope (patients 19, 20, 21, 22, 23, 24 and 25). This analysis did not reveal any somatic mutations.
In summary, a large degree of clinical heterogeneity was evident in the cohort of DCM patients studied regardless of the presence of nuclear envelope abnormalities, LMNA mutation or family history of DCM.
Immunostaining results
To gain further insight into the functional significance of the heterozygous deletion of exons 3–12 of LMNA observed in patient 1, we performed indirect immunofluorescence analysis of endomyocardial samples from this patient as well as a control patient without LMNA mutation, using antibodies directed against both lamin A and C. All cardiomyocyte nuclei were immunostained with antibodies directed against lamin A and C epitopes (Fig. 5). The immunostaining was reduced in patient 1 as compared to the control patient indicating significant attenuation of lamin expression (Fig. 5).
Fig. 5.
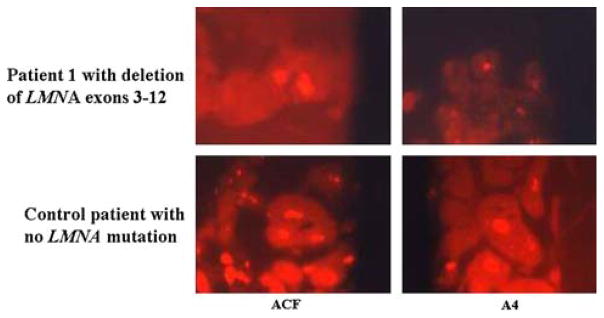
Indirect immunofluorescence analysis of endomyocardial biopsy from patient 1 carrying the LMNA heterozygous exon 3–12 deletion and from a control patient with no LMNA mutation. Endomyocardial biopsy was taken from the right ventricle. Immunostaining was performed using anti-lamin A+C monoclonal antibodies ACF and antibody A4 which detects lamin A only (see “Materials and methods”) and the goat anti-rabbit IgG secondary antibody (original magnification 1,050×)
Discussion
Among heart samples from 25 unrelated DCM patients, we identified major cardiomyocyte nuclear abnormalities in 8 individuals and non-specific nuclear abnormalities in the remaining patients. The presence of nuclear abnormalities, along with the fact that all patients presented with dysrhythmias and/or severe heart failure leading to cardiac transplantation, prompted us to analyze the sequence of LMNA and TMPO, two DCM-causing genes involved in maintaining nuclear envelope architecture.
Direct sequencing allowed the identification of three heterozygous LMNA genomic variations (p.D192G, p.R541S and p.Q353K) in three patients. Since sequencing analysis cannot detect large heterozygous deletions or duplications, we performed multiplex ligation-dependant probe amplification (MLPA) on the LMNA coding sequence in all 25 DCM patients. We found a heterozygous deletion encompassing exons 3–12 of the LMNA gene. This deletion was confirmed using quantitative real-time PCR. All variations were absent in DNA from more than 100 controls. The occurrence of LMNA mutation in our population is high probably due to fact that the studied population presented with cardiac condition requiring undergoing endomyocardial biopsies or cardiac transplantation. Moreover, various arrhythmias were present in all but two patients. Since mutations in LMNA are usually associated with a worse prognosis than any other DCM-associated gene mutation [2, 17, 18, 26, 44, 46, 48] as well as with dysrhythmia [4, 35], our population was probably biased toward LMNA mutations carriers. None of the patients carried a mutation in the TMPO gene.
While rare findings of small deletions in the lamin A/C gene in patients with laminopathies have been reported previously, to our knowledge, this study is the first to document a large deletion encompassing most exons of the lamin A/C gene in a DCM patient. Walter et al. [53] found a 15 amino acid deletion (−3 to 12 nucleotides) in the 5′ end of the LMNA gene in a patient with Emery–Dreifuss muscular dystrophy (EDMD) that resulted in loss of the translation initiator codon. van Tintelen et al. [49] reported a 674 bp deletion encompassing exon 1 and the adjacent non-coding exon in a patient with myocardial fibrosis. The presence of the exon 1 p.Q6Stop LMNA mutation resulting in reduced levels of lamin A/C proteins has also been reported in a family with EDMD [5].
