

Abstract
Of the five muscarinic receptor subtypes, the M5 receptor is the only one detectable in midbrain dopaminergic neurons, making it an attractive potential therapeutic target for treating disorders in which dopaminergic signaling is disrupted. However, developing an understanding of the role of M5 in regulating midbrain dopamine neuron function has been hampered by a lack of subtype-selective compounds. Here, we extensively characterize the novel compound VU0238429 and demonstrate that it acts as a positive allosteric modulator with unprecedented selectivity for the M5 receptor. We then used VU0238429, along with M5 knock-out mice, to elucidate the role of this receptor in regulating substantia nigra pars compacta (SNc) neuron physiology in both mice and rats. In sagittal brain slices that isolate the SNc soma from their striatal terminals, activation of muscarinic receptors induced Ca2+ mobilization and inward currents in SNc dopamine neurons, both of which were potentiated by VU0238429 and absent in M5 knock-out mice. Activation of M5 also increased the spontaneous firing rate of SNc neurons, suggesting that activation of somatodendritic M5 increases the intrinsic excitability of SNc neurons. However, in coronal slices of the striatum, potentiation of M5 with VU0238429 resulted in an inhibition in dopamine release as monitored with fast scan cyclic voltammetry. Accordingly, activation of M5 can lead to opposing physiological outcomes depending on the location of the receptor. Although activation of somatodendritic M5 receptors on SNc neurons leads to increased neuronal firing, activation of M5 receptors in the striatum induces an inhibition in dopamine release.
Keywords: acetylcholine, allosteric, dopamine, M5, mAChR, muscarinic
Introduction
Dysregulation of dopamine release is thought to contribute to multiple CNS disorders including Parkinson's disease (PD), schizophrenia, attention deficit hyperactivity disorder, and others (Purves, 2012). Midbrain dopamine neurons are directly modulated by the neurotransmitter acetylcholine (ACh), with cholinergic innervation targeting both the somatodendritic and synaptic compartments of these neurons (Mena-Segovia et al., 2008; Oldenburg and Ding, 2011). The importance of ACh as a modulator of dopamine neuron function is highlighted by the finding that agents that modulate the cholinergic system exhibit efficacy in treating symptoms associated with PD and schizophrenia (Katzenschlager et al., 2003; Shekhar et al., 2008; Foster et al., 2012). Unfortunately, these nonselective cholinergic modulators have dose-limiting cardiovascular and gastrointestinal side effects that curtail their clinical utility. The muscarinic ACh receptor (mAChR) family consists of five members (M1–M5) that, along with nicotinic receptors, mediate ACh signaling. The M2 and M3 receptor subtypes are thought to mediate the undesired side effects of nonselective cholinergic modulators (Bymaster et al., 2003). Accordingly, it is hypothesized that subtype-selective modulation of M1, M4, and/or M5 could provide clinical efficacy without the peripheral adverse-effect liability. The M5 receptor was the last mAChR subtype to be cloned (Bonner et al., 1988; Liao et al., 1989) and exhibits a low overall expression level (Yasuda et al., 1993) that is restricted to the cerebrovasculature (Yamada et al., 2001) and midbrain dopamine neurons, where it is the only mAChR subtype detectable (Weiner et al., 1990). Studies using M5 knock-out (KO) mice have suggested that M5 is necessary for observing prolonged dopamine release after electrical stimulation of cholinergic inputs from the hindbrain (Forster et al., 2002; Steidl et al., 2011). However, developing a detailed understanding of the role of M5 in regulating dopamine neuron function or as a potential therapeutic target has been hampered by the lack of agents that selectively modulate the M5 receptor subtype. Targeting allosteric sites on receptors that are distinct from the endogenous neurotransmitter binding site has provided unparalleled success in selectively targeting GPCR subtypes that were previously intractable. We have recently reported successful medicinal chemistry efforts based on positive allosteric modulators (PAMs) for the M1 mAChR (Marlo et al., 2009) that resulted in the discovery of multiple PAMs for the human M5 (hM5) receptor (Bridges et al., 2009; Bridges et al., 2010). Of these, VU0238429 stood out as the optimal PAM for in vitro studies based on its hM5 selectivity, potency, and efficacy. Here, we characterize VU0238429 and demonstrate that this compound has robust activity and unprecedented selectivity as an M5 PAM at rat mAChR receptors. Using this compound, along with M5 KO mice, we assessed the ability of M5 activation to modulate dopamine neuron physiology. We report that somatodendritic activation of M5 on dopamine neurons of the substantia nigra pars compacta induced Ca2+ mobilization and inward currents and increased spontaneous firing rates. However, activation of M5 in the striatum reduced electrically evoked dopamine release. These surprising findings suggest that M5 receptor activation engenders opposing effects on dopamine neuron function depending upon the receptor's localization.
Materials and Methods
Materials.
VU0238429 (aka ML129) was developed with support from the Molecular Libraries Probe Production Centers Network (MLPCN) initiative (Bridges et al., 2009; Bridges et al., 2010) and is available to the public (http://www.mc.vanderbilt.edu/centers/mlpcn/obtaining_probes.html). Fluo-4 AM and fluo-4 pentapotassium salt were obtained from Invitrogen. Costar 96-well cell culture plates, V-bottom compound plates, and Hygromycin B were purchased from Corning. Kynurenic acid sodium salt and tetrodotoxin were purchased from Tocris Bioscience. All other compounds were purchased from Sigma-Aldrich. All cell culture reagents were from Invitrogen unless otherwise noted.
Fluorescence-based calcium imaging in cultured cells.
Chinese hamster ovary (CHO) cells stably expressing rat M1, M2, M3, M4, or M5 were grown in standard medium (Ham's F12 medium) supplemented with 10% heat-inactivated FBS, 20 mm HEPES, and 50 μg/ml G418 sulfate and kept at 37°C in a humidified incubator containing 5% CO2. To couple the M2 and M4 receptors to Ca2+ mobilization, these cells also stably expressed the chimeric G-protein Gqi5 and were maintained in the presence of 500 μg/ml Hygromycin B. One day before the assay, cells were plated at 50,000 cells per well in standard growth medium (Ham's F12 medium supplemented with 10% FBS and 20 mm HEPES) in 96-well plates. On the day of the assay, medium was removed and cells were dye loaded for 1 h in Ca2+ assay buffer (HBSS; Invitrogen), 20 mm HEPES, 2.5 mm probenecid (Sigma), pH 7.4, containing 1.8 μm Fluo4-AM. After dye loading, cells were rinsed with Ca2+ assay buffer and maintained at room temperature for the duration of the assay. All experiments were matched in solvent (DMSO) concentration. Ca2+ flux was measured over time as an increase in fluorescence using a FlexStation II (Molecular Devices). The increase in fluorescence over basal was determined for the peak of the response and then normalized to the maximal peak response elicited by ACh.
Radioligand binding.
Membranes were prepared from M5-expressing CHO cells as described previously (Shirey et al., 2008). All binding reactions were performed in assay buffer (100 mm NaCl, 10 mm MgCl2, and 20 mm HEPES, pH 7.4) at room temperature. Reactions contained a nonsaturating concentration (0.1 nm) of [N-methyl-3H]scopolamine methyl chloride ([3H]-NMS; GE Healthcare) and membrane protein (7.5–15 μg) and appropriate concentrations of test compounds or vehicle. Nonspecific binding was determined in the presence of 1 μm atropine. For these experiments, receptor-bound NMS accounted for ≤15% of the total NMS added per well. Reactions were incubated at room temperature for 3 h and terminated by rapid filtration. Filter plates were washed 3 times with ice-cold harvesting buffer (50 mm Tris-HCl and 0.9% NaCl, pH 7.4) using a 96-well Brandel harvester. Plates were dried overnight and then resuspended in microscint scintillation fluid (PerkinElmer) and radioactivity quantified using a TopCount NXT microplate scintillation and luminescence counter (PerkinElmer).
