

Abstract
Myelin membrane, which ensheaths axons, has an unusually high amount of cholesterol. Cholesterol influences membrane fluidity and assembles lipid-rich microdomains within membranes, and some studies have shown that cholesterol is important for myelination. How cholesterol influences the development and differentiation of oligodendrocytes, glial cells that make myelin, is not known nor is clear whether isoprenoids, which also are products of the cholesterol biosynthetic pathway, contribute to myelination. Through a forward genetic screen in zebrafish we discovered that mutation of hmgcs1, which encodes an enzyme necessary for isoprenoid and cholesterol synthesis, causes oligodendrocyte progenitor cells (OPCs) to migrate past their target axons and to fail to express myelin genes. Drawing on a combination of pharmacological inhibitor and rescue experiments, we provide evidence that isoprenoids and protein prenylation, but not cholesterol, are required in OPCs to halt their migration at target axons. On the other hand, cholesterol, but not isoprenoids, is necessary both for axon ensheathment and myelin gene expression. Our data reveal that different products of the cholesterol biosynthetic pathway have distinct roles in oligodendrocyte development and that they together help to coordinate directed migration, axon wrapping, and gene expression.
Keywords: cholesterol, isoprenoid, myelin, prenylation, zebrafish
Introduction
Cholesterol is an important component of cell membranes in animals, where it organizes lipid-rich microdomains and influences membrane fluidity and permeability. Cholesterol is obtained both exogenously from dietary sources, and endogenously from biochemical synthesis. During development, maternal cholesterol is the primary source of fetal cholesterol. However, cholesterol does not cross the blood–brain barrier (BBB), which forms between 12 and 18 weeks of human gestation, and therefore brain cholesterol must be synthesized by neural cells. Consequently, brain development following BBB formation, which includes axogenesis, synaptogenesis, and myelination, might be particularly sensitive to disruption of cholesterol synthesis. Accordingly, cholesterol deficiency has been linked to intellectual disability and autism (Tierney et al., 2006; Aneja and Tierney, 2008; Kanungo et al., 2013).
Cholesterol is synthesized by biochemical reactions that begin with acetyl CoA and acetoacetyl-CoA, which are hydrated to form 3-hydroxy-3-methylglutaryl CoA (Hmg-CoA). This reaction is catalyzed by Hmg-CoA synthase 1, encoded by the gene HMGCS1. Next, in the rate-limiting step for cholesterol synthesis, Hmg-CoA is reduced to mevalonate by Hmg-CoA reductase. Subsequent reactions convert mevalonate to farnesyl pyrophosphate, which is a substrate for divergent pathways that synthesize isoprenoids, used for protein prenylation, and cholesterol.
Neural cells in which cholesterol is likely to be particularly important include oligodendrocytes. Oligodendrocytes produce myelin, a highly specialized membrane that tightly ensheaths axons, providing electrical insulation and promoting rapid, saltatory conduction of nerve impulses. Oligodendrocyte progenitor cells (OPCs) arise from spatially restricted subpopulations of neural precursors during development and then migrate to their target axons. Subsequently, OPCs spirally wrap axons with long extensions of plasma membrane and then differentiate as mature oligodendrocytes by synthesizing the proteins and lipids that endow myelin membrane with its unique characteristics. One of the most prominent of these is cholesterol. Seventy percent of the dry weight of myelin consists of lipids and, of this, cholesterol contributes to >25% of the lipid content (Morell and Jurevics, 1996). Cholesterol associates with myelin proteins (Simons et al., 2000), suggesting that cholesterol contributes to the physical and physiological properties of myelin membrane. Additionally, conditional inactivation in oligodendrocytes of squalene synthase, an enzyme that converts farnesyl diphosphate to squalene subsequent to bifurcation of the isoprenoid and cholesterol synthesis pathways, caused hypomyelination (Saher et al., 2005), indicating that cholesterol is also important for the growth of myelin membrane.
Here we show that the cholesterol biosynthetic pathway is essential for oligodendrocyte development and myelination. Additionally, we define differential requirements for isoprenoid and cholesterol synthesis. From a forward genetic screen in zebrafish, we identified a mutation of hmgcs1, coding for Hmg-CoA synthase 1, which caused OPCs to migrate past their target axons and interfered with myelin gene expression. Using a combination of pharmacological inhibitor and rescue experiments, we found that isoprenoids, but not cholesterol, are required in OPCs to halt their migration at target axons. Conversely, cholesterol is required specifically for oligodendrocyte membrane to wrap axons, in addition to promoting robust myelin gene expression. Therefore, distinct products of the cholesterol biosynthesis pathway have differential functions in oligodendrocyte development.
Materials and Methods
Ethics statement.
The animal work in this study was approved by the Institutional Animal Care and Use Committees of Vanderbilt University and the University of Colorado School of Medicine.
Zebrafish lines and husbandry.
Embryos were raised at 28.5°C in egg water of embryo medium (EM) and staged according to hours postfertilization, days postfertilization, and morphological criteria (Kimmel et al., 1995). The hmgcs1vu57 mutation was uncovered in an ENU mutagenesis screen. Tg(olig2:EGFP)vu12 (Shin et al., 2003) and Tg(nkx2.2a:EGFP-CaaX)vu16 (Ng et al., 2005; Kirby et al., 2006), Tg(sox10:GAL4-VP16, cmlc2:Cerulean)co19, and Tg(4xnrUAS:EGFP-CaaX, cmlc2:EGFP)co18 (see Plasmid construction and generation of transgenic zebrafish, below) fish of either sex were used for this study.
Positional cloning of hmgcs1.
We created a mapping cross by mating vu57± fish, which were from the AB strain, to WIK strain fish and raising the progeny to adulthood. Twenty-four each of 4 d postfertilization (dpf) wild-type and vu57 mutant larvae were collected from crosses of identified vu57± map cross fish and mixed genomic DNA pools were prepared. By bulked segregant analysis using 223 simple sequence-length polymorphism markers, we linked the vu57 mutation to markers z13219, z11911, z22422, z13685, z25783, and z13632, located on chromosome 10. Testing individual mutants revealed that z13632 was most tightly linked. The entire coding region of hmgcs1 was sequenced from PCR products amplified in overlapping fragments from cDNA prepared from 4 dpf vu57 mutant and wild-type larvae.
