

Abstract
Proteolysis by the ubiquitin-proteasome pathway appears to have a complex role in synaptic plasticity, but its various functions remain to be elucidated. Using late phase long-term potentiation (L-LTP) in the hippocampus of the mouse as a model for long-term synaptic plasticity, we previously showed that inhibition of the proteasome enhances induction but blocks maintenance of L-LTP. In this study, we investigated the possible mechanisms by which proteasome inhibition has opposite effects on L-LTP induction and maintenance. Our results show that inhibiting phosphatidyl inositol-3 kinase or blocking the interaction between eukaryotic initiation factors 4E (eIF4E) and 4G (eIF4G) reduces the enhancement of L-LTP induction brought about by proteasome inhibition suggesting interplay between proteolysis and the signaling pathway mediated by mammalian target of rapamycin (mTOR). Also, proteasome inhibition leads to accumulation of translational activators in the mTOR pathway such as eIF4E and eukaryotic elongation factor 1A (eEF1A) early during L-LTP causing increased induction. Furthermore, inhibition of the proteasome causes a buildup of translational repressors, such as polyadenylate-binding protein interacting protein 2 (Paip2) and eukaryotic initiation factor 4E-binding protein 2 (4E-BP2), during late stages of L-LTP contributing to the blockade of L-LTP maintenance. Thus, the proteasome plays a critical role in regulating protein synthesis during L-LTP by tightly controlling translation. Our results provide novel mechanistic insights into the interplay between protein degradation and protein synthesis in long-term synaptic plasticity.
Keywords: activator, late-phase LTP, protein synthesis, proteolysis, repressor, ubiquitin
Introduction
Synaptic plasticity allows the nervous system to store information. Short-term synaptic plasticity and memory depend on changes in neurotransmitter release and alterations in receptors and other pre-existing proteins (Kandel and Schwartz, 1982; Sossin, 2008). Long-term synaptic plasticity and memory, however, require protein synthesis (Byrne et al., 1991; Hernandez and Abel, 2008; Wang and Morris, 2010). In the recent past, several studies have established that protein degradation also plays a role in synaptic plasticity and memory (Hegde, 2010; Fioravante and Byrne, 2011; Jarome and Helmstetter, 2013). The precise roles of protein degradation and how proteolysis relates to protein synthesis are not clear, however. The requirement for protein synthesis has been well studied in a cellular model of long-term synaptic plasticity, late-phase long-term potentiation (L-LTP; Frey et al., 1988; Nguyen et al., 1994). Temporally, L-LTP can be divided into an “induction” phase and a “maintenance” phase. L-LTP induction requires translation of pre-existing mRNAs in dendrites (Kelleher et al., 2004b) while L-LTP maintenance depends on new transcription in the nucleus and on translation of the newly transcribed mRNAs in the cell body (Nguyen et al., 1994).
The protein degradation that is critical for synaptic plasticity occurs primarily through the ubiquitin-proteasome pathway (UPP; Hegde, 2010; Jarome and Helmstetter, 2013). In this pathway, protein substrates are marked by attachment of several ubiquitin molecules and are degraded by a proteolytic complex called the proteasome. Previously, we showed that proteasome inhibition differentially affects L-LTP depending on the site of inhibition within the neuron. Dendritic proteasome inhibition enhances induction of L-LTP whereas nuclear proteasome inhibition blocks maintenance of L-LTP (Dong et al., 2008). Through a series of experiments, we previously obtained evidence indicating that L-LTP induction is enhanced because proteasome inhibition in dendrites stabilizes proteins translated from pre-existing mRNAs and that L-LTP maintenance is blocked by interference with transcription caused by nuclear proteasome inhibition (Dong et al., 2008). Additional experiments revealed that proteasome inhibition also blocks maintenance of L-LTP in isolated dendrites (Dong et al., 2008). Together, these data suggested that proteasome tightly controls translation by modulating both positive and negative regulators of translation.
Local translation of pre-existing mRNAs in dendrites depends on the mammalian target of rapamycin (mTOR) pathway (Steward and Schuman, 2001). Therefore, we hypothesized that the proteasome regulates translation through interaction with the mTOR pathway and tested this idea by inhibiting different parts of the pathway. We found that inhibiting components of the mTOR pathway greatly reduced the enhancement of L-LTP induction caused by the proteasome inhibitor β-lactone. We then focused on four mTOR pathway components: eukaryotic initiation factor 4E (eIF4E; Gingras et al., 1999) and eukaryotic elongation factor 1A (eEF1A; Negrutskii and El'skaya, 1998), two translational activators; and eukaryotic initiation factor 4E-binding protein 2 (4E-BP2; Pause et al., 1994) and polyadenylate-binding protein interacting protein 2 (Paip2; Khaleghpour et al., 2001), two translational repressors. Here, we show that early during L-LTP, proteasome inhibition stabilizes the translational activators eIF4E and eEF1A and that at late stages of L-LTP, proteasome inhibition stabilizes the translational repressors 4E-BP2 and Paip2.
Materials and Methods
Animals.
Mice were obtained from Harlan Laboratories and used for experiments using a protocol approved by the Institutional Animal Care and Use Committee of Wake Forest University Health Sciences.
Extracellular recording.
Using a tissue chopper, transverse hippocampal sections (400 μm) from 6- to 12-week-old C57BL/6 male mice were prepared in an oxygenated and chilled artificial CSF (ACSF) containing 125 mm NaCl, 3 mm KCl, 2.3 mm CaCl2, 1.3 mm MgCl2, 25 mm NaHCO3, 1.25 mm NaH2PO4, and 10 mm glucose, pH 7.4. The slices were submerged at 32°C and superfused with ACSF (1.5 ml/min). All recordings were obtained from slices maintained at 32°C. For recording of field EPSP (fEPSP), in the CA1 region of the hippocampus, a bipolar stimulating electrode was used to stimulate the Schaeffer collateral pathway while recording in the stratum radiatum. The stimulation intensity was adjusted to give ∼35% of the maximal fEPSP slope and the baseline responses were recorded at this intensity. L-LTP was induced by giving four 100 Hz trains spaced 5 min apart (Dong et al., 2008). Extent of LTP was expressed as percentage increase relative to baseline in the fEPSP slope at different time points as stated in figure legends. For experiments with the proteasome inhibitor β-lactone (Cayman Chemical), after recovery, the slices were pre-incubated with the reagent in oxygenated ACSF for 30 min. Inhibition of the proteasome by β-lactone is irreversible and therefore the inhibitory effect of the drug cannot be washed out.
The pharmacological reagents rapamycin, LY294002, 4EGI-1, U0126, U0124, and anisomycin (EMD Millipore), were prepared in dimethylsulfoxide (DMSO). For these reagents the same concentration of DMSO alone was used for controls. All these reagents were applied to hippocampal slices before pre-incubation with β-lactone except anisomycin, which was applied after pre-incubation with β-lactone.
