

Abstract
An emerging paradigm in innate immune signalling is that cell biological context can influence the outcome of a ligand–receptor interaction. In this Review we discuss how Toll-like receptor (TLR) activation and signal transduction are regulated by subcellular compartmentalization of receptors and downstream signalling components. In particular, we focus on the functional specialization of TLRs in the endosomal system. We discuss recent studies that illustrate how basic aspects of the cellular machinery contribute to TLR function and regulation. This emerging area of research will provide important information on how immune signal transduction networks depend on (and in some cases influence) the generic regulators that organize eukaryotic cells.
The past decade has witnessed an incredible expansion in our understanding of the molecular components of innate immunity and their function in host defence1,2. The recognition of microorganisms is mediated by several families of innate immune receptors that collectively survey the extracellular space, endolysosomal compartments and the cytoplasm for signs of infection or tissue damage. These receptors, the specificities of which are fixed in the germline, can recognize diverse pathogenic microorganisms by targeting highly conserved molecular patterns that are common to broad pathogen classes3,4. The Toll-like receptor (TLR) family is the best characterized group of innate immune receptors in terms of known ligands, downstream signalling pathways and functional relevance. There are 10 human TLR family members, each with distinct ligands and functional properties. As these receptors have a central role in linking pathogen recognition to induction of innate immunity, inflammation and adaptive immunity, there is tremendous interest in understanding how TLR activation is regulated.
TLRs recognize a bewildering range of microbial ligands, such as bacterial and fungal cell wall components, bacterial lipoproteins, highly conserved microbial proteins, and bacterial and viral nucleic acids. The molecular basis of such diverse ligand binding remains poorly understood, although the elucidation of several recent structures of ligand–receptor complexes suggest that not all TLRs use the same ligand-binding interface5–7. Recognition of microorganisms is linked to a cascade of events that promote inflammation, activation of innate immune responses and priming of adaptive immune responses. Key to this central role in host defence is the expression of TLRs on antigen-presenting cells, especially macrophages and dendritic cells (DCs)8. When these cells encounter microorganisms or microbial products, TLR activation initiates signal transduction pathways that culminate in potent transcriptional responses9 (BOX 1).
Box 1. Toll-like receptor signal transduction.
Toll-like receptor (TLR) signal transduction is initiated by the recruitment of one or more adaptor proteins. These adaptors (specifically, MYD88 (myeloid differentiation primary response protein 88), TIRAP (TIR domain-containing adaptor protein), TRIF (TIR domain-containing adaptor protein inducing IFNβ; also known as TICAM1) and TRAM (TRIF-related adaptor molecule; also known as TICAM2)) associate with the cytoplasmic domains of TLRs through homophilic interactions between Toll/IL-1 receptor (TIR) domains present in each TLR and each adaptor. All TLR family members use the MYD88 adaptor, except TLR3, which recruits TRIF (see the figure). TLR4 is the only family member that activates both MYD88-dependent and TRIF-dependent signal transduction pathways. The structural or conformational changes that facilitate adaptor binding remain poorly defined, although it seems likely that increased proximity between the cytoplasmic domains of TLRs creates a binding interface for the relevant TIR domain-containing adaptors38. Although the signalling events downstream of MYD88 and TRIF differ, the outcome of each pathway is conceptually similar: nuclear factor-κB, interferon-regulatory factors (IRFs) and other more general transcription factors are activated. In certain cases differential activation of IRF family members leads to distinct transcriptional responses.
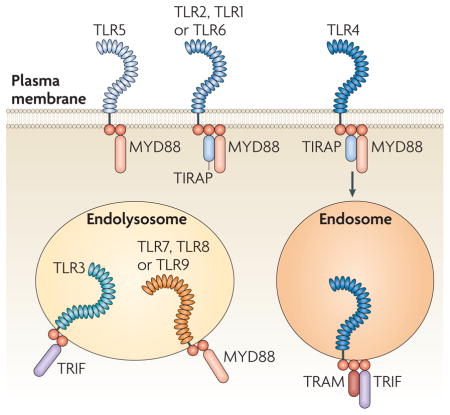
In addition to the induction of distinct signalling pathways, TLRs sample different compartments within cells. The cellular localization of these receptors has important consequences for ligand accessibility and can also affect downstream signalling events. Certain differences in subcellular localization are stable, whereas others are more dynamic (see below). The TLRs involved in the recognition of nucleic acids (TLR3, TLR7, TLR8 and TLR9) are localized within endolysosomal compartments, whereas other TLR family members (TLR1, TLR2, TLR4, TLR5 and TLR6) are found at the cell surface9. The precise nature of the compartment where intracellular TLRs meet internalized ligands remains poorly defined, and it is likely that features of the compartment differ depending on the composition of the internalized cargo and the cell type under consideration. TLRs that are normally present at the surface can also enter the endocytic pathway following their activation10–12. These receptors are rapidly recruited to phagosomes or endosomes containing microbial cargo12. The mechanisms that recruit TLRs to specific cellular compartments are just now being described.