In situ RNA hybridization and immunohistochemistry.
plp1a and mbp (Brösamle and Halpern, 2002) RNA probes were generated using digoxigenin RNA labeling kits (Roche). In situ RNA hybridization was performed as described previously (Hauptmann and Gerster, 2000). For immunohistochemistry, larvae were fixed using 4% paraformaldehyde, embedded, frozen, and sectioned using a cryostat microtome as previously described (Park and Appel, 2003). We used rabbit anti-Sox10 (1:1000; Park et al., 2005), mouse anti-Myc (1:1000, clone 9E10; Covance), mouse anti-acetylated Tubulin (1:5000, Sigma-Aldrich), and Ab-3A10 (1:500, Developmental Studies Hybridoma Bank) as primary antibodies. For fluorescent detection of antibody labeling, we used AlexaFluor 568 goat anti-rabbit and goat anti-mouse conjugates (1:200, Life Technologies). In situ hybridization images were collected using a QImaging Retiga Exi color CCD camera mounted on a compound microscope and imported into Adobe Photoshop. Image manipulations were limited to levels, curve and contrast adjustments. Fluorescence images were collected using a Zeiss Axiovert 200 microscope equipped with a PerkinElmer spinning disk confocal system and Volocity software (PerkinElmer) or a Zeiss LSM 780 confocal microscope and imported into Adobe Photoshop.
Quantitative PCR.
RNA was isolated from 10 to 15 pooled larvae for each control or experimental condition. RNA isolation for each experiment was performed in triplicate. Reverse transcription was performed using iScript Reverse Transcriptase Supermix (no. 170-8840, Bio-Rad Life Science). Real-time qPCR was performed in triplicate for each cDNA sample using an Applied Biosystems StepOne Plus machine and software version 2.1. Taqman gene expression assays were used to detect mpz (Dr03131917_m1), plp1a (Dr03433493_g1), 36k (Dr03438574_g1), mag (DR03102419_m1), and rpl13a (Dr03101115_g1) as an endogenous control. A custom designed assay to detect mbp consisted of the primers: mbp-“A” forward, 5′-GTTCTTCGGAGGAGACAAGAAGAG-3′; mbp-“A” reverse, 5′-GTCTCTGTGGAGAGGAGGATAGATGA-3′; mbp-“a” probe, 5′-AAGGGAAAGGGTTCATT-3′.
hmgcs1 rescue experiments.
psox10:hmgcs1-Myc was created by subcloning hmgcs1-Myc from pCS2-hmgcs1-Myc into pME, which was then used for recombination with p5E-sox10, p3E-polyA, and pDest-cmlc2-GFP using the Tol2 kit (Kwan et al., 2007). The resulting plasmid was injected into newly fertilized eggs in a solution containing 25 ng/μl plasmid, 0.4 m KCl and phenol red. Larvae were sorted GFP+ hearts, marked by the cmlc2-GFP reporter, fixed, sectioned using a cryostat microtome, and processed for immunohistochemistry as described above.
Drug inhibitor and rescue experiments.
Atorvastatin (Cayman Chemical Company), GGTI-2133 (Sigma-Aldrich), Lonafarnib (Cayman Chemical), and Ro 48-8071 (Cayman Chemical) were each dissolved in 100% DMSO at a concentration of 10 mm. Drugs were diluted in EM to make the following working concentrations: Atorvastatin, 2 μm; GGTI-2133, 10 μm; Lonafarnib, 10 μm; Ro 48-8071, 5 μm. Each drug had a final concentration of 0.2% DMSO and 0.2% DMSO in EM was used as a control solution. Drug treatments were initiated at 24 h postf and replaced with fresh drug every 24 h. Water soluble cholesterol (MP Biomedicals, Solon, Ohio) was dissolved in water at a concentration of 10 mg/ml and then diluted in water to a working concentration of 1 mg/ml. 2–3 nl of cholesterol was pressure injected into the yolk of 24 h postfertilization (hpf) embryos. Geranylgeraniol (Santa Cruz Biotechnology) was diluted in 100% DMSO to make a 1 m solution. 0.5–1 nl was pressure injected in the yolk of 24 hpf embryos.
Cholesterol assay.
Fish were collected at 4 dpf, weighed and pooled to equal 15 mg per sample (∼30 larvae). Samples were stored at −80°C before lysis. Samples were lysed in Cholorform:isopropanol:NP-40 (7:11:0.1) with a microhomogenizer. The homogenized tissue was centrifuged at 15,000 × g for 10 min. The resulting organic phase layer was air dried at 50°C and the remaining organic solvent was removed by putting samples under vacuum for 30 min. The resulting lipid pellets were resuspended in 1× Assay Diluent included in the Total Cholesterol Assay Kit (Colormetric; Cell BioLabs). Following the kit protocol, concentrations of cholesterol in samples were determined using a standard curve. Measurements were performed for three biological replicates per group.
Plasmid construction and generation of transgenic zebrafish.
pEXPR-Tol2-sox10:GAL4-VP16, cmlc2:Cerulean was created using one-way Gateway cloning of an entry plasmid containing the 7.2 kb genomic fragment of sox10 (Dutton et al., 2001) and pCH Gtwy G4VP16 (gift from Michael Nonet, Washington University, St. Louis, MO). pEXPR-Tol2-4xnrUAS:EGFP-CaaX, cmlc2:EGFP was created using multisite Gateway cloning (Kwan et al., 2007). To generate transgenic lines, we injected DNA together with transposase RNA into one-cell embryos. Injected fish were raised to adulthood, screened for EGFP or cerulean expression in the heart, and crossed to existing Gal4 or UAS lines to verify faithful expression. Tg(sox10:GAL4-VP16, cmlc2:Cerulean)co19 recapitulated the expression pattern of the previously established line Tg(sox10:mRFP; Kirby et al., 2006). We selected for mosaic expression in F1 and F2 generations of Tg(4xnrUAS:EGFP-CaaX, cmlc2:EGFP)co18, which is likely a consequence of position-effect variegation of UAS transgenes that has been previously described in zebrafish (Akitake et al., 2011).
Time-lapse imaging.
For analysis of axon wrapping, control and Ro 48-8071-treated Tg(nkx2.2a:mEGFP) larvae were anesthetized using 3-aminobenzoic acid ethyl ester (tricaine), immersed in 0.8% low melting temperature agarose, mounted on their sides, and covered with EM in glass bottomed 35 mm Petri dishes. Time-lapse images were captured using a 40× oil-immersion objective mounted on a motorized Zeiss Axiovert 200 microscope equipped with a PerkinElmer spinning disk confocal system and heated stage and chamber to maintain larvae at 28.5°C. Z-image stacks were collected every 5 min and 3D datasets compiled using Sorenson 3 video compression and exported using QuickTime to create movies.
