

Abstract
During normal postnatal mammary gland development and adult remodeling related to the menstrual cycle, pregnancy, and lactation, ovarian hormones and peptide growth factors contribute to the delineation of a definite epithelial cell identity. This identity is maintained during cell replication in a heritable but DNA-independent manner. The preservation of cell identity is fundamental, especially when cells must undergo changes in response to intrinsic and extrinsic signals. The maintenance proteins, which are required for cell identity preservation, act epigenetically by regulating gene expression through DNA methylation, histone modification, and chromatin remodeling. Among the maintenance proteins, the Trithorax (TrxG) and Polycomb (PcG) group proteins are the best characterized. In this review, we summarize the structures and activities of the TrxG and PcG complexes and describe their pivotal roles in nuclear estrogen receptor activity. In addition, we provide evidence that perturbations in these epigenetic regulators are involved in disrupting epithelial cell identity, mammary gland remodeling, and breast cancer initiation.
Keywords: Epithelial cell identity, maintenance proteins, epigenetic control, hormone steroid receptor, breast tumorigenesis
It is currently hypothesized that breast carcinogenesis proceeds through sequential stages, beginning with the dysregulation of normal epithelial cell proliferation; progressing through atypical hyperplasia, carcinoma in situ, and invasive carcinoma; and finally progressing to metastatic disease that may have lethal consequences for the patient[1]. Although accumulating evidence indicates that committed mammary progenitor cells are the bona fide site of breast cancer initiation[2],[3], compelling experimental studies have suggested that terminally differentiated cells could also acquire malignant features (mobility, invasiveness, and resistance to apoptosis) via epithelial-to-mesenchymal transition (EMT)[4],[5].
Normal human mammary glands consist of 6-12 independent ductal systems that form the ductal tree. Each ductal system is composed of a central duct, some peripheral branches, and associated glandular tissue. The entire ductal tree is surrounded by the stroma, a heterogeneous population of cells (fibroblasts, adipose cells, endothelial precursors, and immune system cells) embedded in an extracellular matrix that provides a substrate for cell adhesion and acts as a reservoir of growth factors[6].
Studies in mice have revealed that each peripheral branch is capped by a terminal ductal lobulo-alveolar unit (TDLU) composed of a bilayered and polarized epithelium encompassing a luminal space. The inner luminal epithelium is formed by holocrine cells that possess a specialized apico-basolateral polarity, whereas the outer basal epithelium or myoepithelium is composed of contractile cells that form a basket-like network around the alveoli and ducts[7]. Myoepithelial cells synthesize the basement membrane and act as an active structural barrier between the luminal epithelial cells and the surrounding intra-lobular stroma[8].
Luminal and myoepithelial cells can be distinguished using cell type-specific markers. Luminal epithelial cell-specific markers include E-cadherin; epithelial cell adhesion molecule (EpCAM); mucin; cytokeratins 7, 8, 18, and 19; and CD24. Myoepithelial cell-specific markers include smooth muscle actin (aSMA); membrane metallopeptidase (also known as CD10/CALLA); vimentin; cytokeratins 5/6, 14, and 17; and epidermal growth factor receptor (EGFR).
Estrogen and the Mammary Gland
The development of the mammary gland, unlike that of other human organs, is completed only postnatally when, in response to ovarian hormones (estrogen and progesterone), extensive TDLU proliferation occurs, resulting in branch extension into the surrounding stroma and lobule differentiation. The complete functional development of the lobules occurs only with full-term pregnancy and subsequent lactation, at the end of which the lobules involute but retain a larger number of individual alveoli per lobule than were present before pregnancy[9],[10].
Estrogen receptor and coregulators
Estrogen and progesterone exert their biological functions through specific ligand-activated nuclear receptors. Estrogen bind to two distinct estrogen receptors (ERa and ERβ), which are encoded by genes located on two different chromosomes, whereas progesterone binds to two isoforms of a receptor (PR-A and PR-B) that are transcribed from the same gene by different promoters[11],[12]. In its unliganded state, ER is combined with chaperone proteins from which it dissociates upon hormone binding[13]. Thereafter, the newly formed ligand-receptor complex dimerizes and translocates into the nucleus, where it can bind directly to specific sequences, termed estrogen response elements (EREs), located within the promoter/enhancer regions of their target genes. This complex can also affect gene transcription indirectly through its physical interaction with other transcription factors, such as E2F1, activating protein 1 (AP1), Sp1, Stat3, and nuclear factor-κB (NF-κB)[14],[15] (Figure 1). In addition to ligand availability, ER transcriptional activity depends on the cooperation of specific proteins, collectively called “coregulators,” that act as coactivators or corepressors[16]. ER coactivators, which include steroid receptor coactivator (SRC)/p160 family members, E3 ubiquitin-protein ligases, and the histone acetyltransferase cAMP responsive element binding protein (CREB)-binding protein (CBP)/p300, are recruited directly by ER and enhance ER-mediated gene expression; in breast cancer, the best studied coactivator is amplified in breast cancer (AIB)-1/SRC-3[17]. Conversely, corepressors inhibit gene expression through a direct interaction with unliganded ER or competition with coactivators for ER binding. Most corepressors are widely distributed in human tissues, and the corepressors that are best characterized in breast cancer are nuclear receptor corepressor (N-CoR) and silencing mediator of retinoid and thyroid hormone receptors (SMRT)[18],[19]. These coregulators are not merely “bridging” agents linking ER and the transcription machinery; rather, they perform a wide range of enzymatic activities (e.g., acetylation, methylation, and ubiquitination) that are essential for the appropriate regulation of the transcription process as part of multicomponent protein complexes. Because of their roles in hormone-dependent gene transcription, it is not surprising that coregulator misexpression is a common feature of endocrine-related tumors including breast, uterine, ovarian, and prostate cancers.
Figure 1. The estradiol (E)-estrogen receptor (ER) complex may activate gene transcription directly by binding to specific sequences (estrogen response elements, EREs) within the promoter/enhancer regions of target genes or indirectly by physical interaction with other transcription factors, such as activating protein 1 (AP1).
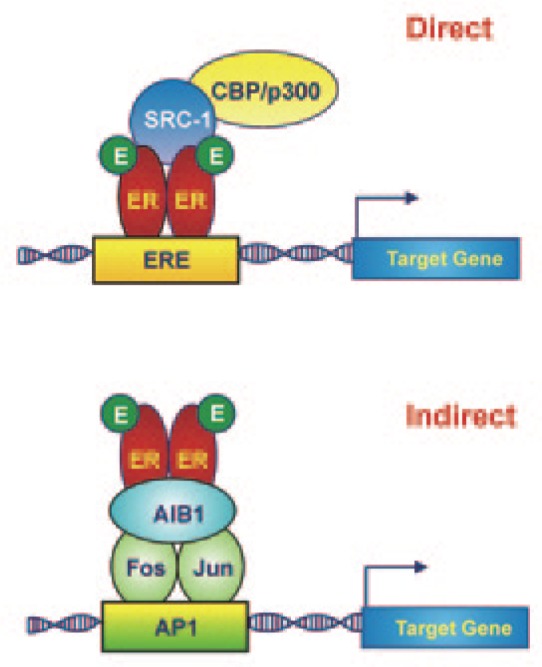
CBP, histone acetyltransferase CREB-binding protein; SRC-1, steroid receptor coactivator-1; AIB1, amplified in breast cancer 1.
Estrogen receptor and pioneer factors
Estrogen receptor and FoxA1
Among the proteins that facilitate the ER-DNA interaction, “pioneer factors” represent a special class of proteins that can associate directly with condensed chromatin independent of and prior to any other transcription factors, allowing chromatin accessibility[20]. Several protein families, including Forkhead box A (FoxA)[21], transducin-like enhancer (TLE)[22], and, to a moderate degree, Gata[23], have been proved to possess this “pioneer” property. Originally described as crucial transcriptional components during development and differentiation, pioneer factors have recently been implicated as mediators of nuclear receptor function under both normal and pathologic conditions[24]. In particular, FoxA1 has been proved to be a master regulator of ER activity due to its ability to bind to DNA and core histones simultaneously, to rearrange chromatin structure by disrupting local internucleosomal interactions and to recruit ER to target gene promoters[25],[26] (Figure 2). The unique “pioneer” capability of FoxA1 is due to the presence of a so-called forkhead box domain, a variant of the helix-turn-helix structure that has two large loops, giving it the appearance of a “winged helix”[27]. Experimental evidence has indicated that FoxA1 moves slowly along the chromatin, scanning it for enhancers with forkhead motifs[28]. Upon finding a forkhead motif, the central helix-turn-helix of FoxA1 makes direct contact with the major groove of the DNA, while each wing interacts with minor grooves adjacent to the target sequence, thus stabilizing the interaction with the DNA[29] and triggering the transcriptional competency of the enhancer.
Figure 2. Forkhead box A1 (FoxA1) is a pioneer factor belonging to a special class of proteins that are able to interact directly with condensed chromatin prior to any other transcription factors.
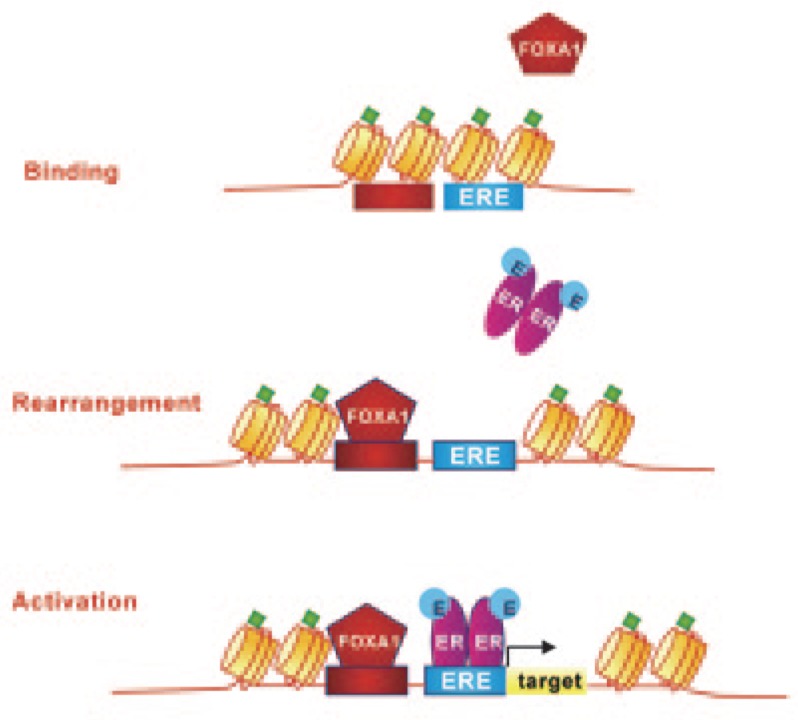
Upon chromatin binding, FoxA1 induces a nucleosomal rearrangement that results in a more relaxed chromatin structure, which is accessible to other transcription regulators, including ERs. Green diamonds represent epigenetic histone modifications (dimethylation of histone H3 at lysine 4, H3K4me2).