Recent studies of innate immunity have been dominated by the identification of the components that define TLR and other signalling pathways. Much less is known about how TLR signalling pathways are integrated into the cellular infrastructure in which they operate, and future studies are likely to focus increasingly on this aspect of TLR function. In this Review we highlight examples of this new paradigm by discussing recent work linking basic cell biological processes and regulation of TLR function. A fundamental principle that seems to govern all aspects of TLR signal transduction is that the quality control mechanisms that ensure the fidelity of signalling initiation rely heavily on the spatial organization of regulators of TLR signal transduction and on the TLRs themselves. We focus on two aspects of TLR regulation in this framework: the compartmentalized activation of TLRs involved in the recognition of nucleic acids and the activation of distinct signalling pathways based on the compartmentalization of TLR signalling components.
Regulation by receptor compartmentalization
The cost of nucleic acid recognition by innate immune receptors
The recognition of nucleic acids as a signature of microbial infection has emerged as a general strategy of mammalian innate immune detection. This specificity is unexpected based on the general concept of innate immune targets — that is, that they are uniquely foreign molecules. Clearly, nucleic acids are not exclusively microbial; indeed, TLRs can be activated inappropriately by self nucleic acids under abnormal circumstances13. Considering this inherent risk of autoimmunity we might ask how nucleic acid recognition provides a selective advantage to the host despite the risk of self reactivity. The answer probably lies with the difficulty in recognizing viruses, which pose a particularly challenging target for recognition by innate immune receptors for at least two reasons14. First, unlike bacteria and fungi, viruses lack unique metabolism or biochemistry because they use host cell machinery to replicate. Second, most viral proteins (although there are notable exceptions) are easily mutated while remaining functional, which makes them poor targets for innate immune recognition. The solution to this dilemma is the recognition of viral nucleic acids by TLR family members.
Because of the risk of autoimmunity associated with the detection of nucleic acids, regulatory mechanisms must exist to determine the origin of the DNA or RNA encountered and to ensure that activation by self nucleic acids is avoided. Examples of breakdown in tolerance illustrate one key mechanism that prevents self recognition: TLRs that recognize nucleic acids are intracellular. Indeed, the delivery of nucleic acids to the intracellular subcompartments is frequently associated with autoimmune disorders15–17. For example, DNA- or RNA-specific autoantibodies deliver nucleic acid-containing immune complexes to intracellular TLRs through Fc receptor-mediated uptake15 (FIG. 1).
Figure 1. Toll-like receptor 7 and Toll-like receptor 9 trafficking and processing regulates receptor activation.

Toll-like receptor 7 (TLR7) and TLR9 are translated into the endoplasmic reticulum (ER), pass through the Golgi and are sorted to the endolysosomal system. The mechanisms responsible for these sorting steps remain undefined, although UNC93B1 is known to have a role. Following its arrival in the endolysosome, TLR9 (and most likely TLR7) is proteolytically cleaved. This cleavage event generates a functionally competent receptor; as a result, receptor activation is limited to these intracellular subcompartments. Delivery of potential ligands to the TLR7- or TLR9-containing subcompartment seems to be a prerequisite for receptor activation; viruses seem to be delivered efficiently, whereas self nucleic acids are generally excluded. An exception to this paradigm is the delivery of self nucleic acids as part of immune complexes through endocytosis mediated by Fc receptors on dendritic cells and macrophages or directly by immunoglobulins on B cells.
The role of nucleic acid-sensing TLRs in autoimmune diseases such as systemic lupus erythematosus has been shown in mouse models and studies of human patients18–23. These data and others support a model in which the intracellular localization of nucleic acid-sensing TLRs may limit access to self nucleic acids and in this way establish the threshold for self and non-self discrimination by these receptors. Implicit to this model is the exclusion of extracellular self nucleic acids from these intracellular subcompartments; however, the mechanisms that normally prevent self ligands from reaching intracellular TLRs remain poorly defined. It is possible that in autoimmune diseases, self nucleic acids are protected from degradation by extracellular and endosomal DNAses by being part of immune complexes, similarly to how viral genomes are thought to be protected by viral capsids. Alternatively, antibody-mediated uptake might deliver ligands more efficiently to TLRs, by simply concentrating otherwise scarce ligands or by accelerating endosomal maturation such that self ligands are not degraded before TLR recognition. Compared with fluid phase endocytosis or scavenger receptor-mediated uptake, Fc receptors may simply dictate a distinct fate for the engulfed cargo. Of course, these possibilities are not mutually exclusive and probably all contribute to the potent stimulatory capacity of immune complexes.
Intracellular localization of nucleic acid-sensing TLRs
As described above, the transport or protection of TLR ligands can influence self and non-self discrimination of nucleic acids, but the requirement for these mechanisms stems from the trafficking properties and subcellular localization of the TLRs themselves. Despite the clear importance of receptor localization in maintaining tolerance to self nucleic acids, the mechanisms controlling the trafficking and localization of TLRs were poorly described until recently.