Acute brain slice preparation.
All of the animals used in the present studies were group housed with food and water available ad libitum. Animals were kept under a 12 h light/dark cycle with lights on from 6:00 A.M. to 6:00 P.M. and were tested during the light phase. All of the experimental procedures were approved by the Vanderbilt University Animal Care and Use committee and followed the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Sagittal brain slices (225–300 μm) containing the substantia nigra pars compacta (SNc), or coronal slices (300–400 μm) containing the striatum were obtained from Sprague Dawley rats of either sex (postnatal days 15–20 [P15-P20]; Charles River Laboratories), male C57BL/6NTac wild-type mice (p30–90; Taconic), or male M5 receptor KO mice (P30–P90; Taconic, with permission from J. Wess, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD). Brain slices were cut using a Leica VT1200S.
Briefly, rats were anesthetized with isoflurane and brains were removed rapidly, submerged, and sliced in ice-cold modified artificial CSF (aCSF) containing the following (in mm): 75 sucrose, 87 NaCl, 2.5 KCl, 7 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 1 kynurenic acid, and 25 d-glucose oxygenated with 95% O2/5% CO2. Slices were then incubated in aCSF containing the following (in mm): 124 NaCl, 4 KCl, 2.45 CaCl2, 1.2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 11 d-glucose at room temperature for at least 1 h before being transferred to the recording chamber.
Slices from mouse brains were prepared in a variation of a reported brain slice methodology for adult mice (Zhao et al., 2011). Mice were anesthetized by intraperitoneal injection of 0.15 ml of ketamine/xylazine (20 mg/2 mg/1 ml) and then transcardially perfused with cold modified aCSF. Mice were then decapitated and the brains were removed, submerged, and sliced in modified aCSF. Slices were initially recovered for 10–15 min at 32°C in protective aCSF containing the following (in mm): 92 N-methyl-d-glucamine, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 d-glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyuruvate, 10 MgSO4, 0.5 CaCl2, and 12 N-acetyl-l-cysteine, pH 7.3, 305 mOsm. After the initial recovery the slices were transferred to aCSF containing 126 mm NaCl, 2.5 mm KCl, 2.45 mm CaCl2, 1.2 mm MgSO4, 1.25 mm NaH2PO4, 26 mm NaHCO3, 12 mm N-acetyl-l-cysteine, and 11 mm d-glucose for at least 1 h before recording. In the recording chamber, slices were perfused with aCSF in the absence of N-acetyl-cysteine. All experiments on acute brain slices were performed at 32°C and drugs were bath applied with a flow rate of 2 ml/min.
Fluorescence-based calcium imaging in brain slices.
Whole-cell recordings were made from visually identified SNc neurons under an Olympus BX50WI upright microscope. A low-power objective (4×) was used to identify the brain region and a 40× water-immersion objective coupled with Hoffman optics was used to visualize the individual neurons of interest. Whole-cell current-clamp or voltage-clamp signals were amplified using an Axon Multiclamp 700B amplifier (Molecular Devices). Patch pipettes were prepared from borosilicate glass (Sutter Instrument) using a Flaming/Brown micropipette puller (Sutter Instrument). The electrode resistance was 3–6 MΩ when filled with the intracellular solution containing the following (in mm): 123 K-gluconate, 7 KCl, 10 HEPES, 0.2 GTP, 2 ATP, and 0.1 Fluo-4 pentapotassium, pH 7.3, 290 mOsm. The identity of SNc neurons was verified by previously established electrophysiological parameters including slow firing rates (1–4 Hz), broad spike half-widths (2.5–4.5 ms), and the presence of a robust hyperpolarization-induced voltage sag (Guzman et al., 2009). Changes in Ca2+ mobilization and inward currents were monitored simultaneously in the presence of 1 μm tetrodotoxin. Electrical signals were low-pass filtered at 3 kHz, digitized at 20 kHz, and acquired using a Clampex9.2/Digidata1440A system (Molecular Devices). Changes in fluorescence were monitored every 2 s, with exposures ranging from 350 to 1000 ms, using a Cool Snap HQ2 camera (Photometrics), Lambda 10–3 shutter, and Lambda LS stand-alone xenon arc lamp and power supply (Sutter Instrument). Increases in Ca2+ mobilization in SNc neurons were reported as changes in relative fluorescence divided by the baseline fluorescence (ΔF/F).
Perforated patch recordings.
Patch pipettes (resistance of 5–7 MΩ) were filled with intracellular solution containing the following (in mm): 123 K-gluconate, 7 KCl, 10 HEPES, 0.1 EGTA, and 1 MgCl2, along with 25 μm CaCl2 and 195 μm amphotericin-B. A small amount of solution not containing amphotericin-B was included in the front of the pipette so that a gigaohm seal could be formed with visually identified SNc neurons. Current-clamp recordings of spontaneous action potentials were made using an Axon Multiclamp 700B amplifier (Molecular Devices). Only recordings with constant firing rates, action potential amplitudes >30 mV, and access resistance >30 MΩ throughout the experiment were analyzed.
Fast scan cyclic voltammetry.
Electrically evoked dopamine overflow was monitored with carbon fiber electrodes with a 5 μm diameter, as described previously (Schmitz et al., 2002). A triangular voltage wave (−400 to +1000 mV at 300 V/s) was applied to fresh cut carbon fiber electrodes every 100 ms. Electrodes were placed 50 μm deep into the dorsolateral striatum and slices were electrically stimulated (30–300 μA) every 2.5 min via a bipolar stimulating electrode placed ∼100 μm from the carbon fiber. Current was recorded with a low-pass Bessell filter at 10 kHz and digitized at 100 kHz. Background-subtracted cyclic voltammograms served both to calibrate the electrodes and to identify dopamine as the substance that was released after electrical stimulation.
Data analysis.
All electrophysiological data were acquired using a Clampex9.2/Digidata1440A system (Molecular Devices). ClampFit, Metafluor (Molecular Devices), MiniAnalysis (Synaptosoft), and Prism (Graphpad) were used for data analysis. The two-tailed Student's t test was used to analyze statistical significance between two data points. The best fit simulation of electrically evoked dopamine overflow was found by nonlinear regression. All averaged data are presented as the mean ± SEM.
Results
Characterization of VU0238429, a positive allosteric modulator at the M5 receptor
To validate the utility of VU0238429 as an M5-selective tool in rodent models, we evaluated the efficacy and selectivity of VU0238429 at the rat mAChR subtypes. Ca2+ mobilization was monitored as a readout of receptor activation in CHO cells stably expressing rat M5 (rM5). VU0238429 (3 μm) or vehicle was added to cells 90 s before addition of an EC20 concentration of ACh (1 nm). For comparison, separate cells were treated with vehicle, followed by a maximally effective concentration of ACh (1 μm). VU0238429 induced a robust potentiation of Ca2+ mobilization induced by an EC20 concentration of ACh (Fig. 1A). Although VU0238429 potentiated ACh-induced responses, it induced little to no Ca2+ mobilization on its own, which is consistent with its activity as a PAM. To establish the potency of VU0238429 at rM5, a concentration response curve (CRC) was generated by establishing the effects of multiple concentrations of VU0238429 on Ca2+ responses induced by an EC20 concentration of ACh. VU0238429 potentiated M5-mediated responses in a concentration-dependent manner with an EC50 value of 2.4 ± 0.9 μm and maximal potentiation equal to 83.2 ± 7.0% of that observed with maximal ACh concentrations (Fig. 1B).