Despite its recognized role as a pioneer factor, the binding capability of FoxA1 can be enabled or restricted by certain chromatin modifications, such as methylation and acetylation. Due to the lack of a classic methylated GC sequence in the forkhead box domain, FoxA1-binding activity occurs primarily at hypomethylated DNA regions. Consequently, heavily methylated DNA regions may have relatively poor FoxA1-binding potential[30]. In addition to DNA hypomethylation, FoxA1-binding activity depends on the methylation status of histone H3, specifically on the mono- and di-methylation of histone H3 at lysine 4 (H3K4me1 and H3K4me2). Experimental evidence has indicated that the demethylation of these histone sites inhibits FoxA1 binding[31], suggesting that H3K4 methylation is necessary to stabilize FoxA1 binding and allow the subsequent recruitment of transcriptional regulators, including sex steroid hormone nuclear receptors. The functional relationship between FoxA1 pioneering activity and ER-binding activity is corroborated by the observation that, in the developing mammary gland, FoxA1 and ER are co-expressed within luminal epithelial cells, especially at the terminal end buds, and that a FOXA1 gene deletion results in the loss of ER expression[32]. Because luminal progenitors give rise to the ductal lineage, FOXA1 deletion and the consequent loss of ER expression may cause the arrest of duct elongation and lobule differentiation in response to hormones during puberty or pregnancy.
Estrogen receptor and Gata-3
As a member of the Gata family, Gata-3 has long been recognized as essential for mammary gland development and maintenance of the terminal differentiation of the luminal lineage in adults[33],[34]. More recently, Gata-3 has been proven to function as a pioneer factor. Co-recruited with FoxA1 to ER cis-regulatory elements, Gata-3 is essential for the ER-mediated transcription of target genes[35]. More specifically, experimental evidence has indicated that ER and Gata-3 participate in a positive feedback loop in which ER expression is required for GATA3 gene transcription[35]. This finding supports that the loss of Gata-3 in the normal mammary gland decreases the ER-expressing luminal population[33],[34].
Dissociation between ER expression and cell proliferation
In addition to ovarian hormones, several other factors orchestrate the development of the human mammary gland via stroma-epithelium interactions[36]–[38]. In fact, ER and PR are expressed only in approximately 30% of luminal cells, and basal cells do not express hormone receptors at all. Furthermore, in the normal mammary gland, ER-positive cells are quiescent and act as “steroid hormone sensors,” producing paracrine/juxtacrine factors to regulate the proliferative activity of adjacent ER-negative epithelial cells. This phenomenon is known as dissociation between steroid receptor expression and cell proliferation and aims to prevent the overgrowth of ER-positive mammary epithelial cells in an antitumor strategy[39]–[42]. The observation that 70% of primary breast cancers are ER-positive, in contrast to normal mammary glands, strongly suggests that this control mechanism is disrupted during breast tumorigenesis and that the enhanced activity of the ER signaling pathway then becomes the major autocrine force driving tumor growth[43].
Maintenance Proteins
During mammary gland development, ovarian hormones and peptide growth factors act in coordination to define a profile that depends on the differential expression of genes that are specifically involved in epithelial cell differentiation. This tissue-specific profile is maintained during cell replication so that the daughter cells will retain the cell type identity of the parental cell (cellular memory), even when cell activity must change in response to intrinsic and extrinsic signals. The preservation of cell identity (i.e., the maintenance of a specific gene expression profile) depends on a class of proteins that are collectively termed maintenance proteins[44]. Previous studies have shown that maintenance proteins act epigenetically by regulating gene expression in a heritable fashion, independent of DNA sequence. This epigenetic control is achieved through DNA methylation, histone modification, and chromatin remodeling[45],[46]. Trithorax (TrxG) and Polycomb (PcG) group proteins are the best characterized maintenance proteins[47],[48].
Discovered in Drosophila, the TrxG and PcG proteins play a crucial role in the epigenetic control of a large number of developmental genes. Genetic studies have established that in Drosophila, the TrxG and PcG proteins exert an antagonistic action on the transcription of homeotic (HOX) genes, a cluster of genes that defines the positional identity of the body segments along the anterior-posterior axis of the fly during early embryogenesis[49]. PcG proteins act as transcriptional repressors that maintain an “off state,” whereas TrxG proteins are transcriptional activators that maintain an “on state.” Mutations in specific PcG and TrxG genes generate flies with homeotic transformations due to the misregulation of developmental master Hox genes[50].
Additional studies performed in other organisms have revealed that TrxG and PcG proteins can act as transcriptional activators or repressors depending on the multi-protein complex in which they are included, and these proteins have been implicated in a variety of normal cellular processes, including genomic imprinting[51], X chromosome inactivation[52], stem cell identity[53], and cell prolifera-tion[54].
TrxG complexes
Based on the function of their key component, the TrxG multi-protein complexes are classified as ATP-dependent chromatin-remodeling complexes that regulate gene transcription in an energy-dependent manner or covalent histone-modifying complexes [55],[56].
ATP-dependent chromatin-remodeling complexes
In all ATP-dependent chromatin-remodeling complexes, the central catalytic subunit belongs to the sucrose non-fermenting 2 (SNF2) family of ATPases; through this subunit, the complex uses the energy of ATP hydrolysis to fuel nucleosome translocation along the DNA, histone H2A/H2B ejection from the nucleosome, entire histone octamer relocation, and histone H2A variant exchange[57],[58]. According to the domain architecture of the catalytic subunit, the complexes are classified into seven families, four of which are preserved in all mammalian cells. In humans, the most prominent of these complexes are the switching-defective (SWI)/SNF and imitation switch (ISWI) complexes[59].
SWI/SNF complex: structure and function
A SWI/SNF complex is composed of a central ATPase subunit, which can be either brahma (BRM) or BRM-related gene 1 (BRG1), and 10-12 proteins, known as BRG1-associated factors (BAFs). Despite their high overall homology (approximately 75% identity at the amino acid level), the BRG1 and BRM sequences diverge at the N-terminal region; these sequences regulate their binding to different transcription factors, and they form mutually exclusive SWI/SNF complexes. Consequently, the SWI/SNF complex may play different and sometimes antagonistic roles depending on whether BRG1 or BRM is the central catalytic subunit[60].
SWI/SNF complexes may exist in two forms, SWI/SNF-BAF or SWI/SNF-polybromo-associated BAF (PBAF), depending on their BAF composition. Both forms contain a highly conserved “core” of BRG1 (sometimes BRM), Baf47/hSNF5/INI1, Baf155, and Baf170, and the additional subunits Baf53, Baf57, Baf60, and β-actin (Figure 3). The two forms are distinguished by the presence of specific subunits: Baf250 in SWI/SNF-BAF or Baf180, Baf200, and bromodomain-containing 7 (Brd7) in SWI/SNF-PBAF[61].
Figure 3. Brahma-related gene 1 (BRG1)-containing chromatin-remodeling complexes.

In addition to the switching-defective (SWI)/sucrose non-fermenting (SNF)-BRG1-associated factors (BAF) and SWI/SNF-polybromo-associated BAF (PBAF) complexes, the catalytic subunit BRG1 (or, less frequently, BRM) can associate with several chromatin-remodeling complexes, including transcription coactivators and corepressors. BRG1 can be found in complexes with transcription coactivators and histone-modifying enzymes such as Williams syndrome transcription factor (WSTF)-including nucleosome assembly complex (WINAC) and nucleosomal methylation activation complex (NUMAC). Conversely, BRG1 can be assembled in complexes known to repress transcription and induce gene silencing, such as the nuclear receptor corepressor (N-CoR) and mSin3A/histone deacetylase (HDAC) complexes. Orange: BRG1 subunit; gold: essential subunits; green: nuclear receptor-associated factors; red: DNA replication factors; yellow: actin-related complexes; blue: factors with other functions.
Several models have been postulated to explain how SWI/SNF modifies chromatin structure, especially the ATP-dependent mobilization of nucleosomes in cis along the DNA and the ejection and insertion of histone octamers[62]. For nucleosome sliding, it has been proposed that after binding to nucleosomal DNA, the SWI/SNF complex disrupts histone-DNA association and makes DNA molecule more accessible to DNA-binding factors. However, the mechanism by which nucleosome ejection and insertion occur has not been completely elucidated. Recent findings suggest that histone ejection may occur not at the nucleosome that is directly bound by the SWI/SNF complex but rather at the adjacent nucleosome following the repositioning of the bound nucleosome[63]. Regardless of the mechanism, the change in chromatin structure induced by the SWI/SNF complex allows a large number of transcription factors, including nuclear steroid receptors, to bind to DNA efficiently[64]–[66].
Although the SWI/SNF complex in S. cerevisiae was identified based on its role in the activation of transcription, experimental evidence indicates that mammalian SWI/SNF complexes contribute to both gene repression and activation. In embryonic stem cells, for instance, BRG1 may act as a transcription repressor of genes associated with differentiation and an enhancer of the core pluripotency transcriptional network[67]. Similarly, the forced overexpression of BRG1 enhances the reprogramming of adult fibroblasts into induced pluripotent stem cells by facilitating Oct4 binding to target genes[68].
It was initially assumed that the whole SWI/SNF complex was required for chromatin remodeling, but biochemical studies have indicated that both BRG1 and BRM may have remodeling activity independent of other SWI/SNF subunits. The addition of core subunits (Baf47, Baf155, and Baf170) is required to achieve chromatin-remodeling activity at near-optimal levels[69], whereas non-essential subunits (Baf53, Baf57 Baf60, and β-actin) aid transcription regulation. In particular, Baf57 is required for hormone steroid receptor activity[70], whereas BAF- and PBAF-specific subunits (Figure 3) are essential for further specifying SWI/SNF complex activity. Baf250a and Baf250b, for instance, are mutually exclusive, and the corresponding Baf250a- and Baf250b-associated complexes have antagonistic effects on cell cycle progression, with Baf250a participating in the repression and Baf250b in the induction of key cell cycle regulators[71]. In addition, Baf250b contributes to the proper expression of genes involved in embryonic stem cell self-renewal[72]. Conversely, the PBAF-specific subunit Baf180 is a critical modulator of p21Cip1 induction and acts as a negative regulator of cell proliferation[73].