The distinct cell biology associated with the recognition of nucleic acids by innate immune receptors was appreciated even before the TLRs responsible for their recognition were identified24. Specifically, oligodeoxynucleotides containing stimulatory CpG motifs were known to enter cells by endocytosis, and the fact that blocking endocytic maturation (with bafilomycin A1 or chloroquine) prevents cells from responding to TLR3, TLR7 and TLR9 ligands has been appreciated for several years24. Identification of the TLR family members that are responsible for nucleic acid recognition confirmed that these receptors are not present on the cell surface but instead localize to endosomes and lysosomes. Whether these receptors reside in specific endocytic subcompartments remains unclear. In fact, reports of the localization of endosomal TLRs vary, with descriptions of localization including the early endosomes, late endosomes, multivesicular bodies, lysosomes, the endoplasmic reticulum (ER) and the plasma membrane24–29. One explanation for these conflicting data might be that the subcompartments where TLRs reside differ among cell types and between TLR family members. Indeed, whether TLR3, TLR7, TLR8 and TLR9 occupy the same subcompartment or have distinct intracellular localizations remains unknown. The extent to which there is dynamic movement between subcompartments is also unexplored. More refined cell biological analyses will be necessary to distinguish between these possibilities.
Most research on the localization of intracellular TLRs has focused on TLR9, although the cell biology of TLR7 and TLR8 is probably similar. One of the more perplexing observations made early in this field was the apparent exclusive localization of TLR9 to the ER in unstimulated cells27,28. The carbohydrates on the mature TLR9 protein lack the modifications that are typically acquired during transit through the Golgi, and microscopy studies have suggested that TLR9 rapidly translocates from the ER to endolysosomes in stimulated cells27,28,30. These data have been used to support a model in which TLR9 traffics from the ER to the endolysosome via an unconventional route, bypassing the Golgi completely27,31. This possibility has been exciting, especially in light of the important role of localization for self and non-self discrimination by TLR7 and TLR9, but the mechanistic details of this unusual process are lacking. Perhaps the most troubling aspect of this model has been the inability to detect the receptor in the endolysosome before cell stimulation. If the model is correct, what signal leads to the translocation of the receptor in response to ligand binding?
Many of these confusing aspects of the TLR9 cell biology can be resolved by recent work. Biochemical analyses of TLR9 in macrophages and DCs led to the discovery of a processed form of the receptor that is exclusively present in endolysosomes32,33. Analysis of the carbohydrates on the receptors suggest that, unlike the ER-resident full-length mature TLR9 protein, the cleaved receptor passes through the Golgi32. Additional experiments have confirmed that the full-length TLR9 protein is sorted in the ER, traffics through the Golgi and is routed to the endolysosome, where it is cleaved by resident proteases (FIG. 1). Similar results have been reported for TLR7 (REF. 32), although another group has reported that TLR7 is not processed33. This processing event has interesting implications for the regulation of receptor function, which we discuss in the following section. For the purpose of understanding TLR7 and TLR9 localization and trafficking, the discovery of this processed receptor resolves many of the troubling issues surrounding unconventional trafficking and ligand accessibility. First, the truncated receptor is present in endolysosomal subcompartments in unstimulated cells, obviating the need for receptor translocation to detect ligand. Second, the receptor exits the ER and traffics through the Golgi via the conventional secretory pathway. Indeed, based on these data, the trafficking and localization of TLR7 and TLR9 are similar to other immunologically relevant proteins that ultimately localize in endosomes and/or lysosomes, such as MHC class II molecules or the invariant chain. One notable exception to the typical nature of TLR7 and TLR9 trafficking is the requirement for UNC93B1, a 12-membrane-spanning protein that resides in the ER31,34,35. TLR3, TLR7 and TLR9 signalling is defective in mice or cells that lack UNC93B1 function, and UNC93B1 associates with TLRs in the ER and facilitates their transport to the endolysosome31,34. Currently, UNC93B1 is the only unique trafficking factor identified for intracellular TLRs. Of course, additional characterization of the machinery controlling trafficking of these receptors may reveal specialized mechanisms, but for now the most surprising feature of this trafficking pathway is how typical it has turned out to be.
Proteolytic regulation of TLR activation
The discovery that TLR7 and TLR9 are proteolytically processed has not only established the localization and trafficking routes of these receptors, but also has contributed to our understanding of how receptor activation is controlled. Several lines of evidence indicate that the cleaved form of TLR9 (and presumably TLR7) is functional and that this depends on its processing. First, protease inhibitors or inhibitors of endosomal acidification prevent the activation of TLR7 and TLR9 (REFS 32,33). Second, the processed form of the receptor can bind CpG-containing oligodeoxynucleotides32,33. Third, MYD88 (myeloid differentiation primary response protein 88) is selectively recruited to the processed receptor, but not the full-length receptor32. Finally, a retrovirus encoding the truncated receptor is sufficient to complement TLR9 deficiency when expressed in DCs33.