Figure 1.
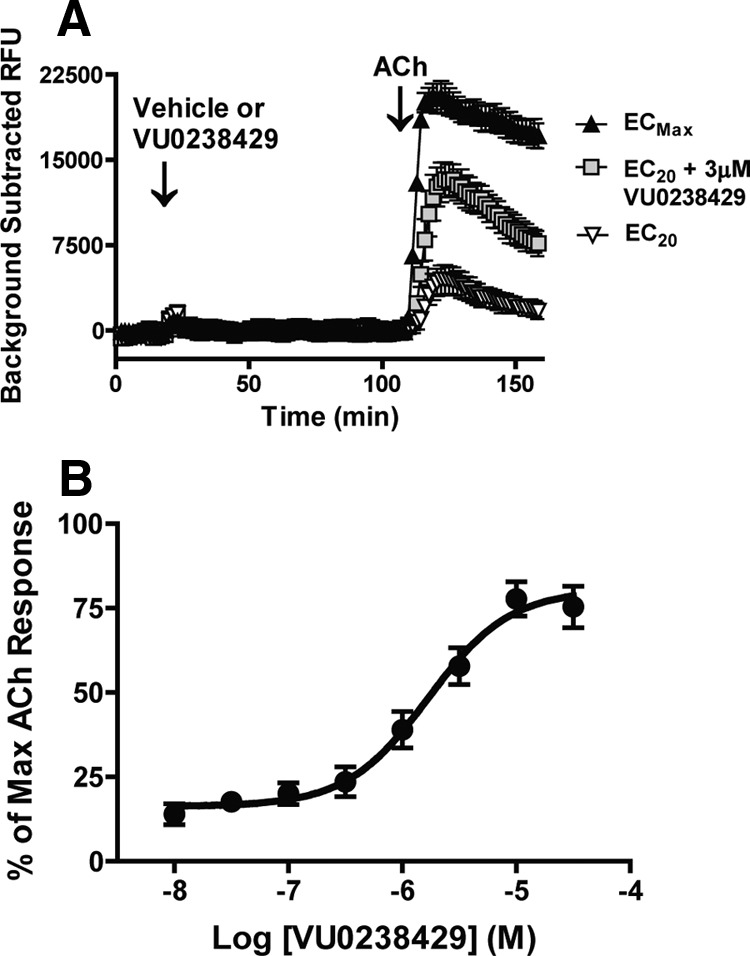
VU0238429 robustly potentiates signaling at the rat M5 receptor. A, Representative traces from cells expressing the rM5 receptor demonstrate that VU0238429 (3 μm) induces little to no effect on Ca2+ mobilization on its own, but robustly potentiates the Ca2+ response induced by an EC20 concentration of ACh. For comparison, separate cells were treated with vehicle followed by either an EC20 (1 nm) or maximally effective (1 μm) concentration of ACh. B, Potency of VU0238429 in cells expressing rat M5 was evaluated by monitoring Ca2+ mobilization induced by various concentrations of VU0238429 followed by an EC20 concentration of ACh (n = 4; EC50 = 2.4 ± 0.9 μm).
To determine whether VU0238429 interacts with the rM5 receptor via an allosteric or orthosteric mechanism, we assessed the ability of VU0238429 to competitively displace the radiolabeled orthosteric antagonist 3H-NMS. Membranes from rM5-expressing cells were harvested and equilibrated with 3H-NMS in the absence or presence of either VU023429 or the orthosteric antagonist atropine. Atropine displaced 3H-NMS with a Ki value of 2.56 ± 0.41 nm, a value consistent with that previously reported at the M5 receptor (Shirey et al., 2008). Conversely, VU0238429, at concentrations sufficient to induce robust potentiation of ACh responses, did not displace 3H-NMS and induced a small (<10%) increase in 3H-NMS binding at higher concentrations (Fig. 2A). The finding that VU0238429 induces positive cooperativity with respect to 3H-NMS binding and does not competitively displace 3H-NMS is consistent with VU0238429 binding to an allosteric site on the M5 receptor to potentiate agonist responses.
Figure 2.
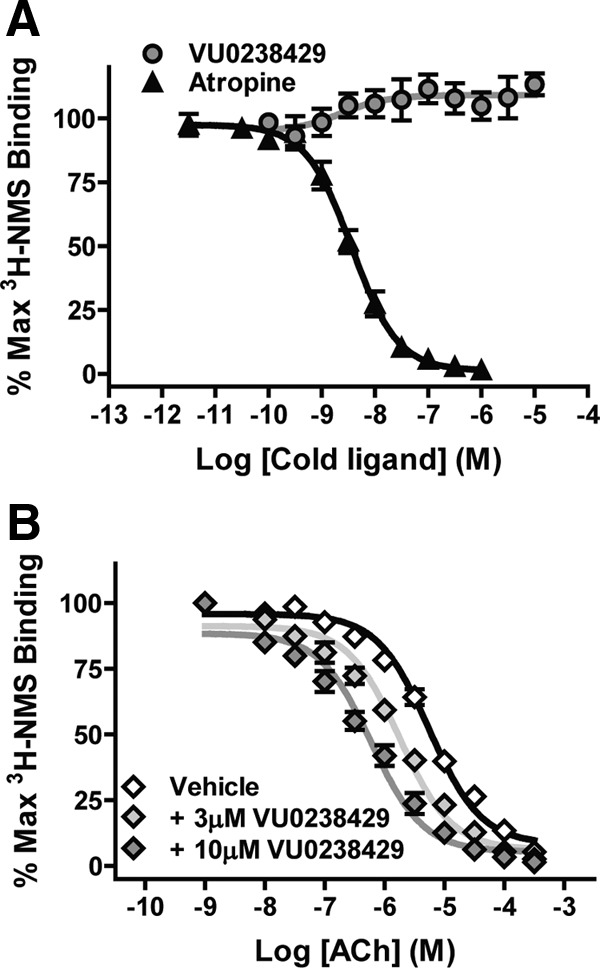
VU0238429 interacts with the rM5 receptor in a manner consistent with an allosteric modulator. A, Cell membranes from rM5-expressing cells were equilibrated with the radiolabeled orthosteric antagonist 3H-NMS. Incubation with the unlabeled orthosteric antagonist atropine (10 μm) displaced 3H-NMS in a concentration-dependent manner. However, incubation with VU0238429 (10 μm) was not able to displace 3H-NMS, suggesting that VU0238429 binds to an allosteric site (n = 3, performed in triplicate). B, The orthosteric agonist ACh can also displace 3H-NMS at the rM5 receptor. Addition of VU0238429 shifts the ability of ACh to displace 3H-NMS in a concentration-dependent manner, suggesting that VU0238429 potentiates ACh signaling, at least in part, through increasing the affinity of the rM5 receptor for ACh (n = 4, performed in triplicate).
To determine whether VU0238429 alters ACh affinity at the orthosteric site, rM5 membranes were equilibrated with 3H-NMS and increasing concentrations of ACh in the absence or presence of VU0238429. Inclusion of ACh displaced 3H-NMS from the M5 receptor with a Ki value of 3.82 ± 0.41 μm. In the presence of 10 μm VU0238429, the ACh displacement curve was left shifted with an observed Ki value of 0.49 ± 0.12 μm. These results suggest that VU0238429 increases ACh signaling at M5 at least in part via increasing the affinity of the receptor for ACh.