Most LMNA mutations that cause DCM are heterozygous point mutations, only some of which have been associated with cardiomyocyte nuclear envelope defects [2, 7, 13, 44, 51]. It has been suggested that mutations causing such abnormalities have a dominant-negative effect on the functions of normal lamin protein. Impaired lamin integration into the nuclear lamina due to the dominant-negative effects of a mutation may cause disruption of the lamina and ultimately compromise nuclear envelope integrity. Our results suggest that, apart from the dominant-negative effects of mutant lamin A/C protein, lamin haplo-insufficiency can also cause nuclear envelope defects and underlie the pathogenesis of DCM. Since the deletion encompasses LMNA exon 3 to 12, it is likely that the observed nuclear abnormalities are not due to the expression of a putative truncated protein composed of exons 1–2 only. It was shown that the potential truncated protein resulting from the Y259X nonsense mutation (exon 4) was not detectable in patient fibroblasts [33]. Similarly, in the mouse model with the LMNA exon 8–11 deletion, the 54-kDa truncated protein resulting from this deletion was not detected [43]. Furthermore, immunostaining revealed reduced lamin A/C in the patient’s cardiomyocyte nuclei. That lamin haploinsufficiency can cause such defects is further corroborated by the fact that a heterozygous LMNA+/− mouse model in which cardiac lamin A/C levels were diminished by 50% compared to wild-type, demonstrated early-onset conduction system disease and late-onset DCM [54]. Furthermore, misshapen cardiomyocyte nuclei and heterochromatin clumping were also observed [54].
The LMNA deletion of exons 3–12 was observed in a patient presenting with major nuclear envelope abnormalities—notably broken nuclear envelope and accumulation of mitochondria within and around the nuclei. This suggests that haplo-insufficiency is the mechanism underlying the observed nuclear abnormalities. Among their many functions, lamins play an integral role in maintaining the mechanical stability of the nucleus; hence we can speculate that the nuclear envelope defects observed in the patient carrying this LMNA deletion results from the reduced levels of lamin A/C protein.
Apart from the observed deletion, one of our patients with nuclear envelope defects carried a p.D192G point mutation in the LMNA gene. When expressed in COS-7 cells, this mutation resulted in a discontinuous nuclear lamina due to the formation of multiple lamin aggregates [44, 45]. These results therefore corroborated the pathological findings from the patient with the mutation.
In our study, not all LMNA mutations were found to cause nuclear envelope abnormalities in patients’ cardiomyocytes. Indeed, two patients with LMNA point mutations demonstrated no specific nuclear envelope abnormalities in the samples taken from explanted heart tissue: one patient carried a p.Q353K mutation, while the second carried the p.R541S mutation. It is possible that the number of abnormal nuclei was so low in these two patients that it resulted in a false-negative finding. Patient age at the time of tissue collection and the protein domain affected by the mutation were considered as possible explanations for the observed discrepancy in nuclear envelope phenotype. Age at the time of tissue collection was 38 and 12 years for the patients carrying mutations p.Q353K and p.R541S, respectively, both of which were associated with normal nuclear envelope ultrastructure. The patients with LMNA deletion and p.D192G mutation, both of whom demonstrated nuclear envelope abnormalities, were 39 and 26 years of age, respectively. Hence, there was no apparent relationship between age and observed nuclear envelope phenotype. It was also considered possible that the presence of nuclear envelope abnormalities may depend upon the protein domain affected by each mutation [12, 32]. The p.D192G mutation, which was associated with major nuclear envelope abnormalities, is located in the central α-helical rod domain at the distal end of coil 1B. This highly conserved region of the lamin A/C protein is critical for the formation of the α-helical coiled-coil dimer, the basic building block for the construction of lamin filaments. In contrast, the p.Q353K mutation in exon 6 affected a highly conserved residue localized in the central rod domain coil 2B fragment. Based on the coil 2B fragment crystal structure, mutations in this domain are unlikely to alter the dimer structure but may interfere with essential molecular interactions occurring in later stages of filament assembly, lamina formation and/or chromatin interaction [42]. Similarly, the p.R541S mutation is located in highly conserved residues in a region of gene shared by lamin A and C isoforms within the carboxyl-terminal end. The buried side chain of R541 participates in the stabilization of the carboxy-terminal β-sandwich through hydrophobic contacts with the core of the domain [22]. Mutations in this domain could therefore destabilize the three-dimensional structure of the C-terminal domain of lamin A/C. This domain also carries sites for many lamin A/C interacting proteins. Therefore, both p.Q353K and p.R541S mutations could theoretically disrupt multiple functions of lamin A and C, including the maintenance of nuclear architecture. Unexpectedly, the ultrastructural phenotype associated with these mutations was indistinguishable from controls. Hence, our results do not support the presence of a relationship between the position of the mutation and the resulting ultrastructural phenotype.