The proteases responsible for the cleavage of TLR7 and TLR9 have not been definitively identified, in part owing to disagreement between several reports. Inhibitors of cathepsin B, cathepsin L and cathepsin K were reported to block TLR9 signalling, and overexpression of cathepsin B, cathepsin S, cathepsin L and cathepsin H could rescue TLR9 responsiveness in BaF3 cells36,37. However, there are conflicting data regarding the phenotype of mice or cells lacking these proteases. Mice deficient in each individual cathepsin do not seem to be markedly impaired in TLR9 function, although one report describes a partial defect in TLR9 signalling in cathepsin L- and cathepsin S-deficient mice33. In the case of cathepsin K, two groups report no defect in TLR9 function and a third group describes a crucial role for this enzyme but only in DCs32,33,36. These differences may be due, at least in part, to differences in methodology, as the third group used a new cathepsin K inhibitor instead of gene-targeted mice. In any case, even if cathepsin K has a role in TLR9 processing, its relevance seems to be restricted to DCs. Setting aside the controversy regarding specific proteases, most work supports the view that the role of any individual protease for processing of TLR9 is largely redundant. In vitro, several lysosomal proteases can cleave TLR9 to generate a truncated receptor of similar size to the receptor observed in cells32. Furthermore, only protease inhibitors with broad specificity or pooled inhibitors of individual proteases block TLR9 processing32,33. Assessing the contribution of individual or even different classes of proteases is complicated by the observation that two distinct processing steps seem to be necessary to generate the final truncated TLR9 receptor33.
The rationale behind TLR7 and TLR9 proteolytic regulation is conceptually analogous to the synthesis and activation of endolysosomal enzymes. In this instance, the destructive potential of proteases is restricted to the endolysosome because enzymes are translated as inactive pro-enzymes that are only processed after arriving in the proteolytic environment of the endolysosome. Similar reasoning may explain the requirement for processing of TLR7 and TLR9. Compartmentalized proteolysis may prevent receptors that leak out to the cell surface from responding inappropriately to self nucleic acid ligands32. In this sense, regulation of these innate receptors is achieved by co-opting a basic cell biological pathway.
Although this model of compartmentalized proteolysis may be teleologically satisfying, it does not explain why activation of the receptor requires removal of the amino-terminal half of its ectodomain. Interestingly, both the full length and processed receptors can bind ligand32,33. It remains possible that the truncated receptor binds ligand with higher affinity than the full-length receptor and that this increased affinity is required for receptor activation32. Alternatively, receptor cleavage may not regulate ligand binding at all. Recent work examining conformational changes associated with TLR9 signalling indicates that preassembled dimers of TLR9 undergo further modifications following activation38. Perhaps truncation of the ectodomain allows receptors to adopt a conformation that is required for signal initiation.
On the basis of either of these tentative models, it is interesting to consider whether other nucleic acid-sensing TLRs are subject to similar regulation. There are conflicting data regarding TLR7 processing and no data on TLR3 or TLR8. As TLR7, TLR8 and TLR9 are similar both in terms of amino acid sequence and potential for self ligand recognition, it would not be surprising if all these receptors were regulated in a comparable manner. By contrast, TLR3 has not been implicated in autoimmune pathology as a result of recognition of self double-stranded RNA. Furthermore, the structure of TLR3 bound to polyinosinic–polycytidylic acid together with mutational analyses implicate residues in the N-terminal region of the ectodomain in ligand binding7,39. These data suggest that the active form of TLR3 is the full-length receptor, although it remains possible that a cleaved form may still be identified.
Distinct signalling through compartmentalization
In search of a comprehensive model to explain TLR signal transduction
A key early discovery in TLR research was the observation that ligand specificity is not the only distinguishing feature of each TLR. In addition to recognizing distinct classes of microorganisms, individual TLRs trigger different signal transduction pathways. For example, some receptors (such as TLR3 and TLR4) induce both a pro-inflammatory and a type I interferon (IFN) response, whereas others (such as TLR2 and TLR5) induce only the pro-inflammatory response40. These differences in signalling output were explained by the discovery of multiple Toll/IL-1R (TIR) domain-containing adaptor proteins, which link activated receptors to the downstream kinases that define a given signalling pathway41–48 (BOX 1). Different adaptors engage different receptors, and the particular adaptor used determines which signalling pathway will be activated. For example, TIRAP (TIR domain-containing adaptor protein) and MYD88 function to induce the activation of a mitogen-activated protein kinase (MAPK)- and nuclear factor-κB (NF-κB)-dependent pro-inflammatory response, whereas TRAM (TRIF-related adaptor molecule; also known as TICAM2) and TRIF (TIR domain-containing adaptor protein inducing IFNβ; also known as TICAM1) activate TBK1 (TANK-binding kinase 1) and IκB kinase-ε (IKKε), which are responsible for type I IFN production. Thus, receptors that induce a type I IFN response engage a TRIF-dependent signalling pathway, whereas those that induce a pro-inflammatory response engage a MYD88-dependent signalling pathway. Although these data provide a generic model to explain TLR signalling, not all available data can be interpreted by this model. For example, this model does not clarify why some TLRs require TIRAP to induce a MYD88-dependent signalling response (for example, TLR2 and TLR4) whereas others do not (for example, TLR5, TLR7 and TLR9)49,50. Similarly, in the IFN-inducing signalling pathway, TLR4 requires TRAM and TRIF, whereas TLR3 requires only TRIF46,47. Why do some receptors require two adaptors to induce a given signalling pathway when others require only one? Furthermore, why can MYD88 induce type I IFN production downstream from some TLRs (TLR7 and TLR9) but not others51,52. In the following sections, we discuss how the cell biological features of certain TLR signalling components can address these questions.