VU0238429 is a subtype-selective potentiator of the rM5 receptor
To determine whether VU0238429 is an rM5-selective modulator, ACh-induced Ca2+ signaling was monitored in cells expressing individual rat mAChR subtypes (rM1–rM5). The M1, M3, and M5 subtypes (but not M2 or M4) endogenously couple to Ca2+ mobilization. To compare the activation of all five subtypes directly in a common assay, cells expressing the rM2 or rM4 receptor coexpressed the mutant G-protein Gqi5 to couple these receptors to Ca2+ mobilization. In cells expressing the rM5 receptor, inclusion of VU0238429 induced a robust leftward shift in the ACh CRC compared with vehicle (Fig. 3A). Increasing concentrations of VU0238429 (3, 10, and 30 μm) induced progressively larger leftward shifts in the ACh EC50 (5.8-, 21.0-, and 29.9-fold changes in the apparent EC50, respectively). Because 10 μm VU0238429 induced a near maximal shift in ACh signaling with little to no observable agonist activity, this concentration was used in all subsequent studies. Although VU0238429 robustly potentiated ACh signaling in rM5-expressing cells, it had little to no effect in cell expressing rM1–rM4 receptors (a ≤2-fold shift in apparent ACh EC50; Fig. 3B).
Figure 3.
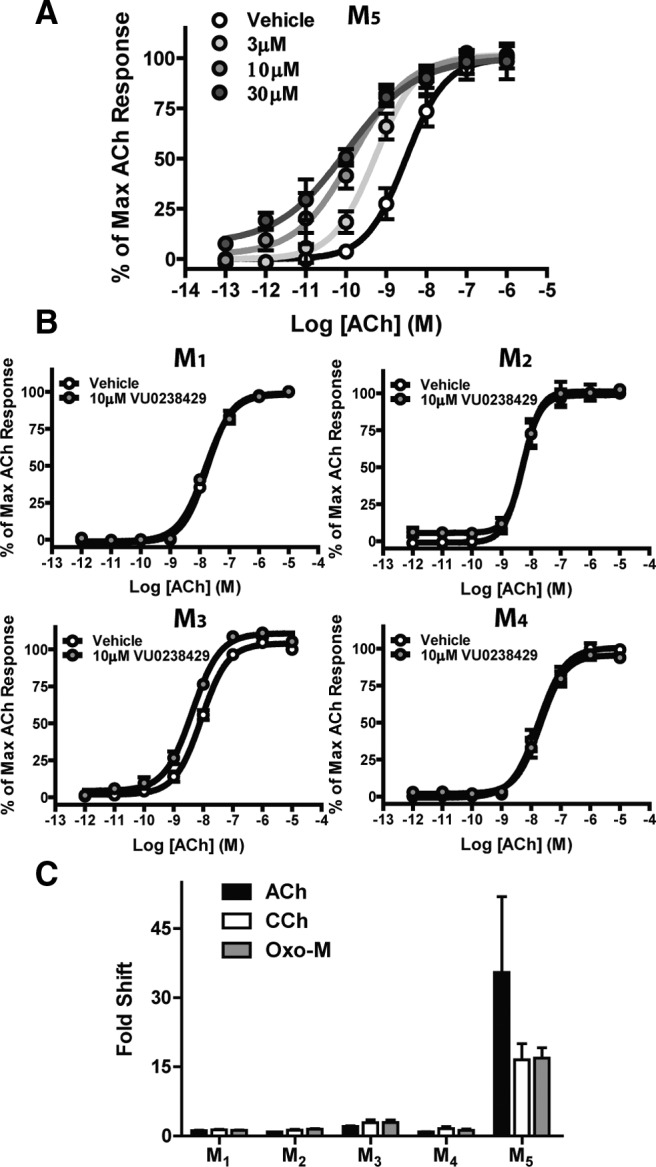
VU0238429 is a robust and selective potentiator of the rM5 receptor. VU0238429 induces a concentration-dependent leftward shift in the ACh CRC when monitoring Ca2+ mobilization in cells expressing the M5 receptor (A), but induces little to no shift in cells expressing M1–M4 (B; n = 3–4 experiments performed in triplicate). VU0238429 shows little to no probe dependence and maintains M5-selectivity when either CCh or Oxo-M is used as an orthosteric agonist (C; n = 3–4 experiments performed in triplicate). Data are normalized to depict the fold shift in CRCs observed with various agonists in cells expressing M1–M5 receptors.
ACh is the endogenous agonist of mAChRs and the positive cooperativity of VU0238429 with ACh is critical for modulating activity in normal physiological settings. However, ACh is rapidly degraded by ACh esterase (AChE), which complicates the use of ACh as an exogenously applied agonist in slice electrophysiology experiments. Oxotremorine-M (Oxo-M) and carbachol (CCh) are two nonselective mAChR agonists that are not susceptible to degradation by AChE, making them excellent tools for studying mAChR function in brain slices. Before using VU0238429 in slice electrophysiology experiments, it was important to determine whether the subtype selectivity observed with VU0238429 and ACh is maintained with other agonists. Inclusion of 10 μm VU0238429 induced a leftward shift in both the Oxo-M and CCh CRCs in cells expressing the rM5 receptor (16.5- and 16.9-fold shifts, respectively) with little to no activity at rM1–rM4 (a <3-fold shift in apparent agonist EC50; Fig. 3C). These results demonstrate that VU0238429 shows little to no probe dependence and maintains rM5 selectivity when CCh, Oxo-M, and ACh are used as mAChR agonists.
M5 receptor mediates Ca2+ mobilization and inward currents in SNc neurons
The M5 receptor is the only subtype of muscarinic receptor that is detectable when measuring mRNA in midbrain dopamine neurons (Weiner et al., 1990). However, to date, there has been no direct evidence that the M5 subtype is functionally expressed in these neurons. To address this issue, patch-clamp recordings were performed in visually identified SNc neurons from acute brain slices prepared from young rats (P14–P20). Cells were voltage clamped at −55 mV and 1 μm tetrodotoxin was included in the bath to inhibit cell firing. Ca2+ mobilization was monitored by loading the cell with a Ca2+-sensitive dye (fluo-4) via the patch pipette. This experimental setup allowed us to simultaneously monitor Ca2+ mobilization and changes in the holding current. Application of the nonselective muscarinic receptor agonist Oxo-M (1 μm) induced a robust and reversible increase in Ca2+ mobilization in all of the cells examined (Fig. 4A,B). Consistent with previous studies (Lacey et al., 1990), application of mAChR agonist also induced a robust and reversible inward current in these neurons (Fig. 4C). Both of these effects were completely blocked by pretreatment with the nonselective mAChR antagonist scopolamine (n = 3; data not shown). These results confirm the presence of a functional mAChR located postsynaptically on SNc neurons.
Figure 4.

Oxo-M induces both Ca2+ mobilization and inward currents in substantia nigra pars compacta neurons. Cells were patched and filled with the Ca2+-sensitive dye fluo-4 and voltage clamped at −55 mV. Brain slices were treated with 1 μm tetrodotoxin to block action potentials and Ca2+ fluorescence and holding currents were then monitored simultaneously. A, Sample images from a representative experiment depicting Ca2+ fluorescence before (A1), during (A2), and after (A3) addition of 1 μm Oxo-M. B, Quantification of Ca2+ fluorescence over time. Data are normalized as ΔF/F as discussed in the Materials and Methods section. C, Simultaneous to the increase in Ca2+ mobilization, a robust inward current was observed upon addition of Oxo-M.