Results
OPC migration and myelin gene expression require hmgcs1 function
In a screen for chemically induced mutations that disrupt oligodendrocyte development we identified mutants for one allele, designated as vu57, that were phenotypically indistinguishable from wild-type at 2 dpf but by 3 dpf had cardiac edema, reduced jaws, brain necrosis (Fig. 1A,B), and OPCs that appeared to occupy ectopic positions within dorsal spinal cord (Fig. 1C,D). To verify the latter observation, we labeled transverse sections of spinal cord with antibody to detect Sox10, which marks cells of the oligodendrocyte lineage (Park et al., 2005). In wild-type larvae, dorsally migrated OPCs settle at the position of the dorsal longitudinal fasciculus (DLF), a white matter fiber tract located at approximately two-thirds the distance from ventral to dorsal spinal cord (Fig. 1E). By contrast, all dorsally migrated OPCs occupied positions dorsal to the position of the DLF in vu57 mutant larvae (Fig. 1F). Quantification revealed that vu57 mutant larvae had a slight deficit of Sox10+ oligodendrocyte lineage cells at 3 dpf, but by 4 dpf the number of Sox10+ was similar to wild-type (Fig. 1G). To determine whether the over migration phenotype might result from absence of DLF axons, we labeled larvae with the antibody Ab-3A10, which binds to a neurofilament-associated antigen. No differences in longitudinal axon tracts were evident between wild-type and vu57 mutant larvae (Fig. 1H,I), indicating that the failure to arrest OPC migration is not a consequence of missing target axons. Finally, to confirm that OPCs were inappropriately positioned relative to their target axons we labeled fixed larvae with Ab-3A10 in combination with anti-GFP antibody. Although in wild-type larvae dorsal OPCs were positioned at the DLF, most OPCs in vu57 mutant larvae were located dorsal to the DLF (Fig. 1J,K).
Figure 1.

Dorsally migrated oligodendrocyte lineage cells occupy ectopic positions within spinal cords of vu57 mutant larvae. A, B, Images of living 3 dpf larvae. The homozygous vu57 mutant larva has cardiac edema (B, c), a small jaw (B, j), and opaque brain (B, b) indicative of dying cells. C, D, Confocal microscope images, viewed from the side, of trunk spinal cords of living 3 dpf larvae. Dorsal is up. olig2:EGFP expression marks cells occupying the pMN domain of ventral spinal cord and dorsally migrated OPCs and oligodendrocytes (arrows). The dashed lines marks the approximate position at which most dorsal OPCs settle in wild-type (C). In mutant larvae, some OPCs occupy ectopic positions in dorsal spinal cord (D). E, F, Transverse sections, dorsal up, at the level of the trunk spinal cord. Oligodendrocyte lineage cells (arrows) are marked by coexpression of olig2:EGFP (green) and Sox10 (red). Asterisks mark the position of the DLF. G, Graph showing the number of oligodendrocyte lineage cells at 3 and 4 dpf in wild-type and vu57 mutant larvae (n = 10 sections from each of 10 larvae for each time point and genotype; ***p = 0.0008, unpaired two-tailed Student's t test). n.s., Not significant. Error bars represent SEM. H, I, Images of 4 dpf wild-type and mutant larvae, viewed from lateral with dorsal up, focused on the trunk spinal cord. Ab-3A10 labeling reveals no differences in axons of the dorsal or ventral longitudinal fascicles (DLF, VLF) or motor nerves (mn). J, K, Lateral images of 4 dpf wild-type and mutant Tg(olig2:EGFP) larvae labeled with Ab-3A10 (red) and GFP (green) antibodies to detect axons and OPCs (arrows), respectively. C, D, H–K, Maximum projections of confocal z-stack optical sections. Scale bars, 10 μm.
To investigate whether the vu57 mutant allele also disrupts oligodendrocyte differentiation, we examined expression of myelin genes using in situ RNA hybridization. In wild-type, both hindbrain and spinal cord expressed plp1a and mbp prominently at early larval stages (Fig. 2A–D). By contrast, very little expression was evident in hindbrains or spinal cords of vu57 mutant larvae at comparable stages (Fig. 2E–H). We confirmed this observation using quantitative RT-PCR. Myelin gene RNA levels were consistently and substantially lower in vu57 mutant larvae than in wild-type larvae (Fig. 2I). These data together indicate that the gene disrupted by the vu57 mutation is required to guide dorsally migrating OPCs to their target axons and for myelin gene expression.
Figure 2.
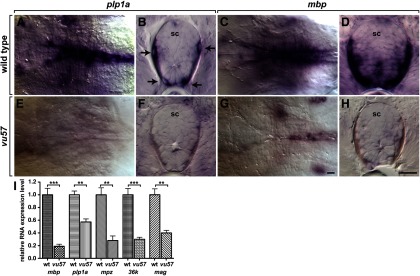
vu57 mutant larvae express abnormally low levels of myelin genes. Images show hindbrains, viewed from dorsal with anterior to the left (A, C, E, G) and transverse sections through spinal cord (sc) with dorsal up (B, D, F, H) of larvae processed for in situ RNA hybridization. Wild-type larvae express the myelin genes plp1a at 3 dpf (A, B) and mbp at 4 dpf (C, D). Arrows indicate oligodendrocytes expressing plp1a. Homozygous vu57 mutant larvae express little plp1a (E, F) or mbp (G, H). I, Graph showing myelin gene RNA expression levels in wild-type and vu57 mutant larvae measured by quantitative RT-PCR (n = 4 biological replicates consisting of 10–15 larvae for each measurement; **p < 0.005, ***p < 0.0005, unpaired two-tailed Student's t test). Error bars represent SEM. Scale bars, 20 μm.
Using bulked segregant analysis with simple sequence length polymorphism markers (Knapik et al., 1998) we mapped the vu57 allele to zebrafish chromosome 10. Subsequent fine mapping revealed that the mutation was closely linked to hmgcs1, which encodes Hmg-CoA synthase 1, an enzyme in the cholesterol and isoprenoid biosynthesis pathway. We considered hmgcs1 as a candidate for two reasons. First, previous work showed that cholesterol is rate-limiting for myelin membrane formation (Saher et al., 2005). Second, EGFP fused to a CaaX membrane localization motif, which requires prenylation for membrane association (Khosravi-Far et al., 1991), was improperly localized to the cytoplasm of oligodendrocytes in vu57 mutant larvae (see below). Sequencing of cDNA obtained from vu57 mutant larvae revealed a T to A transversion predicted to change histidine to glutamine at amino acid position 189 (H189Q, GenBank accession NP_957379; Fig. 3A) within a homodimerization domain identified by the Conserved Domain Database (Marchler-Bauer et al., 2013). Total cholesterol was reduced ∼50% in 4 dpf vu57 mutant larvae relative to wild-type larvae (n = 3 biological replicates consisting of 20–30 larvae per sample; p < 0.05, unpaired parametric t test), consistent with the possibility that the vu57 mutant allele disrupts Hmg-CoA synthase 1 function and diminishes cholesterol synthesis.
Figure 3.