Spatial control of TLR signalling by sorting adaptor proteins
All TIR domain-containing adaptors were initially considered to be soluble cytoplasmic proteins. The TIR domains in each adaptor were thought to serve as localization motifs by facilitating adaptor delivery to membranes through interactions with the TIR domain of a TLR. This model was challenged by several recent articles showing that TIRAP and TRAM are found on intracellular membranes before the initiation of signal transduction53,54 (FIG. 2). TIRAP mainly localizes in the plasma membrane, although a small pool is found on endosomes that are controlled by the GTPase ADP-ribosylation factor 6 (REF. 53). TRAM is also found at the plasma membrane but is enriched on RAB5-positive early endosomes that act as a conduit to the lysosomal system54. TIRAP and TRAM have a similar domain structure; both contain a carboxy-terminal TIR domain and an N-terminal localization motif. Within the localization domain of TIRAP is a phosphoinositide-binding module that selectively interacts with phosphatidylinositol-4,5-bisphospate (PtdIns(4,5)P2). PtdIns(4,5)P2 is concentrated at actin-rich regions of the plasma membrane and functions to recruit TIRAP to this location53. The N-terminus of TRAM is more complex, consisting of a bipartite localization motif that contains both a myristoylation site and a phosphoinositide-binding domain54. In both adaptors, the localization domains are necessary and sufficient to imitate the localization of the wild-type proteins. These data indicate that in the case of TIRAP and TRAM, the TIR domains are not necessary for membrane binding and are therefore not localization motifs. Indeed, in contrast to MYD88, the TIR domain of which is required for recruitment to membrane-associated TIRAP, the localization of TIRAP to the plasma membrane is mediated by its PtdIns(4,5)P2-binding domain.
Figure 2. Schematic of the sorting–signalling adaptor paradigm.

The Toll/IL-1 receptor (TIR) domain-containing adaptors have one of two roles. MYD88 (myeloid differentiation primary response protein 88) and TRIF (TIR domain-containing adaptor protein inducing IFNβ) function as signalling adaptors that biochemically link Toll-like receptors (TLRs) that are found in several organelles to the downstream kinases that define a given signalling pathway. The recruitment of MYD88 to plasma membrane TLRs and of TRIF to endosomal TLRs depends on the sorting adaptors TIRAP (TIR domain- containing adaptor protein) and TRAM (TRIF-related adaptor molecule). Sorting adaptors do not directly engage downstream kinases but instead directly engage signalling adaptors, promoting their delivery to activated receptors. The recruitment of sorting adaptors to specific subcellular locations is controlled by their intrinsic phosphoinositide-binding activity, which restricts their localization to a subset of organelles that contain TLRs. As such, TIRAP and TRAM can recruit their signalling adaptor partners to a subset of organelles. Whether additional sorting adaptors exist to recruit signalling adaptors to the other subcompartments of the cell is currently unknown. IL-1R, interleukin-1 receptor; LPS, lipopolysaccharide; PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphospate; TRAF3, TNFR-associated factor 3.
What is the importance of MYD88 recruitment to the plasma membrane? Using biochemical assays and the MAPPIT system (a variant of the two hybrid assay), it was shown that TIRAP promotes interactions between MYD88 and TLR2 and TLR4 (REF. 55). Moreover, the recruitment of MYD88 to the plasma membrane is functionally important, as an allele of MYD88 that is directed to the plasma membrane (generated by the addition of an N-terminal PtdIns(4,5)P2-binding domain) restored lipopolysaccharide (LPS) responsiveness in TIRAP-deficient macrophages and in fibroblasts deficient for both TIRAP and MYD88 (REF. 53). The finding that simply relocalizing MYD88 to the cell surface is sufficient to bypass the genetic requirement for TIRAP shows that the main function of TIRAP is to recruit MYD88 to the appropriate TLRs. So, rather than acting independently to engage different downstream signalling events, MYD88 and TIRAP act as a functional pair to induce the activation of a single signalling pathway. In this context, TIRAP is unlikely to operate as a signalling adaptor, but instead probably functions to sort a cytoplasmic pool of MYD88 to plasma membrane-bound TLR2 and TLR4. TIRAP can therefore be considered a sorting adaptor protein that recruits its signalling adaptor partner to participate in signal transduction at the plasma membrane53. The classification of TIRAP and MYD88 as sorting and signalling adaptors explains why TIRAP never functions without MYD88, as it requires its signalling adaptor partner to engage the enzymes that define the pathway.