To determine whether these effects were mediated by the M5 receptor, we performed experiments using submaximal Oxo-M concentrations in combination with the M5-selective PAM VU0238429. Bath application of 3 μm Oxo-M induced a Ca2+ mobilization response (0.20 ± 0.03 ΔF/F) that was well below the maximal response elicited by higher Oxo-M concentrations. Addition of 10 μm VU0238429 had no significant effect on Ca2+ mobilization alone, but significantly potentiated the response to 3 μm Oxo-M (0.36 ± 0.07 ΔF/F; Fig. 5C,D). Addition of 3 μm Oxo-M also induced inward currents (70.6 ± 12.4 pA, n = 8; data not shown). However, the inward current induced by this concentration of Oxo-M was near the maximal response. Although these currents were augmented by VU0238429, this effect did not reach statistical significance (84.0 ± 9.6 pA, n = 8; data not shown). Therefore, for studies of inward currents, we reduced the Oxo-M concentration to 500 nm. At this lower concentration, Oxo-M induced a submaximal inward current, as shown in Figure 5, A and B (29.6 ± 1.0 pA), that was significantly potentiated by inclusion of VU02384529 (38.9 ± 1.9 pA; Fig. 5B). Simultaneous recordings of Ca2+ mobilization revealed that 500 nm Oxo-M induced a Ca2+ response (0.22 ± 0.07 ΔF/F; data not shown) that was potentiated by VU0238429 (0.30 ± 0.08 ΔF/F; data not shown); however, this effect did not reach statistical significance. To confirm a role for M5 in mediating these responses, Ca2+ mobilization and inward currents were monitored in SNc neurons from acute slices of either wild-type or M5 KO mice. Similar to experiments in acute rat slices, bath application of 10 μm Oxo-M induced robust inward currents (47.8 ± 5.1 pA) and Ca2+ mobilization (0.21±.06 ΔF/F) in SNc neurons from wild-type mice (Fig. 6A). These Oxo-M-mediated responses were completely absent in M5 KO mice (Fig. 6B), indicating that M5 expression is required for Oxo-M-mediated inward currents and Ca2+ mobilization in SNc neurons. Although SNc neurons from M5 KO mice no longer responded to bath application of Oxo-M, they retained their ability to respond to bath application of the group I mGluR agonist 3,5-dihydroxyphenylglycine (DHPG; Fig. 6B), indicating that SNc neurons from these mice are capable of mediating inward currents and Ca2+ mobilization. These results support previous anatomical studies suggesting that the M5 receptor is the only mAChR subtype expressed on midbrain dopamine neurons (Weiner et al., 1990) and demonstrate that the novel M5 PAM VU0238429 potentiates responses to mAChR agonists in these cells.
Figure 5.

VU0238429 potentiates both Ca2+ mobilization and inward currents induced by submaximal Oxo-M concentrations. A, Sample traces depicting inward currents observed upon addition of 500 nm Oxo-M in the presence or absence of VU0238429. B, VU0238429 significantly increased the magnitude of inward current evoked by addition of 500 nm Oxo-M (n = 6). C, Sample traces depicting Ca2+ responses observed upon addition of 3 μm Oxo-M with or without VU0238429. D, Inclusion of 10 μm VU0238429 significantly increases the peak Ca2+ response observed with 3 μm Oxo-M (n = 8). All averaged data are shown as the mean ± SEM. *Significant difference from control (p < 0.05, two-tailed Student's t test). Experiments were performed in slices prepared from P15–P20 Sprague Dawley rats.
Figure 6.

Oxo-M induces inward currents and Ca2+ mobilization in wild-type, but not M5 KO mice. Sample traces from experiments depicting representative responses upon addition of 10 μm Oxo-M to slices prepared from a wild-type mouse (A) or M5 KO mouse (B). Although Oxo-M-mediated responses were completely absent in M5 KO mice, these neurons retained their ability to respond to 3,5-dihydroxyphenylglycine (DHPG), an agonist of Group I metabotropic glutamate receptors (C, D; n = 4–6). Experiments were performed in slices prepared from P30–P90 wild-type or M5 KO mice.
Activation of the M5 receptor increases the tonic firing rate of SNc neurons
Inward currents and Ca2+ mobilization are often, but not always, associated with increased neuronal excitability. To determine whether M5 activation could increase the spontaneous firing rate of SNc neurons, perforated patch recordings were performed in acute slices from wild-type or M5 KO mice. Consistent with previous reports (Guzman et al., 2009), SNc neurons from wild-type mice fired spontaneous action potentials with a low frequency (2.55 ± 0.16 Hz, n = 17) and tonic firing pattern. Bath application of 10 μm Oxo-M induced a reversible increase in SNc firing rate in wild-type mice (0.72 ± 0.16 Hz increase in firing rate; Fig. 7A,B,E), but had little to no effect in M5 KO mice (−0.02 ± 0.03 Hz change in firing rate; Fig. 7C–E). In wild-type mice, Oxo-M-induced increases in firing rate were concurrent with increases in spike half-widths (Fig. 7F). To determine whether VU0238429 could alter the firing rate, experiments were performed with a submaximal concentration of Oxo-M. In wild-type mice, application of 300 nm Oxo-M induced an increase in firing rate (0.36 ± 0.08 Hz increase, n = 7; data not shown) that was potentiated by pretreatment with VU0238429 (0.53 ± 0.08 Hz increase, n = 7; data not shown), although this effect did not reach statistical significance. Collectively, these results suggest that M5 activation results in an increase in the intrinsic excitability of SNc neurons, which is consistent with somatodendritic M5 activation being stimulatory in these neurons.
Figure 7.

Addition of Oxo-M increases spontaneous firing in SNc neurons from wild-type (WT), but not M5 KO mice. A, In WT mice, addition of 10 μm Oxo-M increased the rate of cell firing, but had little to no effect on firing pattern. B, Time course of a representative experiment depicting a robust and reversible increase in cell firing upon addition of Oxo-M. C, D, Neurons from M5 KO mice display spontaneous tonic firing but do not respond to application of Oxo-M. E, Averaged changes in firing rates upon addition of 10 μm Oxo-M in neurons from either WT or M5 KO mice (n = 5). *Different from WT p < 0.01, Student's t test. F, Increases in firing rate were concurrent with increases in the spike half-width (# denotes different from baseline p < 0.01; paired Student's t test). Experiments were performed in slices prepared from P30–P90 wild-type or M5 KO mice.
Activation of striatal M5 inhibits electrically evoked dopamine overflow
Previous studies using M5 KO mice have suggested that M5 may play a role in regulating dopamine release from SNc terminals in the striatum (Bendor et al., 2010). Here, we monitored dopamine overflow in the dorsolateral striatum of coronal brain slices via fast scan cyclic voltammetry to elucidate whether the M5 receptor can regulate striatal dopamine release. Dopamine release was evoked via application of a single electrical pulse (1 ms) and monitored at a proximal carbon fiber electrode. In coronal brain slices, electrical stimulation induced a background subtracted voltammogram that was consistent with that observed upon calibration of the carbon fiber in dopamine-containing buffer (Fig. 8A). Electrical stimulation induced a rapid and transient increase in extracellular dopamine (Fig. 8B), the magnitude of which was stable when stimulations were applied at intervals of 2.5 min. Consistent with previous studies, application of Oxo-M induced a concentration-dependent reduction in electrically evoked dopamine release. Application of 1 μm Oxo-M to coronal rat brain slices induced a submaximal reduction in dopamine overflow (Fig. 8C), resulting in a reduction of dopamine overflow equal to 65.5 ± 7.4% of baseline.