L-LTP in isolated dendrites.
We separated the dendrites by means of a surgical cut to the slice as described previously by others (Frey and Morris, 1997; Woo and Nguyen, 2003; Cracco et al., 2005) and us (Dong et al., 2008). We placed the hippocampal slices in ice-cold ACSF containing sucrose instead of NaCl and MgCl2 instead of CaCl2 (replacement chemicals were of same molarity), and made a cut below the cell body layer of CA1 region. The slices were then placed in normal ACSF and allowed to recover for 2 h before the experiment. L-LTP was induced as described previously (Vickers et al., 2005; Dong et al., 2008).
Immunohistochemistry.
After being subjected to an L-LTP-inducing protocol (4 × 100 Hz), free floating hippocampal slices were fixed in 4% paraformaldehyde for 1 h followed by five 30 min washes with PBS at room temperature. Control slices for each L-LTP experimental condition were time matched with respect to incubation in ACSF alone or in ACSF containing β-lactone. After washing, slices were blocked in a solution containing 4% normal goat serum (Vector Laboratories), 0.4% Triton X-100, and 0.05% sodium azide in PBS at 4°C for 5 h. Slices were then incubated in blocking solution containing polyclonal primary antibodies against eIF4E (1:80; Bethyl Laboratories), eEF1A (1:80; Abcam), Paip2 (1:50; Proteintech), or 4E-BP2 (1:50; Abcam) at 4°C overnight. Following primary antibody incubation, slices underwent four 20 min washes in 0.2% Triton X-100 in PBS and were incubated in Alexa 488-conjugated goat anti-rabbit secondary antibody (1:300; Invitrogen) and To-Pro-3 (1:500; Invitrogen) at 4°C for 8 h. Following secondary antibody incubation, slices underwent five 30 min washes in 0.2% Triton X-100 in PBS and were mounted with Prolong Gold antifade reagent (Invitrogen). Images were taken with a Carl Zeiss LSM510 laser scanning confocal microscope and analyzed using ImageJ (National Institutes of Health (NIH), Bethesda, MD).
Metabolic labeling of hippocampal proteins with 35S-methionine.
Coronal hippocampal slices were made using a tissue chopper and allowed to recover in oxygenated ACSF at 32°C. After 1 h of recovery, slices were treated with rapamycin (1 μm) or 4EGI-1 (100 μm) for 1 h before treatment with β-lactone (25 μm) for 30 min. All slices were incubated in 15 μCi/ml 35S-methionine (1175 Ci/mmol; PerkinElmer). Because the effect of 4EGI-1 has shown to be reversible (Moerke et al., 2007; McMahon et al., 2011), slices in the 4EGI-1 groups also had 4EGI-1 added during the 35S-methionine incubation. Slices were collected at 30, 60, 90, and 120 min after initiation of metabolic labeling and immediately frozen on dry ice. Slices were then homogenized with radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 150 mm NaCl, 2 mm EDTA, 50 mm NaF) including 1× protease inhibitor cocktail (EMD Millipore). The homogenate was incubated on ice for 1 h, centrifuged at 15,000 × g for 30 min, and the supernatant was collected and processed for Bicinchoninic Acid (BCA) protein estimation assay (Thermo Scientific). After SDS-PAGE of 50 μg protein from each sample, the gels were stained with Coomassie blue stain as a loading control measure. After destaining, gels were incubated for 1 h in En3Hance liquid autoradiography enhancer (PerkinElmer) and then rinsed in ice-cold water for 30 min, dried, and exposed to Kodak BioMax Light film. The autoradiographic images were taken using Gel Doc (Bio-Rad), and optical density was quantified using ImageJ (NIH, Bethesda, MD). Background was subtracted from each measurement, and protein loading differences were controlled for using the optical density from Coomassie blue gel stain. Experimental groups for each time point were normalized to their time-matched control.
Data analysis.
Data were analyzed by using one-way ANOVA followed by a post hoc Tukey test or Student–Newman–Keuls test (pairwise multiple-comparison procedure) as appropriate. In some cases, when the data were not normally distributed, we used Kruskal–Wallis one-way ANOVA on ranks because under such circumstances, this is the most suitable alternative to classical ANOVA and provides higher statistical power compared with ANOVA (Lantz, 2013). For comparison between two groups, Student's t test was used. The values are expressed as mean ± SE. The sample size (n) in each dataset corresponds to the number of animals (not slices) used to collect the data.
Results
Rapamycin pretreatment significantly reduces the β-lactone-mediated increase in the early part of L-LTP in isolated dendrites
Previously we showed that the enhancement in the early part of L-LTP (henceforth referred to as Ep-L-LTP), which encompasses the induction phase, brought about by proteasome inhibition is blocked by prior incubation with rapamycin in intact slices (Dong et al., 2008). We also showed that proteasome inhibition enhances Ep-L-LTP in isolated dendrites in the same way as it does in the intact hippocampal slices (Dong et al., 2008). These data provided indirect evidence that proteasome inhibition enhances Ep-L-LTP by stabilizing the locally translated proteins from the pre-existing mRNA in dendrites via the mTOR pathway. Before commencing to test this idea rigorously, it was important to obtain direct evidence that rapamycin inhibits the enhancement of Ep-L-LTP upon pre-incubation with β-lactone in isolated dendrites. To do this, we placed a surgical cut between the pyramidal cell bodies and dendrites in the stratum radiatum (Fig. 1A) and induced L-LTP after treatment with β-lactone alone or after incubation with rapamycin followed by treatment with β-lactone. We found that rapamycin pretreatment significantly reduces the extent of Ep-L-LTP in isolated dendrites (at 30 min: control, 193 ± 20%, n = 7; rapamycin, 148 ± 9%, n = 8; p < 0.01, t test; Fig. 1B).
Figure 1.

Rapamycin pretreatment significantly reduces β-lactone-mediated enhancement in Ep-L-LTP in isolated dendrites. A, Schematic illustration of surgical isolation of dendrites (modified from Dong et al., 2008). A cut (black bar) is made just below the pyramidal cell body layer in the CA1 region. Recording electrode (Rec) is placed in the dendritic layer of pyramidal cells. Position of the stimulation electrode (Stim), and the layers stratum radiatum and stratum oriens are also indicated. DG, dentate gyrus. B, Treatment of cut slices with rapamycin before β-lactone treatment significantly (p < 0.01) diminishes the magnitude of Ep-L-LTP relative to β-lactone treatment alone. Inset, Representative traces taken at different time points (1 = 30 min; 2 = 180 min) for treatment with β-lactone alone and rapamycin + β-lactone.