The sorting and signalling adaptor concept in TLR signalling also applies to TRAM and TRIF. TRIF is a signalling adaptor, whereas TRAM functions as a sorting adaptor54. Evidence in support of this conclusion was the demonstration that TRAM interacts directly with TRIF and TLR4, whereas TRIF cannot interact strongly with TLR4 (REF. 56). So, TRAM is an obligate intermediate that links the signalling adaptor TRIF to activated TLR4. Subsequent studies revealed that the membrane-binding activity of TRAM was necessary for TRIF-dependent signal transduction by TLR4 (REFS 54,57). Mutations that abolish myristoylation of TRAM result in a protein that is mislocalized to the cytoplasm and unable to participate in signal transduction. Interestingly, a splice variant of TRAM has been described recently in which the bipartite localization motif has been replaced with exons encoding a GOLD (Golgi dynamics) domain (which may be involved in protein–lipid interactions)58. This splice variant, known as TAG, is distinct from TRAM in both subcellular distribution and function. During TLR4 signalling, TAG is recruited to RAB7-positive late endosomes, where it promotes the degradation of TLR4 and the inactivation of the TRAM–TRIF signalling pathway. A TLR adaptor that contains both pro- and anti-inflammatory splice variants is not without precedent. In fact, a splice variant of MYD88, known as MYD88s, is a negative regulator of TLR signalling59. Therefore, splice variants may be a common means of diversifying the functions of adaptor proteins in TLR signalling.
Regarding TAG, it is currently unknown whether the GOLD domain of this protein binds to phosphoinositides, similarly to the bipartite localization motif of TRAM. We speculate that this is a true possibility, as a common feature of both TIRAP and TRAM is that their localization domains can bind lipids53,54. The reason for this similarity may be due to the need to restrict the type of TLR-induced signalling pathway to particular intracellular subcompartments. For example, the ability of TIRAP to bind to PtdIns(4,5)P2 allows this protein to be enriched at the site of TLR4 is important for all internalization (as PtdIns(4,5)P2 endocytic events)60. However, unlike the plasma membrane, endosomes have little or no PtdIns(4,5)P2, and as such, once TLR4 is internalized TIRAP is likely to be released, resulting in the cessation of MYD88-dependent signalling. Thus, although lipid binding promotes the concentration of TIRAP at the initial site of ligand–receptor interactions (the plasma membrane), the specificity for a particular lipid ensures that signal transduction can only occur from this location. Once the receptor is transported to a region of the cell that is not occupied by the adaptor, signalling can no longer take place. This last point may become more relevant in the future, as scientists may be inclined to use the colocalization of a TLR and its cognate ligand as an experimental read-out to identify the subcellular site of signal transduction. However, if the receptor and ligand are in a location that does not contain the downstream regulators, then a signalling event cannot occur. An example of this situation (the TRAM–TRIF signalling pathway) is discussed below.
Endolysosomes as unique IFN-inducing organelles
Although the parallels between the TIRAP–MYD88 and TRAM–TRIF pairs of sorting and signalling adaptors are striking, further studies of TLR4 revealed an intriguing aspect of TLR biology. TLR4 does not induce the simultaneous activation of both MYD88- and TRIF-dependent pathways; instead, TLR4 induces its signalling pathways sequentially54. First, LPS binding to TLR4 at the plasma membrane probably promotes the rapid activation of phosphatidylinositol 5-kinases (by triggering β2 integrins and perhaps other cofactors) that induce the local production of PtdIns(4,5)P2. PtdIns(4,5)P2 mediates the recruitment of TIRAP to the plasma membrane and helps to promote interactions between TIRAP–MYD88 and TLR4, which triggers pro-inflammatory signal transduction. LPS also induces the endocytosis of TLR4, resulting in the internalization of the receptor complex and its delivery to endolysosomes10,11. Interestingly, although receptor endocytosis downregulates the TIRAP–MYD88 signalling pathway11, TLR4 internalization promotes TRAM–TRIF signalling54. This was shown by using inhibitors of dynamin GTPases that abolish LPS-induced receptor endocytosis. In macrophages, dynamin inhibitors completely abolish TRIF-dependent activation of the transcription factor IFN-regulatory factor 3 (IRF3) and consequently the expression of IRF3-dependent genes. The finding that TLR4 internalization promotes TRAM–TRIF signalling was further demonstrated by the observation that a TRAM mutant which localizes to the endosomes retains the ability to participate in TLR4 signal transduction54.
The requirement for delivery to endosomes before TRAM–TRIF signalling indicates that TLR4 is not the only pattern recognition receptor to induce type I IFN production by signalling from the plasma membrane. All other IFN-inducing TLRs signal from endolysosomes40. At least some of these receptors are cleaved to form a signalling-competent receptor by endosomal proteases, a process that helps to restrict their activity to the appropriate cellular subcompartment. What constitutes an appropriate subcompartment? The question of appropriateness can be explained by the obvious benefit of rapid ligand recognition by a TLR. As nucleic acids are not accessible to TLRs until the microorganism is degraded in lysosomes, the appropriate compartments for TLR9 signalling are endolysosomes, as this is the first locale where the ligand is released. However, if ligand access is the only factor that determines where signalling can occur, then what prevents TLR4 from inducing TRAM–TRIF signalling at the plasma membrane? TLR4 clearly encounters its ligand at the plasma membrane and is capable of inducing signal transduction from this location (for example, through TIRAP–MYD88). Again, cell biological studies suggest that there is more to this story.