SWI/SNF complex mutations and malignant transformation
Because of the essential role of the SWI/SNF complex in various and unrelated pathways, it is not surprising that mutations in one or more subunits of the complex can be associated with malignant transformation[74]. Inactivating mutations in the Baf47 subunit, for instance, are present in the majority of malignant rhabdoid tumors, a class of extremely aggressive and lethal cancers of early childhood that arise primarily in the kidney, brain, and other soft tissues[75]. Experimental studies in stable human cell lines generated from malignant rhabdoid tumors have indicated that Baf47 acts as tumor suppressor and that its forced re-expression leads to cell cycle arrest associated with increases in p16INK4a, E2F1, and cyclin D expression[76]. Other SWI/SNF subunits are believed to function as tumor suppressors. Baf155 and Baf250a have recently been associated with a loss of tumor suppressor activity in human cancer cell lines[77] and gynecologic cancers[78], whereas ARID1A, the gene encoding the Baf250a subunit, has been found to be altered in approximately 10% of breast carcinomas[79]. A similar negative activity on cell proliferation has been described for the Baf180 subunit, mutations in which have been identified in a subset of breast cancers [73]. However, the most common cancer-related changes in the SWI/SNF complex are the loss or inactivation of the BRG1 or BRM subunit, which occur in a variety of tumors, including breast cancer[80],[81]. The genetic or epigenetic silencing of the BRG1 subunit is of particular relevance considering that, in addition to the BAF/PBAF complexes, BRG1 is the pivotal component in several other chromatin-remodeling and histone-modifying enzyme complexes, including the Williams syndrome transcription factor (WSTF)-including nucleosome assembly complex (WINAC), N-CoR, nucleosomal methylation activation complex (NUMAC), and mSin3A/histone deacetylase (HDAC) complex[82]–[84] (Figure 3). In particular, when associated with coactivator-associated arginine methyltransferase-1 (CARM1) to form the NUMAC complex, BRG1 can recruit nuclear hormone receptors to EREs and cooperatively activate estrogen-dependent gene transcription[85]–[87]. Notably, in the presence of an estrogen antagonist, BRG1-containing complexes undergo a reversal of functional activity and act as corepressors, recruiting HDAC1, CBP/p300, and prohibitin and significantly inhibiting promoter activation[88].
ISWI complex: structure and function
The second most prominent ATP-dependent chromatin-remodeling complex is ISWI, the human ortholog of the Drosophila nucleosome-remodeling factor (NURF) complex[89]. ISWI is composed of four subunits: a catalytic subunit (SNF2 ligand, SNF2L), a histone-binding component [bromodomain plant homeo domain (PHD)-finger transcription factor (BPTF)], and two highly homologous retinoblastoma-associated proteins, RbAp46 and RbAp48. SNF2L is the energy-transducing component, and BPTF specifically recognizes histone H3 tails that are trimethylated at lysine 4 (H3K4me3), which are present at the transcription start sites of virtually all active genes[90]. RbAp46 and RbAp48 are involved in histone acetylation[91]. In mammary tissue in particular, RbAp46 and RbAp48 have opposing effects on the expression of estrogen-repressed genes; RbAp46 increases the expression of repressed genes, whereas RbAp48 decreases their expression[92]. Because the ability of RbAp48 to inhibit gene expression is most evident in the absence of an ER ligand, it has been proposed that the primary role of this protein might be in the control of estrogen-repressed gene expression in a hormone-free environment. Conversely, the RbAp46 protein should control estrogen-responsive gene expression in the presence of ovarian hormones[93].
Although the ISWI complex has been studied extensively in normal brain development and neurodegenerative diseases[94], little is known about its functional importance in cancer. However, emerging evidence indicates that the inhibition of SNF2L expression may have dramatic effects on the viability of different cancer cell lines[95] and that BPTF deregulation due to a translocation breakpoint may confer a cancer-promoting phenotype in these cells[96]. In addition, RbAp46 and RbAp48 dysregulation has been linked to carcinogenesis in various tissues, including breast tissue[97].
Covalent histone-modifying complexes
Covalent histone-modifying complexes catalyze the addition and removal of post-translational modifications (e.g., methylation, acetylation, phosphorylation, and ubiquitination) on histone pro-teins[56],[98]. By enhancing or reducing chromatin condensation, these modifications affect the binding affinity between histones and DNA, thus enhancing or preventing gene transcription.
Histone methyltransferases: structure and function
Among the proteins within histone-modifying complexes, histone methyltransferases (HMTs) are histone-modifying enzymes that specifically catalyze the transfer of one, two, or three methyl groups to lysine and arginine residues of the histone tail, predominantly those of histones H3 and H4[99]. For this reason, they are sometimes described as “epigenetic writers.” H3K4 methylation is an evolutionarily conserved mark, and the degree of this methylation is biologically significant because proteins that interact with methylated histones are able to distinguish between mono-, di-, and trimethylated lysines. Based on their target residues, HMTs can be subdivided into protein arginine methyltransferases (PRMTs) and protein lysine methyltransferases (PKMTs). PKMTs are further subdivided into su(var)3-9, enhancer of zeste, trithorax (SET) domain-containing, and non-SET domain-containing proteins. All members of the TrxG family with HMT activity contain a SET domain, and in concert with the ISWI class of ATP-dependent chromatin-remodeling enzymes, these proteins recognize disrupted nucleosomal structure and methylate H3K4.
Represented by a single member in yeast, PKMTs diverged and developed increased structural and functional complexity in higher eukaryotes[100]. Humans express at least eight PKMTs, including several members of the TrxG family. MLL1, the founding member of the PKMT family, was discovered in 1991 upon the cloning of the gene involved in human leukemias carrying chromosome band 11q23 translocations, hence the name mixed lineage leukemia (MLL)[101]. Subsequent biochemical and genetic studies provided evidence for the activity of MLL1 in a wide range of physiologic and pathologic cellular processes, including cell proliferation and differentiation, through the regulation of Hox and non-Hox gene expression[102]–[104]. Since the discovery of MLL1, four additional members, MLL2-MLL5, have been identified. Although their detailed mechanisms of action are still largely unknown, some of these proteins interact with nuclear receptors, including ER, to coordinate hormone-dependent gene regulation[105],[106].
Because of their widespread roles in gene regulation, it is not surprising that changes in the expression of MLL genes due to deletion or truncation may have detrimental effects on crucial physiologic processes and may be associated with human diseases, including cancers. The MLL3 gene, for example, is frequently mutated in many types of human solid tumors, including breast cancer, approximately 40% of which show reduced MLL3 protein expression[107]. A similar oncosuppressive activity has been suggested for MLL5, which has been implicated in myeloid leukemia[108]. Biochemical and genetic studies have established that MLL5 acts as an important cell cycle regulator. MLL5 overexpression inhibits cell cycle progression by suppressing the inappropriate expression of S phase-promoting genes and inducing cell cycle arrest at both the G1 and G2/M phases[109].
In addition to the MLL catalytic subunit, all PKMT complexes contain a common accessory subunit core formed by five subunits: Drosophila Ash2-like (Ash2L), retinoblastoma-binding protein 5 (Rbbp5), WD repeat domain 5 (Wdr5), mammalian Dpy-30 homolog (mDpy-30), and CpG-binding protein (CGBP)[110]–[114] (Figure 4). The currently available evidence indicates that these core subunits interact cooperatively to form a structural platform, which is essential for MLL-mediated histone methylation[115]. Ash2L regulates catalytic activity, whereas the multifunctional Wdr5 subunit serves as a binding adaptor for the MLL-SET domain to dimethylated histone H3K4 (H3K4me2), which is subsequently converted to the trimethylated form (H3K4me3). Genetic studies in embryonic stem cells have shown that the depletion of a single core subunit, such as mDpy-30, may alter the H3K4-specific methyltransferase activity of the complex and consequently misregulate the “bivalent domain” status of genes involved in stem cell fate[116]. “Bivalent domains” are pivotal chromatin structures composed of both repressive (H3K27 methylation) and activating (H3K4 methylation) marks that silence developmental genes while preserving stem cells' ability to be activated in response to appropriate developmental cues. Therefore, changes in H3K4-specific methyltransferase activity may strongly affect embryonic stem cell fate specification[117].
Figure 4. Mixed lineage leukemia proteins (MLLs) are the catalytic subunits of covalent histone-modifying complexes and have H3K4-specific methyltransferase activity.

According to a proposed working model, Menin interacts directly with ER through its activation function 2 (AF2) domain, recruiting the MLL complex to the ERE of the target gene. In multi-protein complexes that include MLL3 or MLL4 as the catalytic subunit, the activating signal cointegrator-2 (ASC-2) protein acts as a coactivator in the transactivation of the nuclear estrogen receptor. This crosstalk functions at least in part by mutually facilitating the recruitment of the MLL and ER complexes to ER target genes.
In higher eukaryotes, the subunit core can associate with a broad spectrum of accessory proteins that coordinate the crosstalk between PKMTs and other enzymes and coregulators involved in histone modification (e.g., acetylation, deacetylation, and demethylation), as well as some components of SWI/SNF chromatin-remodeling complexes[118]. The intricate interrelation among HMT complexes, ATP-dependent chromatin-remodeling complexes, and the nuclear ER is particularly relevant to mammary gland development and differentiation. Studies have demonstrated that upon estrogen stimulation, the MLL2-associated HMT complex is recruited to the promoters of ER target genes along with ligand-bound ER, thereby activating transcription[105]. In addition to its direct interaction with MLL2, ER may interact with the HMT complex via other proteins, such as Menin and activating signal cointegrator-2 (ASC-2). It has been demonstrated that Menin, the product of the multiple endocrine neoplasia type 1 (MEN1) tumor suppressor gene, interacts with ER directly through its activation function 2 (AF2) domain and acts as a coregulator, recruiting the MLL2 complex to the promoters of estrogen-responsive genes[119],[120] (Figure 4). Similarly, ASC-2 participates actively in the transactivation of several nuclear receptors including ER as part of a multi-protein complex, termed ASCOM (for ASC-2 complex), that includes MLL3 or MLL4 as its catalytic subunit.