These data suggest that in both intact slices and isolated dendrites, proteasome inhibition stabilizes the proteins whose translation is regulated by the mTOR pathway. Therefore, in the rest of this study we performed a series of experiments to systematically test the role of the mTOR pathway in enhancement of Ep-L-LTP observed with proteasome inhibition.
Phosphatidyl inositol-3 kinase inhibition markedly lowers β-lactone-mediated increase in Ep-L-LTP
In the mTOR pathway, typically an extracellular ligand such as the insulin growth factor binding to its receptor activates phosphatidyl inositol-3 kinase (PI3K), which in turn activates mTOR complex 1 ultimately leading to activation of a translation initiation factor complex containing eIF4E (Hay and Sonenberg, 2004; Zoncu et al., 2011).
To test the role of PI3K, we first characterized the effect of PI3K inhibitor LY294002 on L-LTP. Previous work by others has determined that LY294002 reduces the protein synthesis-independent early LTP (Opazo et al., 2003). Its effect on L-LTP has not been studied, however. Therefore, we first tested the effect of LY294002 on L-LTP. Application of LY294002 (20 μm) before tetanization significantly reduced the amplitude of L-LTP (at 30 min: control, 178 ± 13%, n = 6; LY294002, 131 ± 10%, n = 8; p < 0.0001; at 180 min: control, 138 ± 5%, n = 6; LY294002, 103 ± 5%, n = 8; p < 0.0001, t test; Fig. 2A). We found that pretreatment with LY294002, however, does not affect basal synaptic transmission (Fig. 2B).
Figure 2.

PI3K inhibition reduces normal L-LTP and β-lactone-mediated enhancement of Ep-L-LTP. A, Pretreatment with PI3K inhibitor LY294002 significantly (p < 0.0001) reduces L-LTP. Inset, Representative traces taken at different time points (1 = 30 min; 2 = 180 min) for no treatment “Control” and treatment with LY294002. B, Normal basal synaptic transmission in LY294002-treated hippocampal slices (n = 7) compared with vehicle-treated control (n = 7) slices. The graph shows input–output curves of fEPSP slope (mV/ms) versus stimulus at the Schaffer collateral pathway upon treatment with the vehicle (DMSO) or LY294002. C, Incubation with LY294002 before β-lactone application significantly (p < 0.0001) decreases the enhancement in Ep-L-LTP. Inset, Representative traces taken at different time points (1 = 30 min; 2 = 180 min) for β-lactone alone and LY294002 + β-lactone.
Next, we tested the effect of LY294002 on the β-lactone-mediated increase in Ep-L-LTP. We pre-incubated the slices with LY294002 before applying β-lactone (25 μm). We found that LY294002 significantly reduced the effect of β-lactone on Ep-L-LTP (at 30 min: β-lactone, 199 ± 11%, n = 8; LY294002 + β-lactone, 121 ± 6%, n = 8; p < 0.0001, t test; Fig. 2C).
Disruption of eIF4E-eIF4G interaction greatly decreases the β-lactone-mediated enhancement in Ep-L-LTP
Our previous results provided several lines of evidence that the β-lactone-mediated enhancement in Ep-L-LTP occurs because of stabilization of proteins locally translated from pre-existing mRNAs in dendrites. In addition, the experiments described above showed that rapamycin reduces the enhancement in Ep-L-LTP by proteasome inhibition in isolated dendrites. These results suggest that the mTOR pathway plays a role in β-lactone-mediated augmentation of Ep-L-LTP. To test this hypothesis rigorously, we examined the role of some of the key components of the mTOR pathway downstream of PI3K (Hay and Sonenberg, 2004).
Eukaryotic mRNAs have a special nucleotide, 7-methyl guanosine triphosphate, attached to their 5′ end which is referred to as the cap (Pestova et al., 2007). The process of translational initiation of mRNAs with a cap begins with binding of an initiation factor complex in which interaction between eIF4E and eIF4G plays a critical role (Gingras et al., 1999). Therefore, disrupting this interaction should reduce translation and thus decrease proteasome inhibition-mediated enhancement of Ep-L-LTP. To test this hypothesis, we used a small molecule called 4EGI-1 that inhibits eIF4E-eIF4G interaction (Moerke et al., 2007). Since the effect of 4EGI-1 on L-LTP has not been characterized, we tested its effect on L-LTP. We found that 4EGI-1 (100 μm) significantly inhibits L-LTP (at 30 min: control, 168 ± 7%, n = 8; 4EGI-1, 129 ± 6%, n = 9, p < 0.0001; at 180 min: control, 135 ± 9%, n = 8; 4EGI-1, 114 ± 5%, n = 9; p < 0.0001, t test; Fig. 3A). Additionally, we found that 4EGI-1 does not have any effect on basal synaptic transmission (Fig. 3B).
Figure 3.

An inhibitor of eIF4E-eIF4G interaction decreases normal L-LTP and β-lactone-mediated enhancement of Ep-L-LTP. A, Pretreatment with 4EGI-1, an inhibitor of eIF4E-eIF4G interaction, significantly (p < 0.0001) reduces L-LTP. Inset, Representative traces taken at different time points (1 = 30 min; 2 = 180 min) for no treatment “Control” and treatment with 4-EGI-1. B, Normal basal synaptic transmission in 4EGI-1-treated hippocampal slices (n = 8) compared with vehicle-treated control (n = 7) slices. The graph shows input–output curves of fEPSP slope (mV/ms) versus stimulus at the Schaffer collateral pathway upon treatment with the vehicle (DMSO) or 4EGI-1. C, Incubation with 4EGI-1 before β-lactone significantly (p < 0.0001) decreases the enhancement in Ep-L-LTP. Inset, Representative traces taken at different time points (1 = 30 min; 2 = 180 min) for β-lactone alone and 4EGI-1 + β-lactone.
Next we tested the effect of 4EGI-1 on β-lactone-mediated increase in Ep-L-LTP. We pre-incubated the slices with 4EGI-1 before applying β-lactone. We observed that 4EGI-1 significantly reduced the effect of β-lactone on Ep-L-LTP (at 30 min: β-lactone, 192 ± 19%, n = 8; 4-EGI-1 + β-lactone, 127 ± 10%, n = 10, p < 0.0001, t test; Fig. 3C).
Extracellular signal-regulated kinase inhibition significantly decreases β-lactone-mediated enhancement in Ep-L-LTP
Extracellular signal-regulated kinase (ERK; also called mitogen-activated protein kinase, MAP kinase) plays a role in regulating translation induced by neuronal activity and phosphorylation of some translation factors including eIF4E (Kelleher et al., 2004a). In forskolin-induced L-LTP, upregulation of translation of mRNAs that contain 5′ oligopyrimidine tracts depends on mTOR, ERK, and PI3K (Gobert et al., 2008). Furthermore, ERK upregulates the components of the translation machinery in dendrites, such as eEF1A, that are regulated by the mTOR pathway (Tsokas et al., 2007). These observations suggest cross talk between ERK and the mTOR pathway. Therefore we sought to test whether ERK plays a role in the increase in Ep-L-LTP brought about by proteasome inhibition.