The six patients demonstrating nuclear ultrastructural defects without LMNA or TMPO mutations showed that germ-line mutations in these genes are not a prerequisite for nuclear envelope defects. However, we could not discount the possibility that some of these individuals possessed somatic LMNA or TMPO mutations restricted to diseased cardiac tissue. The fact that only a percentage of observed cardiomyocytes from these patients displayed an abnormal nucleus lends support to this hypothesis. Indeed, somatic mutation occurring in a selective subpopulation of progenitor cell lineage is one of the molecular mechanisms leading to such tissue mosaicism. Furthermore, in other cardiovascular diseases such as idiopathic ventricular tachycardia or idiopathic atrial fibrillation, both germ-line and somatic missense mutations have been linked to the disease [14, 23]. We therefore screened for the presence of LMNA and TMPO mutations in DNA extracted from heart tissues of patients who did not have familial DCM, but were unable to identify any somatic mutations. Another possibility is that mutation in genes encoding lamin A/C-binding proteins are responsible for the major cardiomyocyte nuclear abnormalities observed in these patients [15, 16, 39, 55–57].
We showed here that there is no clear correlation between the presence or position of the LMNA mutation and either clinical phenotype with respect to DCM onset or severity or cardiomyocyte ultrastructural phenotype. We also found no apparent correlation between genotype and either severity of the ultrastructural aberration (i.e., the degree of nuclear envelope disruption), or the proportion of observed cells displaying abnormal nuclei. Furthermore, there was no significant correlation between the severity of the disease and the presence of ultra-structural nuclear envelope defects. Although a quantitative analysis was not possible due to the small sample size, overall, we found no evidence of a relationship between the onset and the severity of DCM, the presence of nuclear abnormalities and the presence or absence of LMNA mutations.
Taken together, our results suggest that lamin A/C haplo-insufficiency, documented by reduced protein level in the patient’s cardiomyocyte nuclear envelope caused by a large deletion in the LMNA gene, can lead to nuclear envelope disruption and underlie the pathogenesis of DCM. However, the finding that two patients without cardiomyocyte nuclear abnormalities had LMNA mutations indicates that LMNA mutations may not necessarily lead to major cardiomyocyte nuclear envelope defects. Furthermore, patients with major nuclear envelope abnormalities may not have LMNA or TMPO mutation. This demonstrates that patients with a clinical suspicion of laminopathy and marked abnormality of cardiomyocyte nuclei can be free of both LMNA and TMPO mutations and may carry a mutation in another gene encoding a protein involved in the maintenance of the nuclear architecture.
Acknowledgments
The work was supported by Canadian Institutes for Health Research operating grants 38054, 65152 and 77685, and by Heart and Stroke Foundation Grants NA 5101 and 6628 awarded to F. Tesson and by an internal grant from the Institute of Cardiology (Warsaw, Poland) no:2.57/VII/03. At the time the study was conducted, N. Sylvius was the recipient of the fellowships awarded by the Heart and Stroke Foundation of Ontario Program Grant 5275 and Astra Zeneca/Canadian Society of Hypertension/CIHR, and Pallavi Gupta was the recipient of Ontario Graduate Scholarship in Science and Technology (OGSST). We acknowledge the important contribution of the Canadian Cardiovascular Genetics Centre (Ottawa, Canada).
Footnotes
Conflict of interest statement The authors declare that they have no conflict of interest.
Contributor Information
Pallavi Gupta, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
Zofia T. Bilinska, 1st Department of Coronary Artery Disease, Institute of Cardiology, Alpejska 42, 04-6 28 Warsaw, Poland
Nicolas Sylvius, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
Emilie Boudreau, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
John P. Veinot, Department of Pathology and Laboratory Medicine, University of Ottawa, Anatomical Pathology, Ottawa Hospital, Ottawa, ON K1Y 4E9, Canada
Sarah Labib, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
Pierrette M. Bolongo, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada
Akil Hamza, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
Tracy Jackson, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
Rafal Ploski, Department of Medical Genetics, Warsaw Medical University, Pawinskiego Street 3c, 02-106 Warsaw, Poland.