In addition to ASC-2, Baf47/hSNF5/INI1, a core subunit of SWI/SNF chromatin-remodeling complexes, has been shown to interact directly with MLL3 and MLL4, suggesting a functional crosstalk between chromatin-remodeling (specifically, SWI/SNF) and covalent histone-modifying complexes (specifically, ASCOM) that promotes the binding of both complexes to ER target genes. Because of their roles as cofactors in estrogen-dependent gene transcription, it is not surprising that a change in the expression of Menin or ASC-2 protein may contribute to neoplastic transformation. Menin is overexpressed in the breast cancer cell line MCF-7, colocalizes with ER, and functions as a direct coactivator of ER-mediated transcription[120]. Similarly, ASC-2 has been found to be amplified and overexpressed in breast, colon, and lung cancers. Further experimental evidence suggests that ASC-2 may interact with several mitogenic transcription factors, including AP-1 and NF-κB, and promote cell proliferation through the enhancement of E2F1-dependent transactivation[121],[122].
Histone demethylases: structure and function
In addition to PKMTs, which act as epigenetic writers, TrxG proteins include some elements deputed to histone demethylation (epigenetic erasers). For many years, histone methylation was believed to be an irreversible modification. On the contrary, recent studies have demonstrated that histone methylation can be dynamically regulated through active demethylation. In particular, genetic studies in Drosophila have demonstrated that little imaginal discs (Lid), a Jumonji C domain-containing TrxG protein, can demethylate the trimethylated form of H3K4, thus removing the activation marks and facilitating gene silencing[123]. The four human orthologs of the Lid protein (RBP2/JARID1A, PLU-1/JARID1B, SMCX, and SMCY) belong to the JARID1/KDM5 protein family and show high sequence conservation. Despite the sequence similarity and the putative functional redundancy among these proteins, experimental evidence indicates that individual members have unique functional properties and divergent expression profiles. Retinoblastoma-binding protein 2 (RBP2), the first identified and best characterized member of the JARID1 family, controls two groups of genes, one involved in cellular differentiation and one implicated in mitochondrial function and RNA/DNA metabolism[124]. More specifically, it is positively involved in the transcriptional regulation of Hox proteins during cell differentiation and in steroid receptor-mediated transcription[125],[126]. Very recent experimental findings have implicated RBP2 in the positive regulation of PR expression in the ER-positive breast cancer cell line MCF-7, showing that RBP2 removes the H3K4 methylation mark from the ERE downstream of the PR transcription start site[127]. PLU-1, the second member of the family, was originally identified in human breast cancer cell lines[128], is required for cell proliferation in the mammary gland[129], and causes the down-regulation of tumor suppressor genes, such as BRCA1, in ER-positive breast cancer cells[130]. Less investigated than RBP-2 and PLU-1 are Smcy homolog X-linked (SMCX) and Smcy homolog Y-linked (SMCY), encoded by two genes located on chromosome X and Y, respectively. SMCX acts at the terminal stage of the neuronal differentiation pathway, as demonstrated by its association with X-linked mental retardation, and SMCY is involved in meiosis during spermatogenesis[131],[132].
PcG complexes
PcG proteins were first discovered in Drosophila, in which they work in a reciprocal manner with TrxG proteins during embryo development[49],[50] and establish a heritable cellular memory system, the disruption of which affects normal embryonic development and eventually triggers neoplastic transformation[133],[134].
Similar to TrxG proteins, PcG proteins form multimeric complexes with several other enzymes. To date, two distinct complexes, polycomb repressive complex (PRC)-1 and PRC-2, have been identified in humans (Figure 5).
Figure 5. Polycomb repressive complex (PRC)-1 and PRC-2 work cooperatively.

Enhancer of zeste homolog 2 (EZH2), the catalytic subunit of PRC-2 with methyltransferase activity, mediates the covalent trimethylation of histone H3 at lysine 27 (H3K27me3). This specific histone modification serves as the docking site for proteins harboring a chromobox domain (especially Cbx4 and Cbx8) that aid in the recruitment of PRC-1 to chromatin through their interactions with H3K27me3. Thereafter, PRC-1 catalyzes the monoubiquitilation of histone H2A at lysine 119 through the enzymatic action of RING1/RING1b and blocks transcriptional elongation by RNA polymerase II.
PcG complexes: structure and function
B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1) is the central component of PRC-1, which comprises one or more proteins harboring a chromobox domain (Cbx2, Cbx4, and Cbx8), one or more homologs of Drosophila polyhomeotic protein (PHC1, PHC2, and PHC3), and the RING1/RING1b proteins. In 1993, melanoma nuclear protein 18 (Mel-18), an additional member of the PRC-1 complex that is structurally related to Bmi-1, was isolated and characterized[135]. Bmi-1 was originally identified as an oncogene that, when overexpressed, collaborates with c-Myc to induce the formation of B-cell lymphomas[136]. Bmi-1 is characterized by a RING domain, which is essential for its formation of a heterodimer with RING1/RING1b subunits and induction of the monoubiquitilation of lysine 119 of histone H2A (H2AK119ub)[137]. The Cbx2, Cbx4, and Cbx8 subunits are essential for the reading of the methylation marks set by PRC-2 (specifically, trimethylation of histone H3 at lysine 27), whereas PHC1 and its paralogs PHC2 and PHC3 are required to maintain the transcriptionally repressed state of the target gene[138].
PRC-2 (also known as the EED-EZH2 complex) contains three essential subunits: a catalytic subunit with methyltransferase activity, enhancer of zeste homolog 2 (EZH2), or, less frequently, its paralog EZH1, and two noncatalytic subunits, suppressor of zeste 12 (SUZ12) and embryonic ectoderm development (EED). Similar to TrxG proteins with methyltransferase activity, EZH2 and EZH1 contain a SET domain, which is essential for histone lysine methylation. In quiescent cells, EZH2, SUZ12, and EED are present at low levels, but their expression increases at the G1/S phase transition, when they up-regulate the expression of certain cell cycle-related factors and concomitantly inhibit the expression of apoptosis-related genes. In addition to these essential components, PRC-2 may contain other non-exclusive subunits, including RpbAp48 or RpbAp46, through which it can bind to certain ATP-dependent chromatin-remodeling complexes [91].
PRC-1 and PRC-2 function cooperatively (Figure 5). Initially, EZH2, the PRC-2 catalytic subunit with methyltransferase activity, mediates the covalent trimethylation of histone H3 at lysine 27 (H3K27me3)[139]. This specific histone modification serves as the docking site for proteins harboring a chromobox domain (especially Cbx4 and Cbx8) that facilitate the recruitment of PRC-1 to chromatin through their interactions with H3K27me3 modifications[140]. Thereafter, PRC-1 catalyzes the monoubiquitilation of lysine 119 at histone H2A through the enzymatic action of RING1/RING1b and blocks the transcriptional elongation of RNA polymerase II[141].
PcG complexes and malignant transformation
Because of their pivotal role as epigenetic silencers, some components of PRC-2 and PRC-1 are involved in a variety of tumors. The overexpression of EZH2 has been associated with ovarian, cervical, colon, renal, oral, gastric, prostatic, and pancreatic cancers[142]–[149], and genetic alterations including polymorphisms and inactivating mutations have been described in lung cancer, lymphoma, and myeloid neoplasms[150],[151]. SUZ12 and EED have been found to be up-regulated in several human tumors, including tumors of the colon and liver[152], malignant pleural mesotheliomas, mantle cell lymphoma, pulmonary carcinomas, and melanoma[153],[154]. Similarly, Bmi-1 and RING1 overexpression are associated with ovarian, endometrial, cervical[155],[156], prostate, bladder, pancreatic, colon, and lung cancers[148],[157]–[160], different types of leukemias and lymphomas[161],[162], neuroblastoma[163], and medulloblastoma[164].
In breast cancer, PcG proteins, especially EZH2, Bmi-1, and SUZ12, are significantly overexpressed even at the very earliest stages of neoplastic transformation[165],[166]. The expression of EZH2 is low or absent in morphologically normal lobules, whereas EZH2 expression progressively increases with increasing severity of epithelial atypias[167]. In a very recent study, we found that atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS) surgical specimens dramatically overexpressed the EZH2 gene compared with histologically normal breast tissue (127% in ADH and 207% in DCIS). Notably, we found that EZH2 overexpression was associated with a well-differentiated, ER-positive luminal phenotype, as indicated by the high expression levels of ER, cytokeratin (CK)-18, CK-19, Mucin 1, and Gata-3, all of which are markers of a terminal luminal phenotype. Considering the physiologic turnover of mammary epithelial cells during a woman's reproductive life (i.e., cyclical switching between differentiation and involution), this finding seems to suggest the persistence of a differentiated phenotype due to a defect in lobular involution. Furthermore, considering that lobular involution has been proved to be associated with a reduced risk of breast cancer and that epidemiologic studies have indicated that women with intraductal epithelial hyperplasia have a greater risk of breast cancer than women without this abnormality[168], the association between EZH2 overexpression and increased breast cancer risk is not surprising. Unfortunately, the mechanism through which EZH2 promotes epithelial hyperplasia is largely unknown. Preliminary experimental evidence indicates that the EZH2 protein interacts directly with ER and β-catenin, linking estrogen activity and the Wnt/β-catenin signaling pathway and interfering in normal mammary gland differentiation[169]. Consistent with the association between EZH2 overexpression and tumor aggressiveness, breast cancer subtypes that are defined as aggressive based on their molecular profile (basal-like, HER2-enriched tumors) or clinical markers (triple-negative tumors) show significantly higher EZH2 expression than subtypes (luminal A, luminal B, and ER+/HER2–) that are generally associated with a favorable prognosis[170].
Studies have demonstrated that Bmi-1 overexpression is also associated with aggressive phenotypes and poor prognosis[171]. The hypothesis that Bmi-1 overexpression increases cell motility and invasive capability is based on the observation that, in highly metastatic breast cancer cells, Bmi-1 repression markedly reduced cellular motility, invasion, and metastatic spread[172]. Bmi-1 is also thought to be involved in early stages of breast tumorigenesis by affecting the self-renewal and proliferation of normal mammary stem cells[173]. Studies have demonstrated that Bmi-1 represses the INK4A/ARF locus, a well-known tumor suppressor region that encodes P16Ink4a and P14Arf, two cyclin-dependent kinase (Cdk) inhibitors[174]. P16Ink4a affects retinoblastoma (Rb) protein activity by inhibiting the Cyclin D/Cdk4/6 complex. In the absence of Bmi-1, P16Ink4a is up-regulated and prevents the binding of Cdk4/6 to Cyclin D, thus inhibiting Rb phosphorylation. Consequently, hypophosphorylated Rb binds to E2F, thus inhibiting the transcription of E2F target genes, including those involved in the G1-S transition. Meanwhile, P14Arf stabilizes P53 by antagonizing mouse double minute 2 (MDM2) and activating P53-dependent transcription, which induces cell cycle arrest in G1 or G2/M and subsequent apoptosis[175]. Experimental evidence has indicated that the Bmi-1-induced down-regulation of P16Ink4a and P14Arf facilitates cell immortalization. Furthermore, in human mammary epithelial cells, the removal of P16Ink4a activity causes EZH2 and SUZ12 to be up-regulated and recruited to HOXA9, a gene that is expressed during normal breast development but epigenetically silenced in breast cancer[176]. Mel-18 is structurally related to but not functionally redundant with Bmi-1. The expression levels of these two genes are negatively correlated in several breast cancer cell lines[177], and Mel-18 expression is a predictor of poor prognosis in patients with breast cancer[178]. Experimental findings have indicated that Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity[179],[180] or competing with Bmi-1 for binding to the INK4A/ARF locus[181].