To test whether ERK plays a role in β-lactone-mediated enhancement of Ep-L-LTP, we pre-incubated the slices with U0126 (20 μm), an inhibitor of MEK, a kinase that activates ERK. As control, we used an inactive analog U0124 (20 μm). We then treated the slices with β-lactone. We found that U0126 (but not the inactive analog U0124) significantly inhibits the β-lactone-mediated increase in Ep-L-LTP (at 30 min: U0126 + β-lactone, 139 ± 15%, n = 8; U0124 + β-lactone, 193 ± 15%; n = 6; p < 0.001, t test; Fig. 4A).
Figure 4.

ERK function is essential for β-lactone-mediated enhancement of Ep-L-LTP in intact slices as well as isolated dendrites. A, In intact slices, pre-incubation with MEK inhibitor U0126 before β-lactone treatment (but not with an inactive analog U0124) significantly (p < 0.001) reduces Ep-L-LTP. Individual traces taken at 30 min are shown in the inset. B, In isolated dendrites, pre-incubation with MEK inhibitor U0126 before β-lactone treatment (but not with an inactive analog U0124) significantly (p < 0.001) reduces Ep-L-LTP. Individual traces taken at 30 min are shown in the inset.
Since the induction of L-LTP depends on protein translation in dendrites, it was important to test whether ERK function is critical for enhancement of Ep-L-LTP in dendrites caused by proteasome inhibition. Therefore, we tested the effect of U0126 and the inactive analog U0124 on hippocampal slices surgically cut to isolate dendrites (Fig. 1A) before treating the slices with β-lactone. We observed that U0126 (but not U0124) significantly inhibits the β-lactone-mediated increase in Ep-L-LTP in isolated dendrites (at 30 min: U0126 + β-lactone, 151 ± 10%, n = 9; U0124 + β-lactone, 180 ± 13%, n = 6; p < 0.001, t test; Fig. 4B).
Blockade of protein synthesis after proteasome inhibition in isolated dendrites prevents the decline in L-LTP maintenance brought about by proteasome inhibition alone
The increase in Ep-L-LTP mediated by β-lactone is not maintained, and by ∼150 min after tetanization it starts to decline below L-LTP levels observed in untreated control slices. Previously we showed that in intact slices this decline is likely to occur because proteasome inhibition blocks the transcription required for L-LTP maintenance (Dong et al., 2008). We did, however, observe the decline in L-LTP with β-lactone even in isolated dendrites. What causes the decline in L-LTP maintenance in isolated dendrites? We hypothesized that initially translation activators are stabilized by β-lactone and in later stages, translation repressors are stabilized. We postulated that the synthesis of these repressors occurs at basal level (i.e., they are not translationally regulated) because earlier biochemical evidence suggested that translational activators are regulated by titratable repressors (Meyuhas, 2000). Therefore, inhibition of their degradation causes their buildup, leading to inhibition of translation at later stages; thus causing blockade of L-LTP maintenance. A corollary of this hypothesis is that, in the absence of the repressor buildup, the initial stabilization of translational activators by proteasome inhibition should be adequate for L-LTP maintenance. If these ideas are correct, then blocking protein synthesis after application of β-lactone to slices should prevent the decline in L-LTP maintenance. To test this hypothesis, we treated the surgically cut hippocampal slices (Fig. 1A) with β-lactone. We then incubated the slices with anisomycin (25 μm) for 1 h after which we induced L-LTP. After inducing L-LTP, we continued anisomycin application through the superfusion fluid for an additional hour (Fig. 5A). We found that application of anisomycin after incubation with β-lactone reversed the decline in L-LTP maintenance in isolated dendrites (at 180 min: β-lactone, 103 ± 4%; β-lactone + anisomycin, 128 ± 12%; control, 131 ± 15%; F(2,16) = 17.578, p = 0.001, one-way ANOVA; β-lactone vs Control: p = 0.004; β-lactone vs β-lactone + anisomycin: p < 0.001; β-lactone + anisomycin vs Control: p = 0.168; post hoc Tukey test; Fig. 5B).
Figure 5.

Reversal of proteasome inhibition-mediated decrease in L-LTP maintenance in isolated dendrites by subsequent inhibition of protein synthesis. A, Schematic outline of the experiment: hippocampal slices were pre-incubated with β-lactone for 30 min and then they were incubated with anisomycin for 60 min. Anisomycin was also added to the ACSF used for superfusion and maintained for 60 min after the first tetanus. B, Treatment with β-lactone alone (open diamonds) blocks L-LTP maintenance in isolated dendrites relative to L-LTP without any treatment, i.e., control (open circles). Application of anisomycin after β-lactone treatment (filled triangles) reverses blockade of L-LTP maintenance and restores maintenance to normal levels seen in L-LTP without any treatment, i.e., control (β-lactone vs β-lactone + anisomycin: p < 0.001; β-lactone + anisomycin vs Control: p = 0.168). Inset, Representative traces taken at different time points (1 = baseline, 2 = 30 min; 3 = 180 min) for β-lactone, β-lactone + anisomycin, and control.
Expression of translational activators peaks early and that of repressors peaks later during L-LTP
To test our hypothesis at the molecular level, we examined the quantities of two positive translational regulators eIF4E and eEF1A, and two negative translational regulators Paip2 and 4E-BP2 in the mTOR pathway. In non-neuronal cell types these proteins are known to be degraded by the UPP (Murata and Shimotohno, 2006; Yoshida et al., 2006; Koiwai et al., 2008; Yanagiya et al., 2012). We chose 4E-BP2 because in hippocampal neurons 4E-BP1 expression is negligible and 4E-BP2 is the predominant isoform (Banko et al., 2005).
We first determined the time course of expression of these proteins. For eIF4E and eEF1A, we induced L-LTP with the 4-train protocol (4 × 100 Hz) and fixed the slices 30, 45, 90, and 120 min after tetanization. We then performed immunohistochemistry with anti-eIF4E and anti-eEF1A antibodies and visualized the immunoreactivity by using confocal laser scanning microscopy. Both eIF4E and eEF1A reached their peak level of expression at 45 min (eIF4E [all time points]: H(4) = 17.996, p = 0.001, n = 6, Kruskal–Wallis one-way ANOVA on ranks; eIF4E [at 45 min]: 562 ± 145%, p < 0.05 compared with its time-matched control and other time points, Student–Newman–Keuls post hoc test; eEF1A [all time points]: H(4) = 15.116, p = 0.004, n = 6, Kruskal–Wallis 0ne-way ANOVA on ranks; eEF1A [at 45 min]: 341 ± 93%, p < 0.05 compared with its time-matched control and other time points, Student–Newman–Keuls post hoc test; Fig. 6).