The vu57 allele is a mutation of hmgcs1. A, Amino acid sequences of the conserved homodimerization domain of Hmgcs1 proteins (gray text). The vu57 allele changes a histidine to glutamine within the homodimerization domain. B–E, Lateral views, with dorsal up, of trunk spinal cords of living 3 dpf larvae. Dorsally migrated OPCs (arrows) occupy positions close to the DLF (dotted line) of wild-type (B). OPCs migrate past the DLF in vu57 mutant larva (C) and vu57 mutant larva injected with mutant hmgcs1 RNA (D) but are close to the DLF in mutant larvae injected with wild-type hmgcs1 RNA (E). F–M, Images of hindbrains viewed from dorsal with anterior to the left, of larvae processed for in situ RNA hybridization. Wild-type larva expresses plp1a (F), whereas a vu57 mutant larva (G) and a vu57 mutant larva injected with mutant hmgcs1 (H) express plp1a only at very low levels. By contrast, a vu57 mutant larvae injected with wild-type hmgcs1 RNA expresses plp1a at levels apparently equivalent to wild-type (I). Compared with wild-type (J), a vu57 mutant larva (K) and a vu57 mutant larva injected with mutant hmgcs1 RNA (L) express little mbp, whereas mbp expression is at high level in a vu57 mutant larva injected with wild-type hmgcs1 RNA (M). Scale bars, 20 μm.
To validate vu57 as a mutation of hmgcs1, we performed rescue experiments by injecting synthetic mRNA into newly fertilized eggs. By comparison to wild-type, in which dorsally migrating OPCs consistently occupied positions near the DLF (Fig. 3B), and to uninjected vu57 mutant controls, in which OPCs migrated past the DLF (Fig. 3C), OPCs occupied ectopic positions in dorsal spinal cords of homozygous vu57 mutant larvae injected with hmgcs1 mRNA engineered to have the predicted vu57 mutant allele (n = 27; Fig. 3D). By contrast, OPCs occupied their normal positions near the DLF in homozygous vu57 mutant larvae injected with wild-type hmgcs1 mRNA (n = 32; Fig. 3E) indicating that wild-type hmgcs1 product can rescue the OPC migration defect of vu57 mutants. Similarly, mutant hmgcs1 mRNA had no effect on plp1a (n = 17; Fig. 3H) or mbp (n = 24; Fig. 3L) expression in mutant larvae whereas wild-type hmgcs1 mRNA strongly rescued plp1a (n = 14; Fig. 3I) and mbp (n = 27; Fig. 3M) expression. We conclude that the vu57 allele is a mutation of hmgcs1 and hereafter refer to it as hmgcs1vu57.
Hmg-CoA synthase 1 catalyzes formation of Hmg-CoA from acetoacetyl-CoA and acetyl-CoA (Fig. 4A). The next enzyme in the pathway, Hmg-CoA reductase, is rate limiting for cholesterol biosynthesis and the target of inhibitory statins (Fig. 4A), used to reduce cholesterol levels. To confirm the requirement for the cholesterol biosynthesis pathway for OPC migration and myelin gene expression, we treated embryos with atorvastatin. Treatment precisely phenocopied both the OPC migration defect (n = 35; Fig. 4B–D), and the plp1a (n = 31) and mbp (n = 17) expression deficit of hmgcs1vu57 mutant larvae (Fig. 4E–K). We conclude that the cholesterol biosynthetic pathway is necessary for OPC migration and myelin gene expression.
Figure 4.

Inhibition of Hmg-CoA reductase phenocopies hmgcs1vu57. A, The cholesterol biosynthetic pathway. Inhibitors used for this study are shown in red. B–D, Lateral views, with dorsal up, of trunk spinal cords of living 3 dpf larvae. Dorsally migrated OPCs occupy positions close to the DLF (dotted line) of wild-type (B). OPCs migrate past the DLF in vu57 mutant larva (C) and a wild-type larvae treated with Atorvastatin (D). E–J, Images of hindbrains viewed from dorsal with anterior to the left, of larvae processed for in situ RNA hybridization. By contrast to wild-type (E) and similar to a hmgcs1vu57 mutant larva (F), a wild-type larva treated with Atorvastatin expresses little plp1a RNA (G). Atorvastatin also blocks mbp expression (H–J). K, Graph showing mbp RNA expression levels in 4 dpf larvae measured by quantitative RT-PCR. DMSO-treated control larvae express mbp at slightly lower level than untreated wild-type. Atorvastatin-treated wild-type larvae express mbp at levels similar to hmgcs1vu57 mutant larvae (n = 4 biological replicates consisting of 10–15 larvae for each measurement; *p = 0.0101, ***p < 0.0005, unpaired two-tailed Student's t test). Error bars represent SEM.
Isoprenoid but not cholesterol biosynthesis is necessary to arrest OPC migration at axon targets in dorsal spinal cord
The cholesterol and isoprenoid biosynthetic pathways diverge downstream of farnesyl pyrophosphate (Fig. 4A). Because protein prenylation is required for germ cell migration in fruit flies and zebrafish (Thorpe et al., 2004; Van Doren et al., 1998) we hypothesized that prenylation regulates OPC migration. To test this hypothesis, we first manipulated the isoprenoid pathway. GGTI-2133 (Vasudevan et al., 1999) and Lonafarnib (Liu et al., 1998) are inhibitors of geranylgeranyl transferase I and farnesyl transferase, respectively (Fig. 4A). Both GGTI-2133 (n = 48) and Lonafarnib (n = 45) phenocopied the OPC migration defect of hmgcs1vu57 mutant larvae (Fig. 5A–D). Furthermore, injection of geranylgeraniol, which can promote protein prenylation by conversion to geranylgeranyl diphosphate (Crick et al., 1994) into hmgcs1vu57 mutant larvae rescued the OPC migration defect (n = 17; Fig. 5E). These data indicate that protein prenylation is necessary for targeted migration of OPCs to dorsal spinal cord axons.
Figure 5.