HOX genes
As extensively described above, TrxG and PcG maintenance proteins are organized in multifactor complexes and play crucial roles in the epigenetic control of a large number of genes. They function as a part of a widely conserved cell memory system that prevents changes in cell identity by maintaining transcription patterns throughout development and in adulthood[182]. Therefore, any perturbation of the levels of TrxG and PcG proteins may have dramatic effects on cell identity, and studies have clearly demonstrated that the deregulated expression of genes involved in stem cell renewal or developmental regulation may trigger neoplastic transformation[183]. The deregulation of stem/progenitor cell self-renewal has also been proposed as a key event in mammary carcinogenesis[184],[185].
Among the genes that play a direct role in cell fate specification and maintenance of cell identity, HOX genes are the best studied. During embryonic development, HOX gene expression controls the identity of various regions along the anterior-posterior axis of the embryo according to the rules of temporal and spatial collinearity, that is, HOX gene expression along the anterior-posterior axis is collinear with the 3′ to 5′ organization of the HOX genes on the chromosome[186]. HOX gene expression varies among normal adult organs. Indeed, each organ, including the mammary gland, shows a specific pattern of HOX gene expression that represents a unique molecular signature. Of the 39 human HOX genes, which are organized into four paralogous clusters (HOX loci A, B, C, D), 17 (HOXA3, HOXA7, HOXA8, HOXA9, HOXA10, HOXA11, HOXB3, HOXB5, HOXB6, HOXB7, HOXC6, HOXC10, HOXC11, HOXC13, HOXD1 HOXD8, and HOXD9) are expressed in the normal adult mammary gland[187] and are differentially expressed in breast cancer[188]. In particular, the HOX genes that are altered in breast cancer mostly correspond to paralogs 1-4 (HOXA1, HOXA3, HOXA5, HOXB2, HOXD3, and HOXD4) and 9-13 (HOXD10, HOXA11, HOXB13, and HOXD13). Among the altered HOX genes, HOXA5 seems to be the most important. Indeed, the HOXA5 protein level is decreased in nearly 70% of all breast carcinomas[189], and experimental studies have indicated that compromised HOXA5 function could limit P53 expression, thus affecting P53 tumor suppressor activity[190]. In addition, HOXA5 gene silencing due to selective hypermethylation of CpG islands in the promoter region also affects PR expression levels and consequently the terminal differentiation of the mammary epithelium[191],[192].
Despite the differential expression of HOX genes between normal and cancer tissues, nothing is known about the dynamics of HOX gene expression, especially during mammary gland turnover. However, because mammary gland development and differentiation depend upon the presence of ovarian hormones, it is reasonable to speculate that there may be a link between HOX gene expression and steroid hormones. Experimental evidence corroborates this hypothesis. Indeed, some studies have demonstrated that several HOX genes, including HOXC10 and HOXC13, are transcriptionally regulated by estrogen via several EREs in their promoter region[193]. Furthermore, HOXC6, which is involved in mammary gland development and milk production, was recently shown to be transcriptionally regulated by estrogen via two putative EREs within its promoter region[194]. Although no direct link between altered HOX gene expression and breast tumorigenesis has been established, it should be hypothesized that an abnormal concentration of steroid hormones, particularly estrogen, could affect HOX gene expression dynamics and contribute to cancer initiation or progression. Indeed, a misexpression of HOX genes could result in a different gene expression profile, leading to changes in cell differentiation or the adoption of an alternative cell identity (i.e., homeotic transformation).
In a similar scenario, the TrxG and PcG maintenance proteins play crucial roles as master regulators of HOX gene expression. At least six independent genome-wide studies have identified common Polycomb targets in vertebrates and flies, most of which are homeobox genes or other developmental transcription factors[195]. In addition, it has been demonstrated that MLL2 and MLL3, the catalytic subunits of covalent histone-modifying complexes with H3K4-specific methyltransferase activities, coordinate with ER to transcriptionally regulate HOXC6 in an estrogen-dependent manner[194], and MLL3 knockdown suppresses the estrogen-induced activation of HOXC13[196].
Concluding Remarks
During normal postnatal mammary gland development and remodeling during the menstrual cycle, pregnancy, and lactation, ovarian hormones and peptide growth factors contribute to the delineation of a definite cell identity by regulating tissue-specific gene expression. This gene expression is maintained during cell replication such that the daughter cells retain the differentiated cell type of the parental cell. The maintenance of this gene expression pattern, termed cellular memory, is critical for the preservation of cell identity, particularly when cells need to change in response to intrinsic and extrinsic signals. The maintenance proteins, which are required for cell identity preservation, act epigenetically by regulating gene expression through DNA methylation, histone modification, and chromatin remodeling[45],[46]. Among the maintenance proteins, those belonging to the TrxG and PcG groups are the best characterized[47],[48], and an increasing body of evidence clearly indicates that their dysregulation may lead to a disruption of cell identity. As chromatin remodeling requires the cooperation of different enzymes acting in different manners (mainly through acetylation and methylation), it is evident that the alteration of any single epigenetic regulator may promote cell identity disruption and consequently affect mammary epithelial morphology and TDLU organization; this may trigger neoplastic transformation. Considering the close relationship between these epigenetic regulators and the nuclear receptors through which ovarian hormones (especially estrogen) act, elucidating how epithelial cell identity is disrupted in response to physiologic stimuli such as ovarian hormones is of paramount importance. Improving our knowledge of the epigenetic control of cell identity-associated gene expression and understanding how perturbations in epigenetic control contribute to breast tumor initiation will provide potential targets for personalized epigenetic therapies aimed to restore the appropriate epigenetic activity and therefore a normal epithelial mammary gland morphology.
References
- 1.Rosen PP. Rosen's Breast Pathology. In: Rosen PP, editor. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 201–248. [Google Scholar]
- 2.Smalley M, Ashworth A. Stem cells and breast cancer: a field in transit. Nat Rev Cancer. 2003;3:832–844. doi: 10.1038/nrc1212. [DOI] [PubMed] [Google Scholar]
- 3.Stingl J, Raouf A, Emerman JT, et al. et al. Epithelial progenitors in the normal human mammary gland. J Mammary Gland Biol Neoplasia. 2005;10:49–59. doi: 10.1007/s10911-005-2540-7. [DOI] [PubMed] [Google Scholar]
- 4.Mani SA, Guo W, Liao MJ, et al. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polyak K, Weinberg RA. Transitions between epithelial and mesen-chymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 6.Fata E, Werb Z, Bissell MJ. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res. 2004;6:1–11. doi: 10.1186/bcr634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Going JJ. Ductal-lobular organisation of human breast tissue, its relevance in disease and a research objective: vector mapping of parenchyma in complete breasts (the Astley Cooper project) Breast Cancer Res. 2006;8:107. doi: 10.1186/bcr1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barsky SH, Karlin NJ. Mechanisms of disease: breast tumor pathogenesis and the role of the myoepithelial cell. Nat Clin Pract Oncol. 2006;3:138–151. doi: 10.1038/ncponc0450. [DOI] [PubMed] [Google Scholar]
- 9.Russo J, Russo IH. Development of the human breast. Maturitas. 2004;49:2–15. doi: 10.1016/j.maturitas.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Brisken C, O'Malley B. Hormone action in the mammary gland. Cold Spring Harb Perspect Biol. 2010;2:a003178. doi: 10.1101/cshperspect.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kastner P, Krust A, Turcotte B, et al. et al. Two distinct estrogen-regulated promoters generate transcripts encoding two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9:1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Echeverria PC, Picard D. Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim Biophys Acta. 2010;1803:641–649. doi: 10.1016/j.bbamcr.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41:263–275. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 16.Lonard DM, O'Malley BW. Nuclear receptor coregulators: judges, juries and executioners of cellular regulation. Mol Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 17.Lahusen T, Henke RT, Kagan BL, et al. et al. The role and regulation of the nuclear receptor co-activator AIB1 in breast cancer. Breast Cancer Res Treat. 2009;116:225–237. doi: 10.1007/s10549-009-0405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jepsen K, Rosenfeld MG. Biological roles and mechanistic actions of corepressor complexes. J Cell Sci. 2002;115:689–698. doi: 10.1242/jcs.115.4.689. [DOI] [PubMed] [Google Scholar]
- 19.Stanya KJ, Kao HY. New insights into the functions and regulation of the transcriptional corepressors SMRT and N-CoR. Cell Div. 2009;4:1–8. doi: 10.1186/1747-1028-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hannenhalli S, Kaestner KH. The evolution of Fox genes and their role in development and disease. Nat Rev Genet. 2009;10:233–240. doi: 10.1038/nrg2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekiya T, Zaret KS. Repression by Groucho/TLE/Grg proteins: genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol Cell. 2007;28:291–303. doi: 10.1016/j.molcel.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cirillo LA, Lin FR, Cuesta I, et al. et al. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 24.Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012;12:381–385. doi: 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- 25.Augello MA, Hickey TE, Knudson KE. FOXA1: master of steroid receptor function in cancer. EMBO J. 2011;30:3885–3894. doi: 10.1038/emboj.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schalch T, Duda S, Sargent DF, et al. et al. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 27.Brennan RG. The winged-helix DNA-binding motif: another helix-turn-helix takeoff. Cell. 1993;74:773–776. doi: 10.1016/0092-8674(93)90456-z. [DOI] [PubMed] [Google Scholar]