Figure 6.
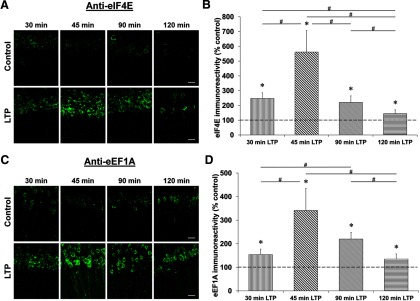
Time course of eIF4E and eEF1A expression after L-LTP induction. Confocal images of eIF4E and eEF1A immunoreactivities in the CA1 region of hippocampal slices 30, 45, 90, and 120 min after the first tetanus and their respective time-matched controls (A and C). Scale bars: 20 μm. Quantification of eIF4E and eEF1A immunoreactivities relative to their time-matched controls shows a peak at 45 min for both proteins (B and D). *p < 0.05 compared with controls (depicted by a dashed line). #p < 0.05 comparison between two given time points as indicated by horizontal lines.
For Paip2 and 4E-BP2, we induced L-LTP with the 4-train protocol and fixed the slices 30, 60, 90, and 120 min after tetanization. We then performed immunohistochemistry with anti-Paip2 and anti-4E-BP2 antibodies and visualized the immunoreactivity by using confocal microscopy. Both Paip2 and 4E-BP2 reached their peak level of expression at 90 min (Paip2 [all time points]: H(4) = 23.672, p = 0.001, n = 6, Kruskal–Wallis one-way ANOVA on ranks; Paip2 [at 90 min]: 323 ± 33%, p < 0.05 compared with its time-matched control and other time points, Student–Newman–Keuls post hoc test; 4E-BP2 [all time points]: H(4) = 13.468, p = 0.009, n = 6, Kruskal–Wallis one-way ANOVA on ranks; 4E-BP2 [at 90 min]: 263 ± 35%, p < 0.05 compared with its time-matched control and other time points, Student–Newman–Keuls post hoc test; Fig. 7).
Figure 7.

Time course of Paip2 and 4E-BP2 expression after L-LTP induction. Confocal images of Paip2 and 4E-BP2 immunoreactivities in the CA1 region of hippocampal slices 30, 60, 90, and 120 min after the first tetanus and their respective time-matched controls (A and C). Scale bars: 20 μm. Quantification of Paip2 and 4E-BP2 immunoreactivities relative to their time-matched controls shows a peak at 90 min for both proteins (B and D). *p < 0.05 compared with controls (depicted by a dashed line). #p < 0.05 comparison between two given time points as indicated by horizontal lines.
Proteasome inhibition augments the quantities of translational activators and repressors
The time course results described above support the idea that early during L-LTP translational activators increase, and at later stages the translational repressors accumulate. If proteasome inhibition were to increase the quantities of these activators and repressors, then the enhancement of Ep-L-LTP and the blockade of L-LTP maintenance by β-lactone could be explained in molecular terms. Therefore, we first tested whether β-lactone treatment enhanced the amount of eIF4E and eEF1A during L-LTP. Since it would be difficult to reliably measure an increase over and above the peak expression level of these proteins, we chose a time point preceding the time when the expression of these proteins peaked during L-LTP. Given that the amounts of eIF4E and eEF1A peak at 45 min, we tested the effect of β-lactone at 30 min. Although β-lactone treatment caused an increase in eEF1A and eIF4E in control slices, in slices with L-LTP induction after incubation with β-lactone, the quantity of these proteins was significantly higher than treatment with β-lactone alone or L-LTP alone. (eIF4E: with L-LTP induction, 257 ± 32%; β-lactone alone, 314 ± 42%; with L-LTP induction after β-lactone, 478 ± 78% relative to time-matched control set at 100%; F(3,24) = 12.680, p = 0.001, n = 7, one-way ANOVA; LTP vs β-lactone + LTP, p < 0.01; β-lactone vs β-lactone + LTP, p < 0.05, Student–Newman–Keuls post hoc test; eEF1A: with L-LTP induction: 152 ± 19%; β-lactone alone, 253 ± 13%; with L-LTP induction after β-lactone, 363 ± 36% relative to time-matched control set at 100%; F(3,20) = 33.168, p = 0.001, n = 6; one-way ANOVA; LTP vs β-lactone + LTP, p < 0.001; β-lactone vs β-lactone + LTP, p < 0.001; Student–Newman–Keuls post hoc test; Fig. 8).
Figure 8.

Proteasome inhibition causes accumulation of eIF4E and eEF1A early during L-LTP. Confocal images showing eIF4E (A) and eEF1A (C) immunoreactivities in the CA1 region of hippocampal slices: untreated (control), subjected to L-LTP induction, treated with β-lactone, and subjected to L-LTP induction after β-lactone treatment. L-LTP slices were fixed 30 min after the initial tetanus and the “Control” and “β-lactone” slices were time-matched to their respective L-LTP counterparts. Scale bars: 20 μm. Quantification of eIF4E (B) and eEF1A (D) immunoreactivities for the four experimental conditions is shown at right. *p < 0.05 and **p < 0.01 compared with controls (depicted by a dashed line). #p < 0.05 and ##p < 0.01 comparison between two given treatments as indicated by horizontal lines.
Next, we investigated the effect of β-lactone on Paip2 and 4E-BP2 at 60 min because these proteins reach their peak expression level at 90 min. While β-lactone treatment increased the amount of Paip2 and 4E-BP2 in control slices, in slices with L-LTP induction after incubation with β-lactone the quantity of these proteins was significantly higher than treatment with β-lactone alone or L-LTP alone (Paip2: with L-LTP induction, 232 ± 23%; β-lactone alone, 193 ± 30%; with L-LTP induction after β-lactone, 345 ± 45% relative to time-matched control set at 100%; F(3,20) = 12.008, p < 0.001, n = 6, one-way ANOVA; LTP vs β-lactone + LTP, p < 0.05; β-lactone vs β-lactone + LTP, p < 0.01, Student–Newman–Keuls post hoc test; 4E-BP2: with L-LTP induction, 207 ± 21%; β-lactone alone, 242 ± 30%; with L-LTP induction after β-lactone, 467 ± 85% relative to time-matched control set at 100%; H(3) = 17.567, p < 0.001, n = 6, Kruskal–Wallis one-way ANOVA on ranks; LTP vs β-lactone + LTP, p < 0.05, n = 6; β-lactone vs β-lactone + LTP, p < 0.05, n = 6; Student–Newman–Keuls post hoc test; Fig. 9).