Michal Walski, Department of Cell Ultrastructure, M Mossakowski Medical Research Centre, Polish Academy of Science, Pawinskiego Street 5, 02-106 Warsaw, Poland.
Jacek Grzybowski, 1st Department of Coronary Artery Disease, Institute of Cardiology, Alpejska 42, 04-6 28 Warsaw, Poland.
Ewa Walczak, Department of Pathology, Institute of Rheumatology, Spartanska 1, 02-637 Warsaw, Poland.
Grzegorz Religa, 2nd Department of Cardiac Surgery, Institute of Cardiology, Alpejska 42, 04-628 Warsaw, Poland.
Anna Fidzianska, Neuromuscular Unit, Medical Research Center, Polish Academy of Science, ul.Pawińskiego 5, 02-097 Warsaw, Poland.
Frédérique Tesson, Faculty of Health Sciences, University of Ottawa, 451 Smyth, Ottawa, ON K1H 8M5, Canada.
References
- 1.Arbustini E, Gavazzi A, Pozzi R, Grasso M, Pucci A, Campana C, Graziano G, Martinetti M, Cuccia M, Salvaneschi L, Martinelli L, Montemartini C, Vigano M. The morphologic spectrum of dilated cardiomyopathy and its relation to immune-response genes. Am J Cardiol. 1989;64:991–995. doi: 10.1016/0002-9149(89)90796-0. [DOI] [PubMed] [Google Scholar]
- 2.Arbustini E, Pilotto A, Repetto A, Grasso M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A, Tavazzi L. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. 2002;39:981–990. doi: 10.1016/s0735-1097(02)01724-2. [DOI] [PubMed] [Google Scholar]
- 3.Arimura T, Hayashi T, Matsumoto Y, Shibata H, Hiroi S, Nakamura T, Inagaki N, Hinohara K, Takahashi M, Manatsu S, Sasaoka T, Izumi T, Bonne G, Schwartz K, Kimura A. Structural analysis of four and half LIM protein-2 in dilated cardiomyopathy. Biochem Biophys Res Commun. 2007;357:162–167. doi: 10.1016/j.bbrc.2007.03.128. [DOI] [PubMed] [Google Scholar]
- 4.Baandrup U, Florio RA, Roters F, Olsen EG. Electron microscopic investigation of endomyocardial biopsy samples in hypertrophy and cardiomyopathy. A semiquantitative study in 48 patients. Circulation. 1981;63:1289–1298. doi: 10.1161/01.cir.63.6.1289. [DOI] [PubMed] [Google Scholar]
- 5.Bécane HM, Bonne G, Varnous S, Muchir A, Ortega V, Hammouda EH, Urtizberea JA, Lavergne T, Fardeau M, Eymard B, Weber S, Schwartz K, Duboc D. High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. J Pac Clin Electrophysiol. 2000;23:1661–1666. doi: 10.1046/j.1460-9592.2000.01661.x. [DOI] [PubMed] [Google Scholar]
- 6.Berger R, Theodor L, Shoham J, Gokkel E, Brok-Simoni F, Avraham KB, Copeland NG, Jenkins NA, Rechavi G, Simon AJ. The characterization and localization of the mouse thymopoietin/lamina-associated polypeptide 2 gene and its alternatively spliced products. Genome Res. 1996;6:361–370. doi: 10.1101/gr.6.5.361. [DOI] [PubMed] [Google Scholar]
- 7.Bilinska ZT, Sylvius N, Grzybowski J, Fidzianska A, Michalak E, Walczak E, Walski M, Bieganowska K, Szymaniak E, Kusmierczyk-Droszcz B, Lubiszewska B, Wagner T, Tesson F, Ruzyllo W. Dilated cardiomyopathy caused by LMNA mutations. Clinical and morphological studies. Polish Heart J. 2006;64:812–818. [PubMed] [Google Scholar]
- 8.Bridger JM, Kill IR, O’Farrell M, Hutchison CJ. Internal lamin structures within G1 nuclei of human dermal fibroblasts. J Cell Sci. 1993;104:297–306. doi: 10.1242/jcs.104.2.297. [DOI] [PubMed] [Google Scholar]
- 9.Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, Hetzer R, Regitz-Zagrosek V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;298:116–120. doi: 10.1016/s0006-291x(02)02374-4. [DOI] [PubMed] [Google Scholar]
- 10.Dechat T, Korbei B, Vaughan OA, Vlcek S, Hutchison CJ, Foisner R. Lamina-associated polypeptide 2 (alpha) binds intranuclear A-type lamins. J Cell Sci. 2000;113:3473–3484. doi: 10.1242/jcs.113.19.3473. [DOI] [PubMed] [Google Scholar]
- 11.Dubosc-Bidot L, Xu P, Charron P, Neyroud N, Dilanian G, Millaire A, Bors V, Komajda M, Villard E. Mutations in the z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res. 2008;77:118–125. doi: 10.1093/cvr/cvm015. [DOI] [PubMed] [Google Scholar]