Figure 8.

VU0238429 enhances Oxo-M-mediated attenuation of electrically evoked dopamine release in the striatum. Dopamine overflow was monitored using fast scan cyclic voltammetry. A, Background-subtracted voltammograms depict the responses observed upon calibration in dopamine-containing buffer (top) or electrically evoked striatal dopamine overflow (middle). B, Representative time course of electrically evoked dopamine overflow; arrow depicts time of stimulation. Electrical stimulations were applied every 2.5 min and data are depicted as the normalized peak dopamine response. C, D, Time courses of representative experiments showing that addition of 1 μm Oxo-M resulted in a reversible inhibition in dopamine release that was further inhibited when applied with VU0238429. E, Averaged data depicting the normalized evoked-dopamine overflow observed with various treatments (n = 5–9). Experiments including 10 μm scopolamine were pretreated 20 min before addition of VU0238429 and 1 μm Oxo-M. Data are depicted as the mean ± SEM. *Significant difference from 1 μm Oxo-M (p < 0.05, one-way ANOVA with a post hoc Bonferroni test). #Significant difference from VU0238429 + 1 μm Oxo-M (p < 0.01, one-way ANOVA with a post hoc Bonferroni test). Experiments were performed in slices prepared from P15–P20 Sprague Dawley rats.
Addition of VU0238429 alone had no effect on electrically evoked dopamine release in brain slices from rats, but significantly enhanced the reduction observed upon application of 1 μm Oxo-M (45.7 ± 5.2% of baseline; Fig. 8D,E). Pretreatment with the nonselective antagonist scopolamine completely blocked any inhibition observed with VU0238429 and 1 μm Oxo-M (Fig. 8E). Accordingly, potentiation of M5 via addition of VU0238429 induces a mAChR-mediated inhibition in the magnitude of evoked dopamine release in the striatum.
To determine whether VU0238429 modulates evoked dopamine overflow in the striatum via the M5 receptor, experiments were performed in both wild-type and M5 KO mice. In wild-type mice, bath application of 10 μm Oxo-M decreased evoked dopamine overflow to 44.7 ± 5.1% of baseline. VU0238429 alone had no effect on dopamine release, but amplified Oxo-M-induced inhibition of dopamine release (Fig. 9B,C). However, VU0238429 did not significantly alter the inhibition of evoked dopamine overflow induced by a lower concentration of Oxo-M in wild-type mice (3 μm; Fig. 9A), suggesting that the more potent effects of Oxo-M on dopamine overflow may be mediated by other mAChR subtypes. Consistent with previous reports (Bendor et al., 2010), Oxo-M (10 μm) reduced electrically evoked dopamine release in M5 KO mice to a greater extent than in than wild-type mice (15.8 ± 3.9% of baseline in M5 KO and 44.7 ± 5.1% of baseline in wild-type mice). Because application of 10 μm Oxo-M induced a nearly maximal inhibition of dopamine release in M5 KO mice, submaximal Oxo-M concentrations of 1 and 3 μm were examined. Addition of 3 μm Oxo-M reduced evoked dopamine release (81.1 ± 15.3% of baseline), but the enhancement of this effect by VU0238429 was absent in M5 KO mice (74.0 ± 14.1% of baseline; Fig. 9E,F). Similarly, inhibition of dopamine overflow by application of 1 μm Oxo-M to striatal slices from M5 KO mice (75.3 ± 4.7 of baseline n = 5) was not significantly altered by VU0238429 (72.4 ± 14.4 n = 5; Fig. 9D,F). The efficacy of VU0238429 in modulating Oxo-M responses in wild-type mice, but not M5 KO mice, suggests that VU0238429 modulates dopamine release via the M5 receptor. Collectively, these results further suggest that activation of M5 in the striatum results in an inhibition of electrically evoked dopamine release.
Figure 9.

VU0238429 enhances Oxo-M-mediated inhibition of dopamine overflow in wild-type but not M5 KO mice. Representative experiments depicting the inhibition of dopamine overflow observed in slices from wild-type mice treated with 3 μm Oxo-M (A) or 10 μm Oxo-M (B) in either the presence or absence of VU0238429. C, Averaged data depicting the normalized percentage inhibition observed with various treatments. Data are graphed as the means ± SEM (n = 3–7). *Different from 10 μm Oxo-M, p < 0.05, one-way ANOVA followed by a post hoc Bonferroni test. Representative experiments depicting the inhibition of dopamine overflow observed in slices from M5 KO mice upon addition of 1 μm Oxo-M (D) or 3 μm Oxo-M (E) in either the presence or absence of VU0238429. F, Averaged data depicting the normalized percentage inhibition observed with various treatments. Data are graphed as the means ± SEM (n = 4–5). Experiments were performed in slices prepared from either wild-type or M5 KO mice at P30–P90.
Discussion
Clinical studies with compounds that directly or indirectly modulate mAChRs have suggested that modulation of these receptors can be efficacious in treating cognitive, behavioral, and psychotic symptoms observed in numerous CNS disorders including schizophrenia, Alzheimer's disease, and PD (Eglen et al., 1999; Katzenschlager et al., 2003; Shekhar et al., 2008). Unfortunately, treatments that broadly modulate mAChR signaling are hampered by dose-limiting adverse effects that are thought to be mediated primarily by peripheral M2 and M3 receptors (Bymaster et al., 2003). To date, elucidating the physiological roles and contributions of individual mAChR subtypes to the clinical efficacy of nonselective mAChR modulators has been challenging due to the paucity of subtype-selective compounds. We now report that VU0238429 is a PAM with unprecedented selectivity for the rat M5 receptor. Previous reports suggested that some GPCR allosteric modulators display species selectivity (Chan et al., 2008; Suratman et al., 2011; Valant et al., 2012). We found that VU0238429 maintained M5 electivity across both human and rat mAChRs when these receptors were individually expressed in cell lines. In addition, VU0238429 had similar effects on DA neuron function in both mice and rats, suggesting that this compound is active at potentiating M5 across these species. Furthermore, the effects of VU0238429 were absent in M5 KO mice. However, although VU0238429 is highly selective for rat M5, further studies examining the subtype selectivity and probe dependence of VU0238429 across all subtypes of mouse mAChRs will be necessary to establish definitively its selectivity in this species. As with previously reported PAMs at other mAChR subtypes (Shirey et al., 2008; Ma et al., 2009; Shirey et al., 2009), VU0238429 does not display any affinity for the orthosteric binding site of the rM5 receptor and, in the assays examined, had little to no agonist activity at the rM5 receptor. Instead, VU0238429 acts at an allosteric site, where it potentiates activation of the receptor by a variety of orthosteric agonists (ACh, Oxo-M, and CCh) with a high degree of M5 selectivity, validating the utility of VU0238429 as a useful tool compound for studies in native tissue. The allosteric mechanism of action of VU0238429 has several advantages compared with traditional orthosteric agonists/antagonists. Because PAMs bind to allosteric sites on the receptor, and not to the highly conserved orthosteric binding site, these compounds have the potential to display unprecedented subtype selectivity. The mechanism of action of allosteric modulators, which necessitates the presence of endogenous agonist for receptor activation, may also provide therapeutic benefits by maintaining the temporal signaling pattern of cholinergic circuits in the brain. The activity dependence observed with VU0238429 is similar to that observed with the benzodiazepine class of GABAA modulators, which provide safe and effective treatment of sleep and anxiety disorders, an outcome not attained with directly acting GABAA agonists (Möhler et al., 2002).