Figure 9.
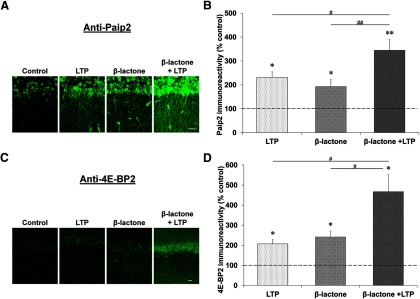
Proteasome inhibition causes accumulation of Paip2 and 4E-BP2 at late stages during L-LTP. Confocal images showing Paip2 (A) and 4E-BP2 (C) immunoreactivities in CA1 region of hippocampal slices: untreated (control), subjected to L-LTP induction, treated with β-lactone, and subjected to L-LTP induction after β-lactone treatment. L-LTP slices were fixed 60 min after the initial tetanus and the “Control” and “β-lactone” slices were time-matched to their respective L-LTP counterparts. Scale bars: 20 μm. Quantification of Paip2 (B) and 4E-BP2 (D) immunoreactivities for the four experimental conditions is shown at right. *p < 0.05 and **p < 0.01 compared with controls (depicted by a dashed line). #p < 0.05 and ##p < 0.01 comparison between two given treatments as indicated by horizontal lines.
Inhibition of mTOR pathway components blocks β-lactone-mediated increase in newly synthesized proteins
Our electrophysiological data indicate that proteasome inhibition by β-lactone has its effects on L-LTP through modulation of the components of the mTOR pathway. To directly test the connection between the β-lactone effect and translation mediated by the mTOR pathway in the hippocampus, we measured the synthesis of new proteins by monitoring 35S-methionine incorporation over time. First, we measured the effect of β-lactone and that of rapamycin. We found that β-lactone causes an increase in 35S-methionine-labeled proteins and pre-incubation with rapamycin blocks this increase at all time points (for example, at 60 min; β-lactone, 158 ± 35%; rapamycin + β-lactone, 96 ± 15%; rapamycin alone, 76 ± 32% relative to control set at 100%; H(3) = 15.299, p < 0.01, n = 6, Kruskal–Wallis one-way ANOVA on ranks; Control vs β-lactone, p < 0.05, n = 6; β-lactone vs rapamycin + β-lactone, p < 0.05, n = 6; Student–Newman–Keuls post hoc test; Fig. 10A,B).
Figure 10.

β-Lactone enhances the quantity of newly translated proteins in hippocampal slices and the enhancement is blocked by rapamycin and 4EGI-1. Autoradiographs (A and C) showing incorporation of 35S-methionine at 30, 60, 90, and 120 min after initiation of metabolic labeling indicating the amount of newly synthesized proteins when the hippocampal slices are treated with β-lactone by itself or after prior treatment with rapamycin (A) or 4EGI-1 (C). Quantification shows that rapamycin (B) and 4EGI-1 (D) block the β-lactone-mediated increase in 35S-methionine-labeled proteins (lanes 2 compared with lanes 3 in A and C and second bars compared with first bars in B and D). Effect of rapamycin alone or 4EGI-1 alone on control slices (lanes 4 in A and C and third bars in B and D) is also shown. *p < 0.05 compared with controls (depicted by a dashed line). #p < 0.05 comparison between two given experimental conditions as indicated by horizontal lines.
Next we performed a time course experiment to test the effect of 4EGI-1, which inhibits interaction between eIF4E and eIF4G. We observed that pre-incubation with 4EGI-1 prevents the increase in the amounts of 35S-methionine-labeled proteins caused by β-lactone at all time points (for example, at 60 min: β-lactone, 157 ± 63%; 4EGI-1+ β-lactone, 53 ± 27%; 4EGI-1 alone, 59 ± 25%; relative to control set at 100%; H(3) = 19.747, p < 0.001, n = 6, Kruskal–Wallis one-way ANOVA on ranks; Control vs β-lactone, p < 0.05, n = 6; β-lactone vs 4EGI-1+ β-lactone, p < 0.05, n = 6; Student–Newman–Keuls post hoc test; Fig. 10 C,D).
Discussion
The role of the UPP in synaptic plasticity, although now generally accepted, is still not clear with respect to mechanistic knowledge. Through a series of electrophysiological experiments, we previously showed disparate roles for proteasome-mediated degradation in dendrites and the nucleus. We found that in dendrites the proteasome limits synaptic plasticity, and therefore proteasome inhibition enhances Ep-L-LTP (which includes the L-LTP induction phase), which depends on local protein translation from pre-existing mRNAs in dendrites. Earlier, we also obtained molecular evidence for an opposite role for the proteasome in the nucleus in facilitating transcription (Dong et al., 2008). Nuclear proteasome inhibition, therefore, blocks L-LTP maintenance by hindering the new gene transcription required for this process. In this study, we provide evidence for an important new role for the proteasome in control of the translation required for L-LTP through modulation of positive as well as negative translational regulators.
It must be noted that although others have investigated the effect of proteasome inhibition on synaptic plasticity, they failed to discover the enhancing effect of proteasome inhibition on what we call Ep-L-LTP. This is most likely because one study used nanomolar concentration of lactacystin (precursor of β-lactone; Fonseca et al., 2006) even though the effective concentration is in the micromolar range (Fenteany et al., 1995; Dick et al., 1996). We previously systematically tested dose responses of β-lactone and found that nanomolar concentrations did not affect either induction or maintenance of L-LTP but micromolar concentrations did (Dong et al., 2008). A second study used MG-132 (Karpova et al., 2006), which is not a highly specific proteasome inhibitor and is known to effectively inhibit other proteases such as calpain and cathepsin B (Chain et al., 1999; Tang and Leppla, 1999). These issues were discussed in-depth in our previous publication (Dong et al., 2008). Our current study provides new molecular evidence to support our earlier electrophysiological results and begins to provide a mechanistic explanation for the role of the proteasome in Ep-L-LTP as well as L-LTP maintenance.
Proteasome inhibition-mediated enhancement of Ep-L-LTP depends on the mTOR pathway and ERK function
Previously, we had an indication that the mTOR pathway might be critical for enhancement of Ep-L-LTP because it was inhibited by rapamycin before treatment with β-lactone (Dong et al., 2008). The results of the present systematic study support a role for the mTOR pathway in mediating Ep-L-LTP increase upon β-lactone treatment. A key signaling molecule that lies upstream of mTOR is PI3K. Our results showed that LY294002, an inhibitor of PI3K, significantly reduced the increase in Ep-L-LTP brought about by β-lactone. Moreover, a molecule that hinders the interaction between eIF4E and eIF4G, 4EGI-1, greatly reduced the enhancement in Ep-L-LTP caused by proteasome inhibition. Also, our 35S-methionine-labeling experiments showed that 4EGI-1 blocks the β-lactone-mediated increase in newly synthesized proteins. Among the translational activators, eIF4G is thought to be a master effector of the mTOR pathway (Thoreen et al., 2012). Thus, our data support the idea that the proteasome limits translation through the mTOR pathway, and therefore, proteasome inhibition enhances mTOR-mediated translation leading to an increase in Ep-L-LTP.