- 12.Favreau C, Dubosclard E, Ostlund C, Vigouroux C, Capeau J, Wehnert M, Higuet D, Worman HJ, Courvalin JC, Buendia B. Expression of lamin A mutated in the carboxyl-terminal tail generates an aberrant nuclear phenotype similar to that observed in cells from patients with Dunnigan-type partial lipo-dystrophy and Emery–Dreifuss muscular dystrophy. Exp Cell Res. 2003;282:14–23. doi: 10.1006/excr.2002.5669. [DOI] [PubMed] [Google Scholar]
- 13.Fidziańska A, Bilińska ZT, Tesson F, Wagner T, Walski M, Grzybowski J, Ruzyllo W, Hausmanowa-Petrusewicz I. Obliteration of cardiomyocyte nuclear architecture in a patient with LMNA gene mutation. J Neurol Sci. 2008;271:91–96. doi: 10.1016/j.jns.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 14.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354:2677–2688. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 15.Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nature Rev Mol Cell Biol. 2005;6:21–31. doi: 10.1038/nrm1550. [DOI] [PubMed] [Google Scholar]
- 16.Haque F, Lloyd DJ, Smallwood DT, Dent CL, Shanahan CM, Fry AM, Trembath RC, Shackleton S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol. 2006;26:3738–3751. doi: 10.1128/MCB.26.10.3738-3751.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hermida-Prieto M, Monserrat L, Castro-Beiras A, Laredo R, Soler R, Peteiro J, Rodriguez E, Bouzas B, Alvarez N, Muniz J, Crespo-Leiro M. Familial dilated cardiomyopathy and isolated left ventricular noncompaction associated with lamin A/C gene mutations. Am J Cardiol. 2004;94:50–54. doi: 10.1016/j.amjcard.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 18.Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, Teraoka K, Chikamori T, Yamashina A, Kimura A. α-B crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;342:379–386. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 19.Karkkainen S, Peuhkurinen K. Genetics of dilated cardiomyopathy. Ann Med. 2007;39:91–107. doi: 10.1080/07853890601145821. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy BK, Barbie DA, Classon M, Dyson N, Harlow E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000;14:2855–2868. doi: 10.1101/gad.842600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ki CS, Hong JS, Jeong GY, Ahn KJ, Choi KM, Kim DK, Kim JW. Identification of lamin A/C (LMNA) gene mutations in Korean patients with autosomal dominant Emery–Dreifuss muscular dystrophy and limb-girdle muscular dystrophy 1B. J Hum Genet. 2002;47:225–228. doi: 10.1007/s100380200029. [DOI] [PubMed] [Google Scholar]
- 22.Krimm I, Östlund C, Gilquin B, Couprie J, Hossenlopp P, Mornon JP, Bonne G, Courvalin J-C, Worman HJ, Zinn-Justin S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy and partial lypodystrophy. Structure. 2002;10:811–823. doi: 10.1016/s0969-2126(02)00777-3. [DOI] [PubMed] [Google Scholar]
- 23.Lerman BB, Dong B, Stein KM, Markowitz SM, Linden J, Catanzaro DF. Right ventricular outflow tract tachycardia due to a somatic cell mutation in G protein subunit alpha i2. J Clin Inves. 1998;101:2862–2868. doi: 10.1172/JCI1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Ben-Shahar TR, Riemer D, Treinin M, Spann P, Weber K, Fire A, Gruenbaum Y. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol Biol Cell. 2000;11:3937–3947. doi: 10.1091/mbc.11.11.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier J, Campbell KH, Ford CC, Stick R, Hutchison CJ. The role of lamin LIII in nuclear assembly and DNA replication, in cell-free extracts of Xenopus eggs. J Cell Sci. 1991;98:271–279. doi: 10.1242/jcs.98.3.271. [DOI] [PubMed] [Google Scholar]