Signalling proteins that act downstream from receptors are typically considered mobile and capable of being recruited to multiple cellular subcompartments to engage active receptors. Interestingly, the ubiquitin ligase TNFR-associated factor 3 (TRAF3), which is used by all IFN-inducing TLRs, seems to be restricted in its mobility54. This protein is not detected at the plasma membrane, but is restricted to an intracellular location (FIG. 2). LPS treatment does not result in the recruitment of TRAF3 to the plasma membrane but induces the internalization of TLR4, TRAM and TRIF into endosomes61. Only after internalization do TRAM and TRAF3 colocalize, and this colocalization occurs at time points that coincide with the production of type I IFNs. So, instead of inducing the recruitment of IFN-inducing proteins to an immobile receptor, TLR4 is mobilized to enter the cell and engage the immobile signalling proteins. TLR4 can therefore be grouped with growth factor receptors and several G protein-coupled receptors, which are also transported to locations where immobile signalling proteins are present62.
The spatial restriction of TRAF3 to an intracellular locale helps to explain why nucleic acid-sensing TLRs such as TLR7 and TLR9 can use MYD88 to induce type I IFN production, whereas TLR2, which also uses MYD88, cannot. Depending on the site where MYD88 is accessed, different downstream effector responses can be induced. As TRAF3 is most easily accessed in endosomes, endosomal TLR7 and TLR9 can most easily use MYD88 to induce type I IFN production. When MYD88 is recruited to receptors found at the plasma membrane, this signalling adaptor is less likely to engage TRAF3 and, consequently, cannot induce the production of type I IFNs. Interestingly, TRAF3 can be recruited to the plasma membrane to interact with tumour necrosis factor receptor (TNFR) family members (such as CD40), but its function in CD40 signalling is different from its function in TLR signalling63. Rather than promoting type I IFN production, as it does for TLRs, TRAF3 acts as a negative regulator of MAPK activation downstream from CD40. How the function of TRAF proteins in TLR signalling relates to their function in other signalling pathways, and how this relates to the subcellular positioning of these enzymes are questions that are being investigated.
Concluding remarks
The examples discussed in this Review illustrate how TLRs have evolved within the cell biological framework such that basic aspects of the cellular machinery contribute to receptor function and regulation. We have tried to highlight how this intrinsic feature of TLR biology serves to promote efficient signalling in certain examples and restrict receptor function in others. Future work will undoubtedly reveal further examples of this paradigm. In the meantime, we highlight a few important questions the answers to which will shape the direction of the emerging field of innate cell biology. How does the localization of TLR signalling proteins relate to the sites of signalling complex assembly? The answer to this question depends on the development of more reliable tools to study the biochemistry of TLR signalling. How common is the sorting–signalling adaptor paradigm? Do sorting adaptors control the recruitment of MYD88 or TRIF to all TLRs? How are self nucleic acids prevented from activating endosomal TLRs, and what is the cost of triggering type I IFN production from the plasma membrane? Can cell-type-specific differences in the subcellular positioning of TLR signalling proteins explain the cell-type-specific responses to TLR ligands? If so, is this the only means of tailoring TLR signalling responses or do cell-type-specific factors exist as well? How do pathogenic microorganisms influence the activity of TLR signalling proteins and is there a common strategy that they use to interfere with these processes? These questions have emerged mainly from cell biological studies of TLR signalling but may also apply to the operation of non-TLR innate signalling pathways that detect microbial infection. As such, we propose that the study of innate immune cell biology extends beyond that of TLRs and should encompass all innate receptors with the goal of understanding how they are integrated into the cellular infrastructure in which they operate.
Acknowledgments
The authors thank members of their laboratories for helpful discussions and the National Institutes of Health, USA (AI072429 to G.M.B., AI072955 to J.C.K.) and the Lupus Research Institute (G.M.B.) for funding support.
Footnotes
DATABASES
UniProtKB: http://www.uniprot.org
MYD88 | TIRAP | TLR1 | TLR2 | TLR3 | TLR4 | TLR5 | TLR6 |
TLR7 | TLR8 | TLR9 | TRAF3 | TRAM | TRIF | UNC93B1
Contributor Information
Gregory M. Barton, Email: barton@berkeley.edu.
Jonathan C. Kagan, Email: jonathan.kagan@childrens.harvard.edu.