Protein prenylation is required to target dorsally migrating OPCs to the DLF. A–J, Lateral views, with dorsal up, of trunk spinal cords of living 3 dpf larvae. Whereas dorsal OPCs (arrows) cluster near the DLF (dotted line) in wild-type (A), OPCs occupy ectopic position in dorsal spinal cord of a hmgcs1vu57 mutant larva (B), a wild-type larva treated with the geranylgeranyl transferase inhibitor GGTI-2133 (C), and a wild-type larva treated with the farnesyl transferase inhibitor Lonafarnib (D). OPCs occupy normal positions near the DLF in a hmgcs1vu57 mutant larva injected with ggol (E). F–J, Images of larvae carrying the Tg(nkx2.2a:EGFP-CaaX) reporter. In wild-type (F), nascent myelin membrane on dorsal, longitudinal axons is revealed by membrane-tethered EGFP (brackets). In a hmgcs1vu57 mutant larva (G) no membrane localization is evident and EGFP appears cytosolic in OPCs (arrows). Membrane localization of EGFP appears normal in a GGTI-2133 treated larvae (H) but absent in a Lonafarnib treated larva (I). Ggol injection into a hmgcs1vu57 mutant larva rescues membrane localization (J). K–T, Images of hindbrains viewed from dorsal with anterior to the left, of larvae processed for in situ RNA hybridization. Similar to wild-type (K) and by contrast to a hmgcs1vu57 mutant (L), wild-type larvae treated with GGTI-2133 (M) and Lonafarnib (N) express plp1a at high level. Ggol injection also rescues plp1a expression in a hmgcs1vu57 mutant larva (O). mbp expression appears similar to wild-type control in inhibitor-treated larvae (P–S) and is also rescued in a hmgcs1vu57 mutant larva by ggol injection (T). U, Graph showing mbp RNA expression levels in 4 dpf larvae measured by quantitative RT-PCR. mbp expression is not affected by GGTI-2133 but is reduced ∼50% by Lonafarnib (n = 4 biological replicates consisting of 10–15 larvae for each measurement; ***p < 0.0005, unpaired two-tailed Student's t test). Error bars represent SEM. Scale bars, 20 μm.
As an additional assessment of isoprenoid formation, we examined localization of EGFP fused to the CaaX peptide, which targets proteins to plasma membrane in a prenylation-dependent manner. In wild-type larvae carrying the Tg(nkx2.2a:EGFP-CaaX) transgene, which marks oligodendrocytes (Kirby et al., 2006), membrane-targeted EGFP reveals nascent myelin membrane ensheathing dorsal spinal cord axons (Fig. 5F). By contrast, EGFP appeared to be localized to the cytosol of oligodendrocytes in hmgcs1vu57 mutant larvae (Fig. 5G), providing additional evidence that isoprenoid synthesis and protein prenylation are deficient in the absence of hmgcs1 function. Notably, whereas EGFP membrane localization appeared to be normal in larvae treated with GGTI-2133 (Fig. 5H), membrane localization was absent in larvae treated with Lonafarnib (Fig. 5I) indicating a specific requirement for farnesylation. As with OPC migration, injection of geranylgeraniol rescued the EGFP membrane localization defect of hmgcs1vu57 mutant larvae (n = 11; Fig. 5J).
We next investigated whether the isoprenoid branch of the cholesterol biosynthetic pathway is necessary for myelin gene expression. Neither GGTI-2133 (n = 29) nor Lonafarnib (n = 16) blocked plp1a expression (Fig. 5K–N). However, hmgcs1vu57 mutant larvae injected with geranylgeraniol expressed plp1a (n = 13) (Fig. 5O), indicating that prenylated proteins might contribute to mechanisms that promote myelin gene expression. Similarly, mbp expression was not obviously inhibited by GGTI-2133 (n = 26) or Lonafarnib (n = 21; Fig. 5P–S) but was rescued in hmgcs1vu57 mutant larvae by geranylgeraniol injection (n = 11; Fig. 5T). To investigate more carefully whether protein prenylation promotes myelin gene expression, we measured mbp RNA levels by quantitative RT-PCR. Whereas mbp transcript levels were similar in control and GGTI-2133 treated larvae, Lonafarnib reduced mbp levels ∼50% (Fig. 5U). Together, these data provide evidence that both geranylgeranylated and farnesylated proteins are necessary for OPC migration to dorsal axons and that farnesylated proteins promote myelination.
In principle, prenylation could be required within OPCs for them to recognize their target axons or within their target axons. To discriminate between these possibilities, we injected plasmid encoding Hmgcs1 fused to the Myc epitope driven by sox10 regulatory DNA into newly fertilized eggs produced by intercrosses of hmgcs1vu57/+;Tg(olig2:EGFP) carriers. We then scored the distribution of olig2:EGFP+ Myc+ cells within transverse sections of spinal cord obtained from 3 dpf larvae. In wild-type larvae, Myc+ OPCs occupied their normal positions in ventral spinal cord and near the DLF (Fig. 6A,C). In hmgcs1vu57 mutant larvae, Myc− OPCs occupied dorsal, medial spinal cord. By contrast, dorsal Myc+ OPCs clustered exclusively at their normal positions near the DLF (Fig. 6B,D). Therefore, hmgcs1 expression in OPCs is sufficient to guide them to their dorsal axon targets, indicating that prenylated proteins expressed by OPCs contribute to directed migration.
Figure 6.

OPC-specific expression rescues the OPC migration defect of hmgcs1vu57 mutant larvae. A, B, Representative images of transverse sections, dorsal up, through trunk spinal cord, showing olig2:EGFP expression (green) and Myc labeling (red). A, Transient expression of sox10:hmgcs1-Myc in a wild-type larva marks an OPC (arrow) that occupies its normal spinal cord position near the DLF (asterisks). B, Transient expression of sox10:hmgcs1-Myc in a hmgcs1vu57 mutant larva marks OPCs that occupy their normal positions. C, Compilation of Myc+ and Myc− OPC distributions in wild-type larvae. Dorsal Myc+ and Myc− OPCs occupy similar positions. For simplicity, ventral wild-type OPCs are not included in the schematic. D, Compilation of Myc+ and Myc− OPC distributions in hmgcs1vu57 mutant larvae. Scale bar, 10 μm.
Cholesterol is required for myelin gene expression and axon wrapping but not OPC migration
The preceding data show that loss of isoprenoid synthesis does not account for the near absence of myelin gene expression in hmgcs1vu57 mutant larvae, thereby implicating a requirement for cholesterol synthesis in gene expression but not migration. Consistent with this, OPCs migrated normally in wild-type larvae treated with Ro 48-8071 (Fig. 7A–C), which inhibits oxidosqualene cyclase (Morand et al., 1997), thereby blocking formation of lanosterol, a necessary intermediate in cholesterol synthesis. Additionally, injection of soluble cholesterol into hmgcs1vu57 mutant embryos (n = 29) failed to rescue the OPC migration defect (Fig. 7D). Therefore, cholesterol is not required for OPCs to stop migration at their target axons.
Figure 7.