Allosteric modulators can potentiate receptor signaling by increasing the affinity and/or efficacy of orthosteric agonists. In functional assays, we found that 10 μm VU0238429 left-shifted the dose–response curve of ACh by 21-fold, whereas in radioligand-binding assays, the same concentration of VU0238429 induced an 8-fold leftward-shift in the ACh affinity at M5. This suggests that although VU0238429 increases the affinity of the M5 receptor for ACh, the magnitude of this change is not sufficient to fully account for the change in potency observed in functional assays. These findings are similar to those previously reported with the M4 PAM VU10010 and, in both cases, the mechanism of action is likely bimodal involving both an increase in agonist affinity and an increase in the efficiency with which the agonist-bound receptor couples to downstream signaling pathways (Shirey et al., 2008).
The M5 receptor is the only mAChR detectable in midbrain dopamine neurons (Weiner et al., 1990). This, coupled with the restricted expression pattern of the M5 receptor, has made it an attractive target with which to selectively modulate dopaminergic function. The M5 gene has been linked to schizophrenia susceptibility in humans (De Luca et al., 2004) and M5 KO mice display blunted responses to drugs of abuse (Basile et al., 2002; Thomsen et al., 2005) and deficits in prepulse inhibition (Thomsen et al., 2007), all findings consistent with the M5 receptor playing an important role in regulating dopaminergic system function. The present studies using a novel subtype-selective M5 PAM in brain slices from both rats and mice provide evidence that M5 directly modulates dopamine neurons. The similarity in results obtained in both young rats (P15–P19) and more mature mice (P30–P90) suggests that M5-mediated modulation of dopamine neuron function is not unique to a single species or developmental stage.
Midbrain dopamine neurons receive two distinct cholinergic inputs from the hindbrain and forebrain. Neurons in the pedunculopontine nuclei (PPN) and the laterodorsal tegmental nuclei of the brainstem innervate dopamine neurons dendrosomatically, whereas interneurons in the striatum or nucleus accumbens innervate proximal dopamine terminals (Mena-Segovia et al., 2008; Oldenburg and Ding, 2011). Electrical stimulation of the PPN induces long-lasting increases in striatal dopamine release in vivo, an effect that is absent in M5 KO mice (Steidl et al., 2011). Similar studies have demonstrated that PPN-evoked striatal dopamine release can be inhibited by local infusion of either mAChR or nAChR antagonists in the SNc (Lester et al., 2010). These previous studies, together with the present results, provide strong evidence that stimulation of PPN cholinergic inputs leads to excitatory effects on SNc dopamine neurons via activation of somatodendrically located M5 receptors, which ultimately induce striatal dopamine release.
In addition to cholinergic inputs from the PPN, dopaminergic transmission is also strongly influenced by cholinergic interneurons located in the striatum. Cholinergic inputs from the hindbrain do not project to the striatum, suggesting that striatal interneurons provide the sole source of cholinergic innervation to striatal neurons and dopaminergic terminals. It has been well established that application of the nonselective mAChR agonist Oxo-M in the striatum can completely inhibit dopamine release induced by proximal electrical stimulation. However, the contribution of various mAChR subtypes to this effect has been of considerable debate (for review, see Rice et al., 2011). Our finding that Oxo-M not only retains efficacy in attenuating dopamine release in M5 KO mice, but actually is more efficacious, is consistent with previous reports (Bendor et al., 2010) suggesting that numerous mAChR subtypes other than M5 play important roles in regulating striatal dopamine release. However, interpreting studies from genetically modified animals can be complicated by developmental and compensatory changes. It has been reported that mice expressing a mutant M5 receptor overexpress the D2 dopamine receptor subtype in the striatum, a key regulator of dopamine release (Wang et al., 2004). The discovery of the M5-selective modulator VU0238429 allowed us to examine directly the role of M5 receptors in nongenetically modified mice and rats. In both mice and rats, VU0238429 significantly potentiated the inhibition of electrically evoked dopamine overflow observed with submaximal concentrations of Oxo-M. The effects of VU0238429 were completely absent when experiments were performed either in the presence of the mAChR antagonist scopolamine or in M5 KO mice, suggesting that VU0238429 is mediating its effects via the M5 receptor. These unexpected results suggest that, in wild-type mice and rats, activation of M5 leads to an inhibition in striatal dopamine release. Because M5 has been detected on dopamine neurons and not on other striatal neurons, these effects are presumably mediated by presynaptically localized M5 receptors. However, we cannot rule out the possibility that M5 could be located on other neurons in the striatum. Further studies will be needed to elucidate the precise mechanism(s) whereby M5 activation results in a reduction of evoked dopamine release. In addition, VU0239429 was only effective at modulating Oxo-M-mediated reductions in dopamine release in wild-type mice at higher concentrations of Oxo-M. These results, combined with the enhanced efficacy of Oxo-M in M5 KO mice, suggest that other mAChR subtypes may influence dopaminergic signaling to a greater extent than M5. Previous studies have demonstrated that Oxo-M-mediated inhibition of dopamine release has been reported to be absent in M2 KO and M4 KO mice (Threlfell et al., 2010), but not in M5 KO mice. Accordingly, although the results reported here suggest that striatal M5 activation reduces striatal dopamine signaling, it is likely that this modulation is less influential than signaling mediated via the M2/M4 subtypes.
This study represents a first step in elucidating the importance of M5 receptor activation in modulating dopaminergic transmission. Discovery of a highly selective M5 modulator has allowed the unprecedented opportunity to potentiate this receptor subtype selectively and determine the significance of M5 activation. Although the studies here have focused on the SNc/striatum, future studies will be necessary to determine whether M5 regulates the VTA/NAc in a similar manner. The finding that somatodendritic M5 activation can stimulate SNc dopamine neuron activity, whereas striatal M5 activation inhibits dopamine release from nigrostriatal synapses, suggests that M5 can have directly opposing effects depending on the receptor's localization. Although having one receptor subtype mediate directly opposing effects may seem contradictory, these two M5 receptor populations are activated by completely independent cholinergic inputs. It is likely that the comparative importance of these two cholinergic inputs to dopamine neuron function will shift as various mental processes are accentuated. Interestingly, a recent study demonstrated that selective ablation of forebrain or hindbrain cholinergic systems produced opposing effects on the locomotion of mice placed in a novel environment, effects that were normalized when both cholinergic systems were ablated. This suggests that these cholinergic systems may differentially regulate both striatal dopaminergic transmission and ultimately exploratory behaviors (Patel et al., 2012). To further elucidate the net consequence of M5 activation on dopaminergic function would require an M5-selective compound with suitable properties for in vivo dosing. Unfortunately, when administered systemically to rats, VU0238429 demonstrated poor exposure with low concentrations in the brain. In the past, our group has had great success in optimizing PAMs and current efforts are under way to identify an M5-selective compound with improved physiochemical properties. The discovery of such compounds will be critical for determining the impact of M5 receptor activation on dopaminergic function under both physiological and pathological conditions.
Footnotes
This work was supported by the National Institutes of Health (Grants R01 MH073676 and F32 MH095285), the Udall Center of Excellence for Parkinson's Disease Research (Grants P50 NS071669 and P50 NS038370), the JPB Foundation, and the Parkinson's Disease Foundation. We thank H. Plumley for technical support.