Our experiments also showed that ERK is crucial for Ep-L-LTP enhancement mediated by proteasome inhibition. There are conflicting reports on the link between the mTOR pathway and ERK during LTP. A study showed that the kinase upstream of mTOR, PI3K, regulates induction of LTP through an ERK-independent mechanism (Opazo et al., 2003). This study, however, did not examine L-LTP. Hence, it is likely that protein synthesis-independent early LTP uses ERK through a different signaling pathway compared with L-LTP. In support of this idea, another study on L-LTP induced by forskolin also found a role for ERK, mTOR, and PI3K (Gobert et al., 2008). We previously showed that proteasome inhibition-mediated Ep-L-LTP enhancement depends on cAMP-dependent protein kinase (PKA) (Dong et al., 2008). Therefore, the signaling pathways that are in play in forskolin-induced L-LTP (which works through activation of PKA) are perhaps similar to the one in the electrical L-LTP paradigm used by us and many others.
Enhancement of Ep-L-LTP by proteasome inhibition likely depends on stabilization of translational activators
Previously we showed that incubation of hippocampal slices with anisomycin before treatment with β-lactone blocks the proteasome inhibition-mediated enhancement in Ep-L-LTP (Dong et al., 2008). Proteasome inhibition likely stabilizes proteins that have a positive influence on L-LTP. Therefore, we hypothesized that the proteins that promote translation might be among these positive regulators of L-LTP. We chose to test our hypothesis by studying eIF4E and eEF1A, both of which are known to be substrates for the UPP in non-neuronal cell types (Murata and Shimotohno, 2006; Koiwai et al., 2008). Our time course studies on the expression of eIF4E and eEF1A showed that the peak induction time (45 min) approximately coincides with the peak of L-LTP induction. We observed increased expression of these proteins both in the cell body and dendritic layers. Proteasome inhibition augments the levels of eIF4E and eEF1A. This likely leads to enhanced translation of other positive regulators of L-LTP, thus leading to an increase in Ep-L-LTP.
Blockade of L-LTP maintenance in dendrites by proteasome inhibition is likely due to stabilization of translational repressors
In isolated dendrites, Ep-L-LTP is enhanced but is not sustained. Since application of anisomycin after treatment of the slices with β-lactone restores normal levels of L-LTP, we reasoned that the decay in L-LTP in isolated dendrites occurs because of slow buildup of translational repressors. Our immunohistochemical experiments on the time course of Paip2 and 4E-BP2 expression support this notion. The peak amount of these negative translational regulators is seen at 90 min, which is before the decay of L-LTP. Application of β-lactone further increases the quantity of these repressors. Therefore, proteasome inhibition-mediated decay in L-LTP maintenance in dendrites is likely due to increased activity of translational repressors such as Paip2 and 4E-BP2.
Possible roles of protein degradation in regulating translation critical for L-LTP induction and maintenance
Based on our earlier results and the present data, it appears that protein degradation by the UPP limits induction of L-LTP (and Ep-L-LTP) by degrading the translational activators such as eIF4E and eEF1A. These activators, as well as the positive regulators of synaptic plasticity whose synthesis these activators stimulate, are likely to be rapidly synthesized and degraded. Hence the new protein synthesis requirement for L-LTP induction reflects the need to overcome degradation to provide a net increase in these proteins. This notion is supported by the results showing that when we applied anisomycin after β-lactone, L-LTP was still induced. We postulate that this seeming protein synthesis independence of L-LTP was due to the fact that the rapidly turning-over proteins, which otherwise would be degraded were now stabilized. This idea is supported by our 35S-methionine-labeling experiments, which show that β-lactone increases the quantity of newly synthesized proteins and inhibition of mTOR pathway components blocks this increase.
Under physiological conditions, stimulations that are sufficient to induce L-LTP likely cause stabilization of these activators perhaps by phosphorylation of specific residues in these proteins. In support of this idea, several proteins are known to be made resistant to degradation by phosphorylation (Hegde, 2004). eIF4E and eEF1A are both known to be degraded by the UPP but the signals that trigger their degradation have not yet been identified. A well studied Ser-209 phosphorylation of eIF4E does not affect its ubiquitination (Murata and Shimotohno, 2006). Thus, phosphorylation on different sites or other post-translational modifications might make eIF4E a target for ubiquitination and degradation. In addition to stabilization of the activators, for the translation required for L-LTP to proceed, the repression by the negative regulators needs to be relieved. Degradation of translational repressors such as 4E-BP2 also might be regulated by phosphorylation, since degradation of a closely related molecule 4E-BP1 is known to be regulated by phosphorylation in non-neuronal cell types (Yanagiya et al., 2012).
Our studies have just begun to identify the elements of translational pathways regulated by ubiquitin proteasome-mediated proteolysis during L-LTP. Degradation of translational activators likely plays a role in setting the threshold for induction of L-LTP in that only strong enough stimuli would cause stabilization of translational activators and allow adequate translation to proceed. It appears that degradation of repressors is also necessary for translation to continue. Eventual stabilization of repressors during late stages of L-LTP likely limits the duration of translation and may be important in preventing saturation of synaptic strengthening. Also, it is becoming clear that too much translation is harmful. For example, excessive protein synthesis that occurs in Drosophila Fragile X mutants blocks long-term memory (Bolduc et al., 2008). Also, increasing the levels of eIF4E in mice results in exaggerated cap-dependent translation and causes synaptic pathology and autism-like behavior (Gkogkas et al., 2013; Santini et al., 2013). Thus, ubiquitin proteasome-mediated protein degradation is likely to be critical for physiological regulation of translation in the nervous system and therefore perturbation of proteolytic regulation of translation might lead to neuronal pathology. Dysregulation of proteolysis, which is often seen in neurodegenerative diseases (Gong et al., 2006; Dennissen et al., 2012), therefore might have an impact on neuronal function through its deleterious effect on protein synthesis as well.
Footnotes
This work was supported by a grant to A.N.H. from National Institute of Neurological Diseases and Stroke (NS065583). We thank Isabel Muzzio (University of Pennsylvania) and Lori McMahon (University of Alabama, Birmingham) for critiques and comments on an earlier version of this manuscript.
The authors declare no competing financial interests.