- 28.Sekiya T, Muthurajan UM, Luger K, et al. et al. Nucleosome binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804–809. doi: 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cirillo LA, Zaret KS. Specific interactions of the wing domains of FOXA1 transcription factor with DNA. J Mol Biol. 2007;366:720–724. doi: 10.1016/j.jmb.2006.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serandour AA, Avner S, Percevault F, et al. et al. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011;21:555–565. doi: 10.1101/gr.111534.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lupien M, Eeckhoute J, Meyer CA, et al. et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernardo GM, Lozada KL, Miedler JD, et al. et al. FOXA1 is an essential determinant of ERa expression and mammary ductal morphogenesis. Development. 2010;137:2045–2054. doi: 10.1242/dev.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asselin-Labat M-L, Sutherland KD, Barker H, et al. et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol. 2007;9:201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 34.Kouros-Mehr H, Slorach EM, Sternlicht MD, et al. et al. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127:1041–1055. doi: 10.1016/j.cell.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eeckhoute J, Keeton EK, Lupien M, et al. et al. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67:6477–6482. doi: 10.1158/0008-5472.CAN-07-0746. [DOI] [PubMed] [Google Scholar]
- 36.Hynes NE, Watson CJ. Mammary gland growth factors: roles in normal development and in cancer. Cold Spring Harb Perspect Biol. 2010;2:a003186. doi: 10.1101/cshperspect.a003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moses H, Barcellos-Hoff MH. TGF-beta biology in mammary development and breast cancer. Cold Spring Harb Perspect Biol. 2011;3:a003277. doi: 10.1101/cshperspect.a003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciarloni L, Mallepell S, Brisken C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci USA. 2007;104:5455–5460. doi: 10.1073/pnas.0611647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke RB, Howell A, Potten CS, et al. et al. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 1997;57:4987–4991. [PubMed] [Google Scholar]
- 40.Mallepell S, Krst A, Chambon P, et al. et al. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci USA. 2006;103:2196–2201. doi: 10.1073/pnas.0510974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asselin-Labat ML, Vaillant F, Sheridan JM, et al. et al. Control of mammary stem cell function by steroid hormone signaling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 42.Grimm SL, Rosen JM. Stop! In the name of transforming growth factor-beta: keeping estrogen receptor-alpha-positive mammary epithelial cells from proliferating. Breast Cancer Res. 2006;8:106. doi: 10.1186/bcr1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan H, Zhong Y, Pan Z. Autocrine regulation of cell proliferation by estrogen receptor-alpha in estrogen receptor-alpha-positive breast cancer cell lines. BMC Cancer. 2009;9:31. doi: 10.1186/1471-2407-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brock HW, Fisher CL. Maintenance of gene expression patterns. Dev Dynamics. 2005;232:633–655. doi: 10.1002/dvdy.20298. [DOI] [PubMed] [Google Scholar]
- 45.Vermaak D, Ahmad K, Henikoff S. Maintenance of chromatin states: an open-and-shut case. Curr Opin Cell Biol. 2003;15:266–274. doi: 10.1016/s0955-0674(03)00043-7. [DOI] [PubMed] [Google Scholar]
- 46.Hake SB, Xiao A, Allis CD. Linking the epigenetic “language” of covalent histone modifications to cancer. Br J Cancer. 2004;90:761–769. doi: 10.1038/sj.bjc.6601575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 48.Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 2010;10:669–682. doi: 10.1038/nrc2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kennison JA. The Polycomb and trithorax group proteins of Drosophila: trans-regulators of homeotic gene function. Annu Rev Genet. 1995;29:289–303. doi: 10.1146/annurev.ge.29.120195.001445. [DOI] [PubMed] [Google Scholar]
- 50.Grimaud C, Negre N, Cavalli G. From genetics to epigenetics: the tale of Polycomb group and trithorax group genes. Chromosome Res. 2006;14:363–375. doi: 10.1007/s10577-006-1069-y. [DOI] [PubMed] [Google Scholar]
- 51.Delaval K, Feil R. Epigenetic regulation of mammalian genomic imprinting. Curr Opin Genet Dev. 2004;14:188–195. doi: 10.1016/j.gde.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 52.Heard E. Delving into the diversity of facultative heterochromatin: the epigenetics of the inactive X chromosome. Curr Opin Genet Dev. 2005;15:482–489. doi: 10.1016/j.gde.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 54.Martinez AM, Cavalli G. The role of polycomb group proteins in cell cycle regulation during development. Cell Cycle. 2006;5:1189–1197. doi: 10.4161/cc.5.11.2781. [DOI] [PubMed] [Google Scholar]
- 55.Johnson CN, Adkins NL, Georgel P. Chromatin remodeling complexes: ATP-dependent machines in action. Biochem Cell Biol. 2005;83:405–417. doi: 10.1139/o05-115. [DOI] [PubMed] [Google Scholar]
- 56.Hassan AH, Neely KE, Vignali M, et al. et al. Promoter targeting of chromatin-modifying complexes. Front Biosci. 2001;6:D1054–D1064. doi: 10.2741/hassan. [DOI] [PubMed] [Google Scholar]
- 57.Eberharter A, Becker PB. ATP-dependent nucleosome remodelling: factors and functions. J Cell Sci. 2004;117:3707–3711. doi: 10.1242/jcs.01175. [DOI] [PubMed] [Google Scholar]
- 58.Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acid Res. 1995;23:2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kassabov SR, Zhang B, Persinger J, et al. et al. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol Cell. 2003;11:391–403. doi: 10.1016/s1097-2765(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 60.Flowers S, Nagl NG, Beck GR, et al. et al. Antagonistic roles for BRM and BRG1 SWI/SNF complexes in differentiation. J Biol Chem. 2009;284:10067–10075. doi: 10.1074/jbc.M808782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roberts CW, Orkin SH. The SWI/SNF complex—chromatin and cancer. Nat Rev Cancer. 2004;4:133–142. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 62.Schnitzler G, Sif S, Kingston RE. Human SWI/SNF interconverts a nucleosome between its base state and a stable remodeled state. Cell. 1998;94:17–27. doi: 10.1016/s0092-8674(00)81217-9. [DOI] [PubMed] [Google Scholar]
- 63.Dechassa ML, Sabri A, Pondugula S, et al. et al. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol Cell. 2010;38:590–602. doi: 10.1016/j.molcel.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fryer CJ, Archer TK. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature. 1998;393:88–91. doi: 10.1038/30032. [DOI] [PubMed] [Google Scholar]
- 65.Belandia B, Orford RL, Hurst HC, et al. et al. Targeting of SWI/SNF chromatin remodeling complexes to estrogen-responsive genes. EMBO J. 2002;21:4094–4103. doi: 10.1093/emboj/cdf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marshall TW, Link KA, Petre-Draviam CE, et al. et al. Differential requirement of SWI/SNF for androgen receptor activity. J Biol Chem. 2003;278:30605–30613. doi: 10.1074/jbc.M304582200. [DOI] [PubMed] [Google Scholar]
- 67.Ho L, Jothi R, Ronan JL, et al. et al. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc Natl Acad Sci USA. 2009;106:5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singhal N, Graumann J, Wu G, et al. et al. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell. 2010;141:943–955. doi: 10.1016/j.cell.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 69.Phelan ML, Sif S, Narlikar GJ, et al. et al. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell. 1999;3:247–253. doi: 10.1016/s1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- 70.Garcia-Pedrero JM, Kiskinis E, Parker MG, et al. et al. The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J Biol Chem. 2006;281:22656–22664. doi: 10.1074/jbc.M602561200. [DOI] [PubMed] [Google Scholar]
- 71.Nagl NG, Jr, Wang X, Patsialou A, et al. et al. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007;26:752–763. doi: 10.1038/sj.emboj.7601541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan Z, Wang Z, Sharova L, et al. et al. BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells. 2008;26:1155–1165. doi: 10.1634/stemcells.2007-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xia W, Nagase S, Montia AG, et al. et al. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008;68:1667–1674. doi: 10.1158/0008-5472.CAN-07-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson BG, Roberts WM. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 75.Versteege I, Sevenet N, Lange J, et al. et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 76.Versteege I, Medjkane S, Rouillard D, et al. et al. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002;21:6403–6412. doi: 10.1038/sj.onc.1205841. [DOI] [PubMed] [Google Scholar]
- 77.DelBove J, Rosson G, Strobeck M, et al. et al. Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics. 2011;6:1444–1453. doi: 10.4161/epi.6.12.18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guan B, Wang TL, Shih IM. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71:6718–6727. doi: 10.1158/0008-5472.CAN-11-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang X, Nagl NG, Jr, Flowers S, et al. et al. Expression of p250 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer. 2004;112:636–642. doi: 10.1002/ijc.20450. [DOI] [PubMed] [Google Scholar]
- 80.Medina PP, Sanchez-Cespedes M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics. 2008;3:64–68. doi: 10.4161/epi.3.2.6153. [DOI] [PubMed] [Google Scholar]
- 81.Wong AK, Shanahan F, Chen Y, et al. et al. BRG1, a component of SWI/SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–6177. [PubMed] [Google Scholar]
- 82.Oya H, Yokoyama A, Yamaoka I, et al. et al. Phosphorylation of Williams syndrome transcription factor by MAPK induces a switching between two distinct chromatin remodeling complexes. J Biol Chem. 2009;284:32472–32482. doi: 10.1074/jbc.M109.009738. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83.Underhill C, Qutob MS, Yee SP, et al. et al. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J Biol Chem. 2000;275:40463–40470. doi: 10.1074/jbc.M007864200. [DOI] [PubMed] [Google Scholar]
- 84.Xu W, Cho H, Kadam S, et al. et al. A methylation-mediator complex in hormone signaling. Genes Dev. 2004;18:144–156. doi: 10.1101/gad.1141704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trotter KW, Archer TK. The BRG1 transcriptional coregulator. Nuclear Recept Signal. 2008;6:e004. doi: 10.1621/nrs.06004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aoyagi S, Trotter KW, Archer TK. ATP-dependent chromatin remodeling complexes and their role in nuclear receptor-dependent transcription in vivo. Vitam Horm. 2005;70:281–307. doi: 10.1016/S0083-6729(05)70009-1. [DOI] [PubMed] [Google Scholar]