Cholesterol is required for myelin gene expression but not OPC migration. A–D, Lateral views, with dorsal up, of trunk spinal cords of living 3 dpf larvae. Similar to wild-type (A) and by contrast to hmgcs1vu57 (B), OPCs (arrows) occupy their normal positions near the DLF (dashed line) in a wild-type larva treated with oxidosqualene cyclase inhibitor Ro 48-8071 (C). OPCs occupied ectopic positions in dorsal spinal cord of a hmgcs1vu57 mutant larva injected with cholesterol (D). E–L, Images of hindbrains viewed from dorsal with anterior to the left, of larvae processed for in situ RNA hybridization. Ro 48-8071 treatment of a wild-type larva phenocopies the deficit of plp1a expression in a hmgcs1vu57 mutant larva (E–G). By contrast, a hmgcs1vu57 mutant larva injected with cholesterol expresses an apparently normal level of plp1a (H). mbp expression is similarly blocked by Ro 48-8071 and rescued in a hmgcs1vu57 mutant larva by cholesterol injection (I–L). Scale bars, 20 μm.
Notably, Ro 48-8071 treatment of wild-type embryos (n = 24) reduced plp1a expression to levels similar to hmgcs1vu57 mutants (Fig. 7E–G), indicating that cholesterol is essential for myelin gene expression. Consistent with this, injection of soluble cholesterol into hmgcs1vu57 mutant larvae produced a strong rescue of plp1a expression (n = 14; Fig. 7H). Similarly, inhibition of cholesterol synthesis reduced mbp expression (n = 14; Fig. 7I–K) and cholesterol injection into mutant larvae produced a moderate to strong rescue of mbp expression (n = 72; Fig. 7L). Together, these data provide strong evidence that cholesterol is required for robust myelin gene expression but not OPC migration.
Cholesterol is highly enriched within myelin membrane ensheathing axons (Muse et al., 2001; Dietschy, 2009). Whether cholesterol is necessary for oligodendrocyte membrane to ensheath axons is not known. To investigate this, we treated Tg(nkx2.2a:EGFP-CaaX) embryos with Ro 48-8071 from 24 to 65 hpf and then performed time-lapse imaging. In control larvae (n = 8), oligodendrocytes formed nascent myelin sheaths on axons in the DLF (Fig. 8A). In R0 48-8071 treated larvae (n = 8), OPCs migrated normally and continuously extended and retracted long membrane processes, similar to the behavior of OPCs before axon ensheathment and myelination (Kirby et al., 2006). However, OPC processes failed to wrap axons (Fig. 8B). To validate this finding, we repeated the drug treatment on larvae having the transgenic combination Tg(sox10:GAL4-VP16, cmlc2:Cerulean);Tg(4xnrUAS:EGFP-CaaX, cmlc2:EGFP), which expresses membrane-tethered EGFP in a subset of oligodendrocytes, thereby facilitating quantification of axon wrapping. In control larvae, oligodendrocytes formed numerous compact internodes on axons (Fig. 8C). By contrast, oligodendrocytes in Ro 48-8071 treated larvae had many highly branched membrane processes but ensheathed few axons (Fig. 8D). Quantification confirmed this, showing that whereas control oligodendrocytes formed an average of ∼12 internodes, oligodendrocytes in Ro 48-8071-treated larvae averaged only ∼1 internode per cell (n = 31 cells in 13 control larvae and 43 cells in 20 Ro 48–8071 treated larvae; p = 0.0001, unpaired two-tailed Mann–Whitney test). Finally, we investigated whether cholesterol injection could rescue the axon wrapping hmgcs1 mutant larvae. In transverse sections of wild-type larvae carrying the Tg(nkx2.2a:EGFP-CaaX) transgene, axon wrapping was evident as green circles encompassing acetylated-Tubulin+ axons (Fig. 8E,E′). Although oligodendrocyte processes were evident in hmgcs1 mutant larvae, no axons were wrapped (Fig. 8F,F′). By contrast, axon wrapping was apparent in mutant larvae injected with cholesterol (Fig. 8G,G′). Similarly, imaging of living larvae revealed no evidence of axon ensheathment by oligodendrocyte processes in hmgcs1 mutant larvae (Fig. 8H), whereas cholesterol injection restored ensheathment (Fig. 8I). We conclude that cholesterol is necessary for axon wrapping in addition to myelin gene expression.
Figure 8.

Cholesterol promotes axon ensheathment by myelin membrane. A, B, Images captured from time-lapse movies of Tg(nkx2.2a:EGFP-CAAX) larvae focused on the trunk spinal cord, viewed from lateral with dorsal up. Developmental time is indicated on the left. Several myelinating oligodendrocytes (A1–A3) are evident in the control larva. At 65 hpf, nascent myelin membrane has formed on axons and myelin sheaths lengthen over time. In the Ro 48-8071 treated larva, numerous processes extending from oligodendrocytes (B4, B5) are evident but do not initiate sheath formation (B). Oligodendrocytes are numbered B4–B6. C, D, Representative images of Tg(sox10:GAL4-VP16, cmlc2:Cerulean);Tg(4xnrUAS:EGFP-CaaX, cmlc2:EGFP) larvae focused on the trunk spinal cord, viewed from lateral with dorsal up. C, Image of a single oligodendrocyte (asterisk) with numerous internodes in a control-treated larva. D, Image of a single oligodendrocyte in a Ro 48-8071 treated larva with numerous membrane processes but no internodes. (E–G) Transverse sections through trunk spinal cord with dorsal up. Oligodendrocytes express nkx2.2a:EGFP-CaaX (green) and axons are marked by anti-acetylated Tubulin staining (red). E–G, Insets are magnified in E′–G′. Axons wrapped by oligodendrocyte membrane in wild-type and cholesterol injected hmgcs1 mutant larvae are indicated by arrows. H, I, Images of living larvae, viewed from the side. Oligodendrocytes are marked by nkx2.2a:EGFP-CaaX expression. Cholesterol injection rescues axon wrapping. Scale bars, 10 μm.
Discussion
Development of myelinating oligodendrocytes entails distinct steps including specification of OPCs from neural precursors, OPC division, OPC migration to target axons, ensheathment of axons by oligodendrocyte membrane, and myelin synthesis (Miller, 2002; Emery, 2010). These steps must be precisely coordinated, for example, myelin synthesis occurs only after initiation of axon ensheathment, but the molecular mechanisms that guide the progression of oligodendrocyte lineage cells from birth to myelination are poorly understood. Here we show that products of the cholesterol biosynthetic pathway are essential for discrete features of oligodendrocyte development.
OPCs are produced by spatially restricted subpopulations of neural precursors during development and must therefore migrate to their target axons in white matter. Implicit in this behavior is the ability to stop migration upon reaching their targets. Migration of OPCs to ectopic positions in dorsal spinal cord of hmgcs1vu57 mutant zebrafish larvae indicates that mutants lack either a stop signal or the ability to respond to one. Our pharmacological inhibitor and rescue experiments firmly establish that halting OPC migration requires protein prenylation, and not cholesterol, in OPCs.