References
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–1145. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Baxter DA, Buonomano DV, Cleary LJ, Eskin A, Goldsmith JR, McClendon E, Nazif FA, Noel F, Scholz KP. Neural and molecular bases of nonassociative and associative learning in Aplysia. Ann N Y Acad Sci. 1991;627:124–149. doi: 10.1111/j.1749-6632.1991.tb25918.x. [DOI] [PubMed] [Google Scholar]
- Chain DG, Casadio A, Schacher S, Hegde AN, Valbrun M, Yamamoto N, Goldberg AL, Bartsch D, Kandel ER, Schwartz JH. Mechanisms for generating the autonomous cAMP-dependent protein kinase required for long-term facilitation in Aplysia. Neuron. 1999;22:147–156. doi: 10.1016/S0896-6273(00)80686-8. [DOI] [PubMed] [Google Scholar]
- Cracco JB, Serrano P, Moskowitz SI, Bergold PJ, Sacktor TC. Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus. 2005;15:551–556. doi: 10.1002/hipo.20078. [DOI] [PubMed] [Google Scholar]
- Dennissen FJ, Kholod N, van Leeuwen FW. The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog Neurobiol. 2012;96:190–207. doi: 10.1016/j.pneurobio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Dick LR, Cruikshank AA, Grenier L, Melandri FD, Nunes SL, Stein RL. Mechanistic studies on the inactivation of the proteasome by lactacystin: a central role for clasto-lactacystin beta-lactone. J Biol Chem. 1996;271:7273–7276. doi: 10.1074/jbc.271.13.7273. [DOI] [PubMed] [Google Scholar]
- Dong C, Upadhya SC, Ding L, Smith TK, Hegde AN. Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem. 2008;15:335–347. doi: 10.1101/lm.984508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Byrne JH. Protein degradation and memory formation. Brain Res Bull. 2011;85:14–20. doi: 10.1016/j.brainresbull.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nägerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-X. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert D, Topolnik L, Azzi M, Huang L, Badeaux F, Desgroseillers L, Sossin WS, Lacaille JC. Forskolin induction of late-LTP and upregulation of 5′ TOP mRNAs translation via mTOR, ERK, and PI3K in hippocampal pyramidal cells. J Neurochem. 2008;106:1160–1174. doi: 10.1111/j.1471-4159.2008.05470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hegde AN. Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog Neurobiol. 2004;73:311–357. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Hegde AN. The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem. 2010;17:314–327. doi: 10.1101/lm.1504010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez PJ, Abel T. The role of protein synthesis in memory consolidation: progress amid decades of debate. Neurobiol Learn Mem. 2008;89:293–311. doi: 10.1016/j.nlm.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Helmstetter FJ. The ubiquitin-proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol Learn Mem. 2013;105:107–116. doi: 10.1016/j.nlm.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH. Molecular biology of learning: modulation of transmitter release. Science. 1982;218:433–443. doi: 10.1126/science.6289442. [DOI] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knöpfel T, Behnisch T. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci. 2006;26:4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004a;116:467–479. doi: 10.1016/S0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004b;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Khaleghpour K, Kahvejian A, De Crescenzo G, Roy G, Svitkin YV, Imataka H, O'Connor-McCourt M, Sonenberg N. Dual interactions of the translational repressor Paip2 with poly(A) binding protein. Mol Cell Biol. 2001;21:5200–5213. doi: 10.1128/MCB.21.15.5200-5213.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koiwai K, Maezawa S, Hayano T, Iitsuka M, Koiwai O. BPOZ-2 directly binds to eEF1A1 to promote eEF1A1 ubiquitylation and degradation and prevent translation. Genes Cells. 2008;13:593–607. doi: 10.1111/j.1365-2443.2008.01191.x. [DOI] [PubMed] [Google Scholar]
- Lantz B. The impact of sample non-normality on ANOVA and alternative methods. Br J Math Stat Psychol. 2013;66:224–244. doi: 10.1111/j.2044-8317.2012.02047.x. [DOI] [PubMed] [Google Scholar]
- McMahon R, Zaborowska I, Walsh D. Noncytotoxic inhibition of viral infection through eIF4F-independent suppression of translation by 4EGi-1. J Virol. 2011;85:853–864. doi: 10.1128/JVI.01873-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem. 2000;267:6321–6330. doi: 10.1046/j.1432-1327.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Murata T, Shimotohno K. Ubiquitination and proteasome-dependent degradation of human eukaryotic translation initiation factor 4E. J Biol Chem. 2006;281:20788–20800. doi: 10.1074/jbc.M600563200. [DOI] [PubMed] [Google Scholar]
- Negrutskii BS, El'skaya AV. Eukaryotic translation elongation factor 1 alpha: structure, expression, functions, and possible role in aminoacyl-tRNA channeling. Prog Nucleic Acid Res Mol Biol. 1998;60:47–78. doi: 10.1016/s0079-6603(08)60889-2. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Belsham GJ, Gingras AC, Donzé O, Lin TA, Lawrence JC, Jr, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- Pestova TV, Lorsch JR, Hellen CU. The mechanism of translation initiation in eukaryotes. In: Mathews M, Sonenberg N, Hershey JWB, editors. Translational control in biology and medicine. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 2007. pp. 87–128. [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2013;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS. Defining memories by their distinct molecular traces. Trends Neurosci. 2008;31:170–175. doi: 10.1016/j.tins.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- Tang G, Leppla SH. Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infect Immun. 1999;67:3055–3060. doi: 10.1128/iai.67.6.3055-3060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27:5885–5894. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers CA, Dickson KS, Wyllie DJ. Induction and maintenance of late-phase long-term potentiation in isolated dendrites of rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;568:803–813. doi: 10.1113/jphysiol.2005.092924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Morris RG. Hippocampal-neocortical interactions in memory formation, consolidation, and reconsolidation. Annu Rev Psychol. 2010;61:49–79. C1–4. doi: 10.1146/annurev.psych.093008.100523. [DOI] [PubMed] [Google Scholar]
- Woo NH, Nguyen PV. Protein synthesis is required for synaptic immunity to depotentiation. J Neurosci. 2003;23:1125–1132. doi: 10.1523/JNEUROSCI.23-04-01125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagiya A, Suyama E, Adachi H, Svitkin YV, Aza-Blanc P, Imataka H, Mikami S, Martineau Y, Ronai ZA, Sonenberg N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol Cell. 2012;46:847–858. doi: 10.1016/j.molcel.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Yoshida K, Kozlov G, Lim NS, De Crescenzo G, Pang Z, Berlanga JJ, Kahvejian A, Gehring K, Wing SS, Sonenberg N. Poly(A) binding protein (PABP) homeostasis is mediated by the stability of its inhibitor, Paip2. EMBO J. 2006;25:1934–1944. doi: 10.1038/sj.emboj.7601079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]