- 87.Al-Dhaheri M, Wu J, Skliris GP, et al. et al. CARM1 is an important determinant of ERα-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer Res. 2011;71:2118–2128. doi: 10.1158/0008-5472.CAN-10-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang B, Chambers KJ, Faller DV, et al. et al. Reprogramming of the SWI/SNF complex for co-activation or co-repression in prohibitin-mediated estrogen receptor regulation. Oncogene. 2007;26:7153–7157. doi: 10.1038/sj.onc.1210509. [DOI] [PubMed] [Google Scholar]
- 89.Barak O, Lazzaro MA, Lane WS, et al. et al. Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J. 2003;22:6089–6100. doi: 10.1093/emboj/cdg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wysocka J, Swigut T, Xiao H, et al. et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 91.Kuzmichev A, Nishioka K, Erdjument-Bromage H, et al. et al. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Creekmore A, Walt KA, Schultz-Norton JR, et al. et al. The role of retinoblastoma associated proteins 46 and 48 in estrogen receptor α mediated gene expression. Mol Cell Endocrinol. 2008;291:79–86. doi: 10.1016/j.mce.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ginger MR, Gonzalez-Rimbau MF, Gay JP, et al. et al. Persistent changes in gene expression induced by estrogen and progesterone in the rat mammary gland. Mol Endocrinol. 2001;15:1993–2009. doi: 10.1210/mend.15.11.0724. [DOI] [PubMed] [Google Scholar]
- 94.Mu X, Springer JE, Bowser R. FAC1 expression and localization in motor neurons of developing, adult, and amyotrophic lateral sclerosis spinal cord. Exp Neurol. 1997;146:17–24. doi: 10.1006/exnr.1997.6508. [DOI] [PubMed] [Google Scholar]
- 95.Ye Y, Xiao Y, Wang W, et al. et al. Inhibition of expression of the chromatin remodeling gene, SNF2L, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol Cancer Res. 2009;7:1984–1999. doi: 10.1158/1541-7786.MCR-09-0119. [DOI] [PubMed] [Google Scholar]
- 96.Buganim Y, Goldstein I, Lipson D, et al. et al. A novel translocation breakpoint within the BPTF gene is associated with a pre-malignant phenotype. PLoS ONE. 2010;5:e9657. doi: 10.1371/journal.pone.0009657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thakur A, Rahman KW, Wu J, et al. et al. Aberrant expression of X-linked genes RbAp46, Rsk4, and Cldn2 in breast cancer. Mol Cancer Res. 2007;5:171–181. doi: 10.1158/1541-7786.MCR-06-0071. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev. 2003;17:2733–2740. doi: 10.1101/gad.1156403. [DOI] [PubMed] [Google Scholar]
- 99.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 100.Malik S, Bhaumik SR. Mixed lineage leukemia: histone H3 lysine 4 methyltransferases from yeast to human. FEBS J. 2010;277:1805–1821. doi: 10.1111/j.1742-4658.2010.07607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ziemin-van der Poel S, McCabe NR, Gill HJ, et al. et al. Identification of a gene, MLL, that spans the breakpoint in11q23 translocations associated with human leukemias. Proc Natl Acad Sci USA. 1991;88:10735–10739. doi: 10.1073/pnas.88.23.10735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Milne TA, Briggs SD, Brock HW, et al. et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 103.Liu H, Takeda S, Cheng EH, et al. et al. Biphasic MLL takes helm at cell cycle control: implications in human mixed lineage leukemia. Cell Cycle. 2008;7:428–435. doi: 10.4161/cc.7.4.5426. [DOI] [PubMed] [Google Scholar]
- 104.Milne TA, Dou Y, Martin ME, et al. et al. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102:14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mo R, Rao SM, Zhu YJ. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem. 2006;281:15714–15720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 106.Lee S, Kim DH, Goo YH, et al. et al. Crucial roles for interactions between MLL3/4 and INI1 in nuclear receptor transactivation. Mol Endocrinol. 2009;23:610–619. doi: 10.1210/me.2008-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang XX, Fu L, Li X, et al. et al. Somatic mutations of the mixed-lineage leukemia 3 (MLL3) gene in primary breast cancers. Pathol Oncol Res. 2011;17:429–433. doi: 10.1007/s12253-010-9316-0. [DOI] [PubMed] [Google Scholar]
- 108.Emerling BM, Bonifas J, Kratz CP, et al. et al. MLL5, a homolog of Drosophila trithorax located within a segment of chromosome band 7q22 implicated in myeloid leukemia. Oncogene. 2002;21:4849–4854. doi: 10.1038/sj.onc.1205615. [DOI] [PubMed] [Google Scholar]
- 109.Cheng F, Liu J, Zhou SH, et al. et al. RNA interference against mixed lineage leukemia 5 resulted in cell cycle arrest. Int J Biochem Cell Biol. 2008;40:2472–2481. doi: 10.1016/j.biocel.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 110.Nakamura T, Mori T, Tada S, et al. et al. ALL-1 is a histone methyl-transferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 111.Steward MM, Lee JS, O'Donovan A, et al. et al. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complex. Nat Struct Mol Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- 112.Odho Z, Southall SM, Wilson JR. Characterization of a novel WDR5-binding site that recruits RbBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J Biol Chem. 2010;285:32967–32976. doi: 10.1074/jbc.M110.159921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang X, Lou Z, Dong X, et al. et al. Crystal structure of the C-terminal domain of human DPY-30-like protein: a component of the histone methyltransferase complex. J Mol Biol. 2009;390:530–537. doi: 10.1016/j.jmb.2009.05.061. [DOI] [PubMed] [Google Scholar]
- 114.Southall SM, Wong PS, Odho Z, et al. et al. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 115.Dou Y, Milne TA, Ruthenberg AJ, et al. et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- 116.Jiang H, Shukla A, Wang X, et al. et al. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell. 2011;144:513–525. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bernstein BE, Mikkelsen TS, Xie X, et al. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 118.Suganuma T, Workman JL. Crosstalk among histone modifications. Cell. 2008;135:604–607. doi: 10.1016/j.cell.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 119.Dreijerink KM, Mulder KW, Winkler GS, et al. et al. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006;66:4929–4935. doi: 10.1158/0008-5472.CAN-05-4461. [DOI] [PubMed] [Google Scholar]
- 120.Imachi H, Murao K, Dobashi H, et al. et al. Menin, a product of the MEN1 gene, binds to estrogen receptor to enhance its activity in breast cancer cells: possibility of a novel predictive factor for tamoxifen resistance. Breast Cancer Res Treat. 2010;122:395–407. doi: 10.1007/s10549-009-0581-0. [DOI] [PubMed] [Google Scholar]
- 121.Lee SK, Na SY, Jung SY, et al. et al. Activating protein-1, nuclear factor-kappaB, and serum response factor as novel target molecules of the cancer-amplified transcription coactivator ASC-2. Mol Endocrinol. 2000;14:915–925. doi: 10.1210/mend.14.6.0471. [DOI] [PubMed] [Google Scholar]
- 122.Kong HJ, Yu HJ, Hong S, et al. et al. Interaction and functional cooperation of the cancer-amplified transcriptional coactivator activating signal cointegrator-2 and E2F-1 in cell proliferation. Mol Cancer Res. 2003;1:48–58. [PubMed] [Google Scholar]
- 123.Lee N, Zhang J, Klose RJ, et al. et al. The trithorax group protein Lid is a histone H3 trimethyl-Lys4 demethylase. Nat Struct Mol Biol. 2007;14:341–343. doi: 10.1038/nsmb1216. [DOI] [PubMed] [Google Scholar]
- 124.Lopez-Bigas N, Kisiel TA, Dewaal DC, et al. et al. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell. 2008;31:520–530. doi: 10.1016/j.molcel.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Benevolenskaya EV, Murray HL, Branton P, et al. et al. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005;18:623–635. doi: 10.1016/j.molcel.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 126.Chan SW, Hong W. Retinoblastoma-binding protein 2 (Rbp2) potentiates nuclear hormone receptor-mediated transcription. J Biol Chem. 2001;276:28402–28412. doi: 10.1074/jbc.M100313200. [DOI] [PubMed] [Google Scholar]
- 127.Stratmann A, Haendler B. The histone demethylase JARID1A regulates progesterone receptor expression. FEBS J. 2011;278:1458–1469. doi: 10.1111/j.1742-4658.2011.08058.x. [DOI] [PubMed] [Google Scholar]
- 128.Lu PJ, Sundquist K, Baeckstrom D, et al. et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–15645. doi: 10.1074/jbc.274.22.15633. [DOI] [PubMed] [Google Scholar]
- 129.Catchpole S, Spencer-Dene B, Hall D, et al. et al. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol. 2011;38:1267–1277. doi: 10.3892/ijo.2011.956. [DOI] [PubMed] [Google Scholar]
- 130.Scibetta AG, Santangelo S, Catchpole S, et al. et al. Identification of genes specifically down-regulated by the breast cancer associated transcriptional repressor PLU-1/JARID1B. Mol Cell Biol. 2007;27:7220–7235. doi: 10.1128/MCB.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jensen LR, Amende M, Gurok U, et al. et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Akimoto C, Kitagawa H, Matsumoto T, et al. et al. Spermatogenesis-specific association of SMCY and MSH5. Genes Cells. 2008;13:623–633. doi: 10.1111/j.1365-2443.2008.01193.x. [DOI] [PubMed] [Google Scholar]
- 133.Schuettengruber B, Chourrout D, Vervoort M, et al. et al. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 134.Richly H, Aloia L, Di Croce L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2011;2:e204. doi: 10.1038/cddis.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ishida A, Asano H, Hasegawa M, et al. et al. Cloning and chromosome mapping of the human Mel-18 gene which encodes a DNA-binding protein with a new “RING-finger” motif. Gene. 1993;129:249–255. doi: 10.1016/0378-1119(93)90275-8. [DOI] [PubMed] [Google Scholar]
- 136.Jacobs JJ, Scheijen B, Voncken JW, et al. et al. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13:2678–2690. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Joazeiro CA, Weissman AM. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000;102:549–552. doi: 10.1016/s0092-8674(00)00077-5. [DOI] [PubMed] [Google Scholar]
- 138.Levine SS, Weiss A, Erdjument-Bromage H, et al. et al. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol. 2002;22:6070–6078. doi: 10.1128/MCB.22.17.6070-6078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 140.Bezsonova I, Walker JR, Bacik JP, et al. et al. Ring1B contains a ubiquitin-like docking module for interaction with Cbx proteins. Biochemistry. 2009;48:10542–10548. doi: 10.1021/bi901131u. [DOI] [PubMed] [Google Scholar]
- 141.Stock J, Giadrossi S, Casanova M, et al. et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9:1428–1435. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- 142.Li H, Cai Q, Godwin AK, et al. et al. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res. 2010;8:1610–1618. doi: 10.1158/1541-7786.MCR-10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fang J, Zhang M, Li Q. Enhancer of zeste homolog 2 expression is associated with tumor cell proliferation and invasion in cervical cancer. Am J Med Sci. 2011;342:198–204. doi: 10.1097/MAJ.0b013e31821335a9. [DOI] [PubMed] [Google Scholar]