- 26.Mercuri E, Brown SC, Nihoyannopoulos P, Poulton J, Kinali M, Richard P, Piercy RJ, Messina S, Sewry C, Burke MM, McKenna W, Bonne G, Muntoni F. Extreme variability of skeletal and cardiac muscle involvement in patients with mutations in exon 11 of the lamin A/C gene. Muscle Nerve. 2005;31:602–609. doi: 10.1002/mus.20293. [DOI] [PubMed] [Google Scholar]
- 27.Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative research group of the European Human and Capital Mobility Project on familial dilated cardiomyopathy. Eur Heart J. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- 28.Moir RD, Montag-Lowy M, Goldman RD. Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol. 1994;125:1201–1212. doi: 10.1083/jcb.125.6.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moir RD, Spann TP, Herrmann H, Goldman RD. Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J Cell Biol. 2000;149:1179–1792. doi: 10.1083/jcb.149.6.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moir RD, Yoon M, Khuon S, Goldman RD. Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J Cell Biol. 2000;15:1155–1168. doi: 10.1083/jcb.151.6.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 32.Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, Desguerre I, Mayer M, Ferrer X, Briault S, Hirano M, Worman HJ, Mallet A, Wehnert M, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30:444–450. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- 33.Muchir A, van Engelen BG, Lammens M, Mislow JM, McNally E, Schwartz K, Bonne G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp Cell Res. 2003;291:352–362. doi: 10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Rowan RA, Masek MA, Billingham ME. Ultrastructural morphometric analysis of endomyocardial biopsies. Idiopathic dilated cardiomyopathy, anthracycline cardiotoxicity, and normal myocardium. Am J Cardiovasc Pathol. 1998;2:137–144. [PubMed] [Google Scholar]
- 35.Perrot A, Hussein S, Ruppert V, Schmidt HH, Wehnert MS, Duong NT, Posch MG, Panek A, Dietz R, Kindermann I, Böhm M, Michalewska-Wludarczyk A, Richter A, Maisch B, Pankuweit S, Ozcelik C. Identification of mutational hot spots in LMNA encoding lamin A/C in patients with familial dilated cardiomyopathy. Basic Res Cardiol. 2009;104:90–99. doi: 10.1007/s00395-008-0748-6. [DOI] [PubMed] [Google Scholar]
- 36.Schiender M, Franziska J, Sanz J, Kanel TV, Gallati S. Detection of exon deletions (CFTR) by relative quantification on the lightcycler. Clin Chem. 2006;52:2005–2012. doi: 10.1373/clinchem.2005.065136. [DOI] [PubMed] [Google Scholar]
- 37.Scholz D, Diener W, Schaper J. Altered nucleus/cytoplasm relationship and degenerative structural changes in human dilated cardiomyopathy. Cardioscience. 1994;5:127–138. [PubMed] [Google Scholar]
- 38.Shimi T, Pfleghaar K, Kojima S, Pack CG, Solovei I, Goldman AE, Adam SA, Shumaker DK, Kinjo M, Cremer T, Goldman RD. The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22:3409–3421. doi: 10.1101/gad.1735208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shumaker DK, Kuczmarski ER, Goldman RD. The nucleoskeleton: lamins and actin are major players in essential nuclear functions. Curr Opin Cell Biol. 2003;15:358–366. doi: 10.1016/s0955-0674(03)00050-4. [DOI] [PubMed] [Google Scholar]
- 40.Smythe C, Jenkins HE, Hutchison CJ. Incorporation of the nuclear pore basket protein nup153 into nuclear pore structures is dependent upon lamina assembly: evidence from cell-free extracts of Xenopus eggs. EMBO J. 2000;19:3918–3931. doi: 10.1093/emboj/19.15.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spann TP, Goldman AE, Wang C, Huang S, Goldman RD. Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J Cell Biol. 2002;156:603–308. doi: 10.1083/jcb.200112047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strelkov SV, Schumacher J, Burkhard P, Aebi U, Herrmann H. Crystal structure of the human lamin A coil 2B dimer: implications for the head-to-tail association of nuclear lamins. J Mol Biol. 2004;343:1067–1080. doi: 10.1016/j.jmb.2004.08.093. [DOI] [PubMed] [Google Scholar]