Among prenylated proteins with known roles in cell migration are Ras-like GTPases (Seabra, 1998; Charest and Firtel, 2007). Because membrane association is necessary for GTPase activity, a failure of prenylation would result in an absence of Ras-like GTPase function and potentially disrupt OPC migration. We note that whereas inhibition of either farnesylation or geranylgeranylation could phenocopy the OPC migration defect, injection of geranylgeraniol into hmgcs1 mutants could rescue OPC migration. One possible explanation is that Ras proteins can be geranylgeranylated in the absence of farnesylation (Whyte et al., 1997), and this might rescue their functions. Other potentially relevant targets of prenylation are G-protein γ subunits (Muntz et al., 1992), which mediate signaling by G-protein coupled receptors (GPCR). Notably, GPCR function has been implicated in halting OPC migration in mice. Mouse OPCs express the chemokine receptor Cxcr2, which is a GPCR, and oligodendrocytes were disproportionately enriched at the periphery of the spinal cord in Cxcr2 mutant animals (Tsai et al., 2002), consistent with a failure to arrest migration of OPCs at their normal positions. This phenotype is remarkably similar to that of prenylation-deficient zebrafish described in this paper. Therefore, we propose that the principal targets of prenylation that are required to arrest OPC migration are G-protein γ subunits, relaying signals from GPCRs activated by chemokines encountered within white matter.
Mutation of hmgcs1 also reduced myelin gene expression to very low levels, even in ventral spinal cord where oligodendrocytes occupied their normal positions. Our pharmacological inhibitor and rescue experiments show that cholesterol is required for robust myelin gene expression. Previous investigation of the role of cholesterol in myelination used mice in which squalene synthase activity, which is necessary for cholesterol synthesis, was specifically removed in oligodendrocytes (Saher et al., 2005). Some CNS regions of these mice were severely hypomyelinated, whereas other regions appeared to have normal myelin. Oligodendrocytes accumulated cholesterol, despite the inability to synthesize it, indicating that they were able to acquire it from another source, probably astrocytes. hmgcs1 mutant zebrafish lack the ability to synthesize any cholesterol and their early development is likely enabled by maternal deposits, which we assume become depleted by 3 dpf. Therefore, our data showing that hmgcs1 mutant larvae are almost entirely deficient for expression of myelin genes confirm that cholesterol is essential for oligodendrocyte differentiation and myelination.
Remarkably, myelin integrity and node structure appeared normal in mice lacking squalene synthase in oligodendrocytes (Saher et al., 2005), indicating that axon wrapping occurred normally. By contrast, our in vivo imaging revealed that OPCs failed to wrap axons in zebrafish larvae treated with an inhibitor of cholesterol synthesis. We speculate that this distinction also results from relative differences in cholesterol levels available to OPCs in these mouse and zebrafish models. Whereas cholesterol is essential for axon wrapping in zebrafish, small amounts of cholesterol acquired from astrocytes might enable squalene synthase-deficient OPCs to wrap axons in mice.
How might cholesterol facilitate axon wrapping and myelin gene expression? Cholesterol organizes lipid microdomains, sometimes called lipid rafts, within plasma membranes (Lingwood and Simons, 2010). These microdomains are platforms for molecules that can regulate the cytoskeleton and signal transduction pathways (Simons and Toomre, 2000), including those that promote myelination. For example, Fyn kinase, a member of the Src-family of nonreceptor tyrosine kinases, is myristoylated and palmitoylated, which targets it to lipid microdomains. Fyn-deficient mice have a deficit of myelinated axons (Umemori et al., 1994; Sperber and McMorris, 2001) and Fyn activity can recruit cytoskeletal elements to oligodendrocyte processes (Klein et al., 2002). Therefore, failure to localize Fyn might help explain the myelination failure of hmgcs1 mutant zebrafish. However, recent in vivo analysis in zebrafish indicated that Fyn activity positively influences the number of myelin segments an oligodendrocyte makes on axons but is not required for axon wrapping (Czopka et al., 2013). Thus, loss of Fyn localization is not sufficient to account for the lack of axon wrapping in cholesterol deficient zebrafish.
Cholesterol-rich microdomains might also promote assembly of signaling complexes that drive myelin gene expression. For example, receptor tyrosine kinases can translocate to membrane microdomains upon activation, where they interact with other molecules to initiate signal transduction (Romanelli and Wood, 2008). Among RTKs found localized to microdomains in various cell types are some that promote oligodendrocyte differentiation and myelination, including PDGFR-α and ErbB (Nagy et al., 2002; Decker and ffrench-Constant, 2004). Given the extremely low level of myelin gene expression in hmgcs1 mutant larvae, we speculate that cholesterol-rich microdomains consolidate multiple signaling complexes that function together to stimulate robust myelination.
Sterol metabolism disorders, resulting from mutations of genes that encode enzymes of the cholesterol biosynthetic pathway, are associated with an array of neurodevelopmental disorders (Kanungo et al., 2013). For example, Smith-Lemli-Opitz Syndrome (SLOS), which results from disruption of 7-dehydrocholesterol reductase, is associated with brain structural defects, intellectual disability and autism spectrum disorder (Kelley and Hennekam, 2000; Tierney et al., 2006). Although white matter abnormalities have been noted in some SLOS patients (Lee et al., 2013), the nature of the defect is not understood nor is it clear whether it results from a deficit of cholesterol or an increase in its precursor, 7-dehydrocholesterol. Statins interfere with oligodendrocyte differentiation, myelination, and remyelination in cell culture and rodent models (Klopfleisch et al., 2008; Maier et al., 2009; Miron et al., 2009; Smolders et al., 2010), but whether these defects result from deficits of isoprenoids, cholesterol, or both has not been known. By establishing that different features of oligodendrocyte development have distinct requirements for isoprenoids and cholesterol, our work should help clarify the brain abnormalities resulting from sterol metabolism defects and illuminate specific risks associated with statin therapy.
Footnotes
This work was supported by NIH Grant RO1 NS046668 to B.A., National Multiple Sclerosis Society Postdoctoral Fellowship (FG 2024-A-1) and National Institutes of Health T32 MH015442 Fellowship to J.H.H., and a gift from the Gates Frontiers Fund. The University of Colorado Anschutz Medical Campus Zebrafish Core Facility is supported by NIH Grant P30 NS048154. The Ab-3A10 antibody, developed by T.M. Jessell, J Dodd, and S. Benner-Morton, was obtained from the Developmental Studies Hybridoma Band developed under the auspices of the National Institutes of Child Health and Human Development and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, Iowa. We thank members of the Appel laboratory for discussions, and Mary Goll, Michael Nonet, and Chi-Bin Chien for Gateway plasmids.
The authors declare no competing financial interests.