- 144.Fussbroich B, Wagener N, Macher-Goeppinger S, et al. et al. EZH2 depletion blocks the proliferation of colon cancer cells. PLoS One. 2011;6:e21651. doi: 10.1371/journal.pone.0021651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wagener N, Holland D, Bulkescher J, et al. et al. The enhancer of zeste homolog 2 gene contributes to cell proliferation and apoptosis resistance in renal cell carcinoma cells. Int J Cancer. 2008;123:1545–1550. doi: 10.1002/ijc.23683. [DOI] [PubMed] [Google Scholar]
- 146.Kidani K, Osaki M, Tamura T, et al. et al. High expression of EZH2 is associated with tumor proliferation and prognosis in human oral squamous cell carcinomas. Oral Oncol. 2009;45:39–46. doi: 10.1016/j.oraloncology.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 147.Choi JH, Song YS, Yoon JS, et al. et al. Enhancer of zeste homolog 2 expression is associated with tumor cell proliferation and metastasis in gastric cancer. APMIS. 2010;118:196–202. doi: 10.1111/j.1600-0463.2009.02579.x. [DOI] [PubMed] [Google Scholar]
- 148.van Leenders GJ, Dukers D, Hessels D, et al. et al. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol. 2007;52:455–463. doi: 10.1016/j.eururo.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 149.Toll AD, Dasgupta A, Potoczek M, et al. et al. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum Pathol. 2010;41:1205–1209. doi: 10.1016/j.humpath.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 150.Yoon KA, Gil HJ, Han J, et al. et al. Genetic polymorphisms in the polycomb group gene EZH2 and the risk of lung cancer. J Thorac Oncol. 2010;5:10–16. doi: 10.1097/JTO.0b013e3181c422d9. [DOI] [PubMed] [Google Scholar]
- 151.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 152.Kirmizis A, Bartley SM, Farnham PJ. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol Cancer Ther. 2003;2:113–121. [PubMed] [Google Scholar]
- 153.Kemp CD, Rao M, Xi S, et al. et al. Polycomb repressor complex-2 is a novel target for mesothelioma therapy. Clin Cancer Res. 2012;18:77–90. doi: 10.1158/1078-0432.CCR-11-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Martín-Pérez D, Sánchez E, Maestre L, et al. et al. Deregulated expression of the polycomb-group protein SUZ12 target genes characterizes mantle cell lymphoma. Am J Pathol. 2010;177:930–942. doi: 10.2353/ajpath.2010.090769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Honig A, Weidler C, Häusler S, et al. et al. Overexpression of polycomb protein BMI-1 in human specimens of breast, ovarian, endometrial and cervical cancer. Anticancer Res. 2010;30:1559–1564. [PubMed] [Google Scholar]
- 156.Zhang X, Wang CX, Zhu CB, et al. et al. Overexpression of Bmi-1 in uterine cervical cancer: correlation with clinicopathology and prognosis. Int J Gynecol Cancer. 2010;20:1597–1603. [PubMed] [Google Scholar]
- 157.Qin ZK, Yang JA, Ye YL, et al. et al. Expression of Bmi-1 is a prognostic marker in bladder cancer. BMC Cancer. 2009;9:61. doi: 10.1186/1471-2407-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Song W, Tao K, Li H, et al. et al. Bmi-1 is related to proliferation, survival and poor prognosis in pancreatic cancer. Cancer Sci. 2010;101:1754–1760. doi: 10.1111/j.1349-7006.2010.01577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Du J, Li Y, Li J, et al. et al. Polycomb group protein Bmi1 expression in colon cancers predicts the survival. Med Oncol. 2010;27:1273–1276. doi: 10.1007/s12032-009-9373-y. [DOI] [PubMed] [Google Scholar]
- 160.Vrzalikova K, Skarda J, Ehrmann J, et al. et al. Prognostic value of Bmi-1 oncoprotein expression in NSCLC patients: a tissue microarray study. J Cancer Res Clin Oncol. 2008;134:1037–1042. doi: 10.1007/s00432-008-0361-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bhattacharyya J, Mihara K, Yasunaga S, et al. et al. BMI-1 expression is enhanced through transcriptional and posttranscriptional regulation during the progression of chronic myeloid leukemia. Ann Hematol. 2009;88:333–340. doi: 10.1007/s00277-008-0603-8. [DOI] [PubMed] [Google Scholar]
- 162.Dukers DF, van Galen JC, Giroth C, et al. et al. Unique polycomb gene expression pattern in Hodgkin's lymphoma and Hodgkin's lymphoma-derived cell lines. Am J Pathol. 2004;164:873–881. doi: 10.1016/S0002-9440(10)63175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cui H, Hu B, Li T, et al. et al. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am J Pathol. 2007;170:1370–1378. doi: 10.2353/ajpath.2007.060754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Leung C, Lingbeek M, Shakhova O, et al. et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 165.Ding L, Erdmann C, Chinnaiyan AM, et al. et al. Identification of EZH2 as a molecular marker for a precancerous state in morphologically normal breast tissues. Cancer Res. 2006;66:4095–4099. doi: 10.1158/0008-5472.CAN-05-4300. [DOI] [PubMed] [Google Scholar]
- 166.Li X, Gonzalez ME, Toy K, et al. et al. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol. 2009;175:1246–1254. doi: 10.2353/ajpath.2009.090042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kleer CG, Cao Q, Varambally S, et al. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transfor-mation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Milanese TR, Hartmann LC, Sellers TA, et al. et al. Age-related lobular involution and risk of breast cancer. J Natl Cancer Inst. 2006;98:1600–1607. doi: 10.1093/jnci/djj439. [DOI] [PubMed] [Google Scholar]
- 169.Shi B, Liang J, Yang X, et al. et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27:5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Holm K, Grabau D, Lövgren K, et al. et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol Oncol. 2012;6:494–506. doi: 10.1016/j.molonc.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang Y, Zhe H, Ding Z, et al. et al. Cancer stem cell marker Bmi-1 expression is associated with basal-like phenotype and poor survival in breast cancer. World J Surg. 2012;36:1189–1194. doi: 10.1007/s00268-012-1514-3. [DOI] [PubMed] [Google Scholar]
- 172.Guo BH, Feng Y, Zhang R, et al. et al. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011;10:10. doi: 10.1186/1476-4598-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu S, Dontu G, Mantle ID, et al. et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Silva J, Garcia JM, Pea C, et al. et al. Implication of polycomb members Bmi-1, Mel-18, and Hpc-2 in the regulation of p16INK4a, p14ARF, h-TERT, and c-Myc expression in primary breast carcinomas. Clin Cancer Res. 2006;12:6929–6936. doi: 10.1158/1078-0432.CCR-06-0788. [DOI] [PubMed] [Google Scholar]
- 175.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 176.Reynolds PA, Sigaroudinia M, Zardo G, et al. et al. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281:24790–24802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- 177.Riis ML, Lüders T, Nesbakken AJ, et al. et al. Expression of BMI-1 and Mel-18 in breast tissue—a diagnostic marker in patients with breast cancer. BMC Cancer. 2010;10:686. doi: 10.1186/1471-2407-10-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Guo BH, Zhang X, Zhang HZ, et al. et al. Low expression of Mel-18 predicts poor prognosis in patients with breast cancer. Ann Oncol. 2010;21:2361–2369. doi: 10.1093/annonc/mdq241. [DOI] [PubMed] [Google Scholar]
- 179.Guo WJ, Zeng MS, Yadav A, et al. et al. Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and downregulating Akt activity in breast cancer cells. Cancer Res. 2007;67:5083–5089. doi: 10.1158/0008-5472.CAN-06-4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee JY, Jang KS, Shin DH, et al. et al. Mel-18 negatively regulates INK4a/ARF-independent cell cycle progression via Akt inactivation in breast cancer. Cancer Res. 2008;68:4201–4209. doi: 10.1158/0008-5472.CAN-07-2570. [DOI] [PubMed] [Google Scholar]
- 181.Maertens GN, El Messaoudi-Aubert S, Racek T, et al. et al. Several distinct polycomb complexes regulate and co-localize on the INK4a tumor suppressor locus. PLoS ONE. 2009;4:e6380. doi: 10.1371/journal.pone.0006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lee TI, Jenner RG, Boyer LA, et al. et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jacobs JJ, van Lohuizen M. Polycomb repression: from cellular memory to cellular proliferation and cancer. Biochim Biophys Acta. 2002;1602:151–161. doi: 10.1016/s0304-419x(02)00052-5. [DOI] [PubMed] [Google Scholar]
- 184.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspec-tives on current status and future directions: CR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 185.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 187.Lewis MT. Homeobox genes in mammary gland development and neoplasia. Breast Cancer Res. 2000;2:158–169. doi: 10.1186/bcr49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cantile M, Pettinato G, Procino A, et al. et al. In vivo expression of the whole HOX gene network in human breast cancer. Eur J Cancer. 2003;39:257–264. doi: 10.1016/s0959-8049(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 189.Henderson GS, van Diest PJ, Burger H, et al. et al. Expression pattern of a homeotic gene, HOXA5, in normal breast and in breast tumors. Cell Oncol. 2006;28:305–313. doi: 10.1155/2006/974810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Raman V, Martensen SA, Reisman D, et al. et al. Compromised HOXA5 function can limit p53 expression in human breast tumors. Nature. 2000;405:974–979. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- 191.Raman V, Tamori A, Vali M, et al. et al. HOXA5 regulates expression of the progesterone receptor. J Biol Chem. 2000;275:26551–26555. doi: 10.1074/jbc.C000324200. [DOI] [PubMed] [Google Scholar]
- 192.Tommasi S, Karm DL, Wu X, et al. et al. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res. 2009;11:R14. doi: 10.1186/bcr2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Daftary GS, Taylor HS. Endocrine regulation of HOX genes. Endocr Rev. 2006;27:331–355. doi: 10.1210/er.2005-0018. [DOI] [PubMed] [Google Scholar]
- 194.Ansari KI, Hussain I, Shrestha B, et al. et al. HOXC6 is transcriptionally regulated via coordination of MLL histone methylase and estrogen receptor in an estrogen environment. J Mol Biol. 2011;411:334–349. doi: 10.1016/j.jmb.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ringrose L. Polycomb comes of age: genome-wide profiling of target sites. Curr Opin Cell Biol. 2007;19:290–297. doi: 10.1016/j.ceb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 196.Ansari KI, Kasiri S, Hussain I, et al. et al. MLL histone methylases in estrogen-dependent regulation of HOXC13. FEBS J. 2009;276:7400–7411. doi: 10.1111/j.1742-4658.2009.07453.x. [DOI] [PubMed] [Google Scholar]