- 43.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sylvius N, Bilinska ZT, Veinot JP, Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan KL, Tang AS, Dyack S, Grzybowski J, Ruzyllo W, McBride H, Tesson F. In vivo and in vitro examination of the functional significances of novel lamin gene mutations in heart failure patients. J Med Genet. 2005;42:639–647. doi: 10.1136/jmg.2004.023283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sylvius N, Hathaway A, Boudreau E, Gupta P, Labib S, Bolongo P, Rippstein P, Mcbride H, Bilinska ZT, Tesson F. Specific contributions of lamin A and lamin C in the development of laminopathies. Exp Cell Res. 2008;314:2362–2375. doi: 10.1016/j.yexcr.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, Boucek MM, Lascor J, Moss AC, Li WL, Stetler GL, Muntoni F, Bristow MR, Mestroni L. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–780. doi: 10.1016/s0735-1097(02)02954-6. [DOI] [PubMed] [Google Scholar]
- 47.Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, Cavanaugh J, Graw SL, Ruegg P, Feiger J, Zhu X, Ferguson DA, Bristow MR, Gotzmann J, Foisner R, Mestroni L. Thymopoietin (lamina-associated polypeptide2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–574. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 48.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005;83:79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- 49.van Tintelen JP, Tio RA, Kerstjens-Frederikse WS, van Berlo JH, Boven LG, Suurmeijer AJH, White SJ, den Dunnen JTD, te Meerman GJ, Vos YJ, van der Hout AH, Osinga J, van den Berg MP, van Veldhuisen DJ, Buys CHC, Hofstra RMW, Pinto YM. Severe myocardial fibrosis caused by a deletion of the 5′ end of the lamin A/C gene. J Am Coll Cardiol. 2007;49:2430–2439. doi: 10.1016/j.jacc.2007.02.063. [DOI] [PubMed] [Google Scholar]
- 50.Vaughan OA, Whitefield WGF, Hutchinson CJ. Functions of nuclear lamins. Protoplasma. 2000;211:1–7. [Google Scholar]
- 51.Verga L, Concardi M, Pilotto A, Bellini O, Pasotti M, Repetto A, Tavazzi L, Arbustini E. Loss of lamin A/C expression revealed by immuno-electron microscopy in dilated cardiomyopathy with atrioventricular block caused by LMNA gene defects. Virchows Arch. 2003;443:664–671. doi: 10.1007/s00428-003-0865-4. [DOI] [PubMed] [Google Scholar]
- 52.Vlcek S, Dechat T, Foisner R. Nuclear envelope and nuclear matrix: Interactions and dynamics. Cell Mol Life Sci. 2001;58:1758–1765. doi: 10.1007/PL00000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walter MC, Witt TN, Weigel BS, Reilicha P, Richard P, Pongratza D, Bonne G, Wehnert MS, Lochmuller H. Deletion of the LMNA initiator codon leading to a neurogenic variant of autosomal dominant Emery–Dreifuss muscular dystrophy. Neuromuscular Disord. 2005;15:40–44. doi: 10.1016/j.nmd.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 54.Wolf CM, Wang L, Alcalai R. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol. 2008;44:293–303. doi: 10.1016/j.yjmcc.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zastrow MS, Flaherty DB, Benian GM, Wilson KL. Nuclear titin interacts with A- and B-type lamins in vitro and in vivo. J Cell Sci. 2006;119:239–249. doi: 10.1242/jcs.02728. [DOI] [PubMed] [Google Scholar]
- 56.Zastrow MS, Vlcek S, Wilson KL. Proteins that bind Atype lamins: Integrating isolated clues. J Cell Sci. 2004;117:979–987. doi: 10.1242/jcs.01102. [DOI] [PubMed] [Google Scholar]
- 57.Zhong N, Radu G, Ju W, Brown WT. Novel progerin-interactive partner proteins hnRNP E1, EGF, Mel 18, and UBC9 interact with lamin A/C. Biochem Biophys Res Commun. 2005;338:855–861. doi: 10.1016/j.bbrc.2005.10.020. [DOI] [PubMed] [Google Scholar]