References
- 1.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 3.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 5.Jin MS, et al. Crystal structure of the TLR1–TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Kim HM, et al. Crystal structure of the TLR4–MD-2 complex with bound endotoxin antagonist eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, et al. Structural basis of Toll-Like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nature Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 9.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 10.Akashi S, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4–MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Husebye H, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBo J. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Underhill DM, et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 13.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nature Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barton GM. Viral recognition by Toll-like receptors. Semin Immunol. 2007;19:33–40. doi: 10.1016/j.smim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Boule MW, et al. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin–immunoglobulin G complexes. J Exp Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau CM, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leadbetter EA, et al. Chromatin–IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 18.Berland R, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–440. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 19.Christensen SR, et al. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christensen SR, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 21.Lande R, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 22.Pisitkun P, et al. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian S, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hacker H, et al. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBo J. 1998;17:6230–6240. doi: 10.1093/emboj/17.21.6230. This paper is the first to describe the requirement for internalization of TLR9 ligands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ewaschuk JB, et al. Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect Immun. 2007;75:2572–2579. doi: 10.1128/IAI.01662-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda K, et al. Spatiotemporal regulation of MyD88–IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 27.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nature Immunol. 2004;5:190–198. doi: 10.1038/ni1028. This paper is the first to suggest that TLR9 is an ER-resident protein with unique trafficking properties. [DOI] [PubMed] [Google Scholar]
- 28.Leifer CA, et al. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173:1179–1183. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumoto M, et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. 2003;171:3154–3162. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 30.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nature Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 31.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–238. doi: 10.1038/nature06726. This paper implicates UNC93B1 in the trafficking of TLR9 from the ER to endolysosomes. [DOI] [PubMed] [Google Scholar]
- 32.Ewald SE, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park B, et al. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nature Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. References 32 and 33 report that TLR9 is cleaved in its extracellular domain prior to activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brinkmann MM, et al. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tabeta K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nature Immunol. 2006;7:156–164. doi: 10.1038/ni1297. This paper shows that UNC93B1 is necessary for function of the intracellular TLRs. [DOI] [PubMed] [Google Scholar]
- 36.Asagiri M, et al. Cathepsin K-dependent Toll-like receptor 9 signaling revealed in experimental arthritis. Science. 2008;319:624–627. doi: 10.1126/science.1150110. This paper reports a role for cathepsin K in TLR9 signalling. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto F, et al. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun. 2008;367:693–699. doi: 10.1016/j.bbrc.2007.12.130. This paper provides the first evidence that cathepsins are involved in TLR9 activation. [DOI] [PubMed] [Google Scholar]
- 38.Latz E, et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nature Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- 39.Bell JK, Askins J, Hall PR, Davies DR, Segal DM. The dsRNA binding site of human Toll-like receptor 3. Proc Natl Acad Sci USA. 2006;103:8792–8797. doi: 10.1073/pnas.0603245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawai T, Akira S. Innate immune recognition of viral infection. Nature Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 41.Fitzgerald KA, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 42.Fitzgerald KA, et al. LPS–TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J Exp Med. 2003;198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nature Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 44.Medzhitov R, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 45.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-β induction. Nature Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto M, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nature Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 48.Yamamoto M, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 49.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto M, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 51.Hemmi H, Kaisho T, Takeda K, Akira S. The roles of Toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J Immunol. 2003;170:3059–3064. doi: 10.4049/jimmunol.170.6.3059. [DOI] [PubMed] [Google Scholar]
- 52.Kawai T, et al. Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nature Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 53.Kagan J, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. This paper provides the first evidence that TLR adaptors can interact with phosphoinositides, and the authors put forward the sorting adaptor hypothesis to distinguish the functions of TIRAP and MYD88 in TLR signalling. [DOI] [PubMed] [Google Scholar]
- 54.Kagan J, et al. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nature Immunol. 2008;9:361–368. doi: 10.1038/ni1569. This paper provides the first evidence that TLR4 induces its two signalling pathways sequentially, by a process coordinated around the endocytosis of the receptor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peter U, Frank P, Rudi B, Jan T. MAPPIT analysis of TLR adaptor complexes. FEBS Lett. 2007;581:629–636. doi: 10.1016/j.febslet.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 56.Oshiumi H, et al. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to Toll-like receptor 4 TICAM-1 that induces interferon-β. J Biol Chem. 2003;278:49751–49762. doi: 10.1074/jbc.M305820200. [DOI] [PubMed] [Google Scholar]
- 57.Rowe DC, et al. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc Natl Acad Sci USA. 2006;103:6299–6304. doi: 10.1073/pnas.0510041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palsson-McDermott EM, et al. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nature Immunol. 2009;10:579–586. doi: 10.1038/ni.1727. [DOI] [PubMed] [Google Scholar]
- 59.Janssens S, Burns K, Vercammen E, Tschopp J, Beyaert R. MyD88S, a splice variant of MyD88, differentially modulates NF-κB- and AP-1-dependent gene expression. FEBS Lett. 2003;548:103–107. doi: 10.1016/s0014-5793(03)00747-6. [DOI] [PubMed] [Google Scholar]
- 60.De Matteis MA, Godi A. PI-loting membrane traffic. Nature Cell Biol. 2004;6:487–492. doi: 10.1038/ncb0604-487. [DOI] [PubMed] [Google Scholar]
- 61.Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. 2008;368:94–99. doi: 10.1016/j.bbrc.2008.01.061. This paper shows that endosomes are the sites where the TRAM and TRAF3 signalling proteins converge to promote type I IFN production. [DOI] [PubMed] [Google Scholar]
- 62.Gould GW, Lippincott-Schwartz J. New roles for endosomes: from vesicular carriers to multi-purpose platforms. Nature Rev Mol Cell Biol. 2009;10:287–292. doi: 10.1038/nrm2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuzawa A, et al. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]