Over the past year, C.W.L. consulted for Abbott and P.J.C. consulted for Pfizer. C.W.L. and P.J.C. also receive research support that includes salary support from Bristol Myers Squibb and Astra Zeneca and are inventors on multiple composition of matter patents protecting allosteric modulators of GPCRs. The remaining authors declare no competing financial interests.
References
- Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, Yamada M, Wess J. Deletion of the M5 muscarinic acetylcholine receptor attenuates morphine reinforcement and withdrawal but not morphine analgesia. Proc Natl Acad Sci U S A. 2002;99:11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor J, Lizardi-Ortiz JE, Westphalen RI, Brandstetter M, Hemmings HC, Jr, Sulzer D, Flajolet M, Greengard P. AGAP1/AP-3-dependent endocytic recycling of M5 muscarinic receptors promotes dopamine release. EMBO J. 2010;29:2813–2826. doi: 10.1038/emboj.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner TI, Young AC, Brann MR, Buckley NJ. Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron. 1988;1:403–410. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- Bridges TM, Marlo JE, Niswender CM, Jones CK, Jadhav SB, Gentry PR, Plumley HC, Weaver CD, Conn PJ, Lindsley CW. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges TM, Kennedy JP, Cho HP, Breininger ML, Gentry PR, Hopkins CR, Conn PJ, Lindsley CW. Chemical lead optimization of a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) lead. Part I: development of the first highly selective M5 PAM. Bioorg Med Chem Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE, Nathanson NM, McKinzie DL, Felder CC. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci. 2003;17:1403–1410. doi: 10.1046/j.1460-9568.2003.02588.x. [DOI] [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJ, Bymaster FP, Felder CC. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci U S A. 2008;105:10978–10983. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca V, Wang H, Squassina A, Wong GW, Yeomans J, Kennedy JL. Linkage of M5 muscarinic and alpha7-nicotinic receptor genes on 15q13 to schizophrenia. Neuropsychobiology. 2004;50:124–127. doi: 10.1159/000079102. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Choppin A, Dillon MP, Hegde S. Muscarinic receptor ligands and their therapeutic potential. Curr Opin Chem Biol. 1999;3:426–432. doi: 10.1016/S1367-5931(99)80063-5. [DOI] [PubMed] [Google Scholar]
- Forster GL, Yeomans JS, Takeuchi J, Blaha CD. M5 muscarinic receptors are required for prolonged accumbal dopamine release after electrical stimulation of the pons in mice. J Neurosci. 2002;22:RC190. doi: 10.1523/JNEUROSCI.22-01-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DJ, Jones CK, Conn PJ. Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discov Med. 2012;14:413–420. [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenschlager R, Sampaio C, Costa J, Lees A. Anticholinergics for symptomatic management of Parkinson's disease. Cochrane Database Syst Rev. 2003;2:CD003735. doi: 10.1002/14651858.CD003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Calabresi P, North RA. Muscarine depolarizes rat substantia nigra zona compacta and ventral tegmental neurons in vitro through M1-like receptors. J Pharmacol Exp Ther. 1990;253:395–400. [PubMed] [Google Scholar]
- Lester DB, Rogers TD, Blaha CD. Acetylcholine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther. 2010;16:137–162. doi: 10.1111/j.1755-5949.2010.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CF, Themmen AP, Joho R, Barberis C, Birnbaumer M, Birnbaumer L. Molecular cloning and expression of a fifth muscarinic acetylcholine receptor. J Biol Chem. 1989;264:7328–7337. [PubMed] [Google Scholar]
- Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danziger A, Regan C, Flick R, Pascarella D, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Sur C, Kinney G, Seabrook GR, Ray WJ. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, Austin CA, Williams MB, Kim K, Williams R, Orton D, Brown HA, Lindsley CW, Weaver CD, Conn PJ. Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharmacol. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena-Segovia J, Winn P, Bolam JP. Cholinergic modulation of midbrain dopaminergic systems. Brain Res Rev. 2008;58:265–271. doi: 10.1016/j.brainresrev.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- Oldenburg IA, Ding JB. Cholinergic modulation of synaptic integration and dendritic excitability in the striatum. Curr Opin Neurobiol. 2011;21:425–432. doi: 10.1016/j.conb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JC, Rossignol E, Rice ME, Machold RP. Opposing regulation of dopaminergic activity and exploratory motor behavior by forebrain and brainstem cholinergic circuits. Nat Commun. 2012;3:1172. doi: 10.1038/ncomms2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D. Neuroscience. Ed 5. Sunderland, MA: Sinauer Associates; 2012. [Google Scholar]
- Rice ME, Patel JC, Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011;198:112–137. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Schmauss C, Sulzer D. Altered dopamine release and uptake kinetics in mice lacking D2 receptors. J Neurosci. 2002;22:8002–8009. doi: 10.1523/JNEUROSCI.22-18-08002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, Jadhav SB, Menon UN, Xiang Z, Watson ML, Christian EP, Doherty JJ, Quirk MC, Snyder DH, Lah JJ, Levey AI, Nicolle MM, Lindsley CW, Conn PJ. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidl S, Miller AD, Blaha CD, Yeomans JS. M(5) muscarinic receptors mediate striatal dopamine activation by ventral tegmental morphine and pedunculopontine stimulation in mice. PLoS One. 2011;6:e27538. doi: 10.1371/journal.pone.0027538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suratman S, Leach K, Sexton P, Felder C, Loiacono R, Christopoulos A. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol. 2011;162:1659–1670. doi: 10.1111/j.1476-5381.2010.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Woldbye DP, Wörtwein G, Fink-Jensen A, Wess J, Caine SB. Reduced cocaine self-administration in muscarinic M5 acetylcholine receptor-deficient mice. J Neurosci. 2005;25:8141–8149. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Wörtwein G, Fink-Jensen A, Woldbye DP, Wess J, Caine SB. Decreased prepulse inhibition and increased sensitivity to muscarinic, but not dopaminergic drugs in M5 muscarinic acetylcholine receptor knock-out mice. Psychopharmacology. 2007;192:97–110. doi: 10.1007/s00213-006-0682-y. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Clements MA, Khodai T, Pienaar IS, Exley R, Wess J, Cragg SJ. Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum. J Neurosci. 2010;30:3398–3408. doi: 10.1523/JNEUROSCI.5620-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Felder CC, Sexton PM, Christopoulos A. Probe dependence in the allosteric modulation of a G protein-coupled receptor: implications for detection and validation of allosteric ligand effects. Mol Pharmacol. 2012;81:41–52. doi: 10.1124/mol.111.074872. [DOI] [PubMed] [Google Scholar]
- Wang H, Ng K, Hayes D, Gao X, Forster G, Blaha C, Yeomans J. Decreased amphetamine-induced locomotion and improved latent inhibition in mice mutant for the M5 muscarinic receptor gene found in the human 15q schizophrenia region. Neuropsychopharmacology. 2004;29:2126–2139. doi: 10.1038/sj.npp.1300502. [DOI] [PubMed] [Google Scholar]
- Weiner DM, Levey AI, Brann MR. Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc Natl Acad Sci U S A. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Lamping KG, Duttaroy A, Zhang W, Cui Y, Bymaster FP, McKinzie DL, Felder CC, Deng CX, Faraci FM, Wess J. Cholinergic dilation of cerebral blood vessels is abolished in M(5) muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:14096–14101. doi: 10.1073/pnas.251542998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Development of antisera selective for m4 and m5 muscarinic cholinergic receptors: distribution of m4 and m5 receptors in rat brain. Mol Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]