References
- Akitake CM, Macurak M, Halpern ME, Goll MG. Transgenerational analysis of transcriptional silencing in zebrafish. Dev Biol. 2011;352:191–201. doi: 10.1016/j.ydbio.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneja A, Tierney E. Autism: the role of cholesterol in treatment. Int Rev Psychiatry. 2008;20:165–170. doi: 10.1080/09540260801889062. [DOI] [PubMed] [Google Scholar]
- Brösamle C, Halpern ME. Characterization of myelination in the developing zebrafish. Glia. 2002;39:47–57. doi: 10.1002/glia.10088. [DOI] [PubMed] [Google Scholar]
- Charest PG, Firtel RA. Big roles for small GTPases in the control of directed cell movement. Biochem J. 2007;401:377–390. doi: 10.1042/BJ20061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick DC, Waechter CJ, Andres DA. Utilization of geranylgeraniol for protein isoprenylation in C6 glial cells. Biochem Biophys Res Commun. 1994;205:955–961. doi: 10.1006/bbrc.1994.2758. [DOI] [PubMed] [Google Scholar]
- Czopka T, Ffrench-Constant C, Lyons DA. Individual oligodendrocytes have only a few hours in which to generate new myelin sheaths in vivo. Dev Cell. 2013;25:599–609. doi: 10.1016/j.devcel.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker L, ffrench-Constant C. Lipid rafts and integrin activation regulate oligodendrocyte survival. J Neurosci. 2004;24:3816–3825. doi: 10.1523/JNEUROSCI.5725-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM. Biological chemistry: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390:287–293. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton KA, Pauliny A, Lopes SS, Elworthy S, Carney TJ, Rauch J, Geisler R, Haffter P, Kelsh RN. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development. 2001;128:4113–4125. doi: 10.1242/dev.128.21.4113. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Hauptmann G, Gerster T. Multicolor whole-mount in situ hybridization. Methods Mol Biol. 2000;137:139–148. doi: 10.1385/1-59259-066-7:139. [DOI] [PubMed] [Google Scholar]
- Kanungo S, Soares N, He M, Steiner RD. Sterol metabolism disorders and neurodevelopment-an update. Dev Disabil Res Rev. 2013;17:197–210. doi: 10.1002/ddrr.1114. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi-Far R, Lutz RJ, Cox AD, Conroy L, Bourne JR, Sinensky M, Balch WE, Buss JE, Der CJ. Isoprenoid modification of rab proteins terminating in CC or CXC motifs. Proc Natl Acad Sci U S A. 1991;88:6264–6268. doi: 10.1073/pnas.88.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kirby BB, Takada N, Latimer AJ, Shin J, Carney TJ, Kelsh RN, Appel B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat Neurosci. 2006;9:1506–1511. doi: 10.1038/nn1803. [DOI] [PubMed] [Google Scholar]
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;22:698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfleisch S, Merkler D, Schmitz M, Klöppner S, Schedensack M, Jeserich G, Althaus HH, Brück W. Negative impact of statins on oligodendrocytes and myelin formation in vitro and in vivo. J Neurosci. 2008;28:13609–13614. doi: 10.1523/JNEUROSCI.2765-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapik EW, Goodman A, Ekker M, Chevrette M, Delgado J, Neuhauss S, Shimoda N, Driever W, Fishman MC, Jacob HJ. A microsatellite genetic linkage map for zebrafish (Danio rerio) Nat Genet. 1998;18:338–343. doi: 10.1038/ng0498-338. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lee RW, Conley SK, Gropman A, Porter FD, Baker EH. Brain magnetic resonance imaging findings in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2013;161:2407–2419. doi: 10.1002/ajmg.a.36096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Lipari P, Malkowski M, Ferrari E, Nielsen L, Prioli N, Dell J, Sinha D, Syed J, Korfmacher WA, Nomeir AA, Lin CC, Wang L, Taveras AG, Doll RJ, Njoroge FG, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998;58:4947–4956. [PubMed] [Google Scholar]
- Maier O, De Jonge J, Nomden A, Hoekstra D, Baron W. Lovastatin induces the formation of abnormal myelin-like membrane sheets in primary oligodendrocytes. Glia. 2009;57:402–413. doi: 10.1002/glia.20769. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41:D348–52. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67:451–467. doi: 10.1016/S0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Miron VE, Zehntner SP, Kuhlmann T, Ludwin SK, Owens T, Kennedy TE, Bedell BJ, Antel JP. Statin therapy inhibits remyelination in the central nervous system. Am J Pathol. 2009;174:1880–1890. doi: 10.2353/ajpath.2009.080947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morand OH, Aebi JD, Dehmlow H, Ji YH, Gains N, Lengsfeld H, Himber J. Ro 48–8.071, a new 2,3-oxidosqualene:lanosterol cyclase inhibitor lowering plasma cholesterol in hamsters, squirrel monkeys, and minipigs: comparison to simvastatin. J Lipid Res. 1997;38:373–390. [PubMed] [Google Scholar]
- Morell P, Jurevics H. Origin of cholesterol in myelin. Neurochem Res. 1996;21:463–470. doi: 10.1007/BF02527711. [DOI] [PubMed] [Google Scholar]
- Muntz KH, Sternweis PC, Gilman AG, Mumby SM. Influence of gamma subunit prenylation on association of guanine nucleotide-binding regulatory proteins with membranes. Mol Biol Cell. 1992;3:49–61. doi: 10.1091/mbc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse ED, Jurevics H, Toews AD, Matsushima GK, Morell P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J Neurochem. 2001;76:77–86. doi: 10.1046/j.1471-4159.2001.00015.x. [DOI] [PubMed] [Google Scholar]
- Nagy P, Vereb G, Sebestyén Z, Horváth G, Lockett SJ, Damjanovich S, Park JW, Jovin TM, Szöllosi J. Lipid rafts and the local density of ErbB proteins influence the biological role of homo- and heteroassociations of ErbB2. J Cell Sci. 2002;115:4251–4262. doi: 10.1242/jcs.00118. [DOI] [PubMed] [Google Scholar]
- Ng AN, de Jong-Curtain TA, Mawdsley DJ, White SJ, Shin J, Appel B, Dong PD, Stainier DY, Heath JK. Formation of the digestive system in zebrafish: III. Intestinal epithelium morphogenesis. Dev Biol. 2005;286:114–135. doi: 10.1016/j.ydbio.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Park HC, Appel B. Delta-Notch signaling regulates oligodendrocyte specification. Development. 2003;130:3747–3755. doi: 10.1242/dev.00576. [DOI] [PubMed] [Google Scholar]
- Park HC, Boyce J, Shin J, Appel B. Oligodendrocyte specification in zebrafish requires notch-regulated cyclin-dependent kinase inhibitor function. J Neurosci. 2005;25:6836–6844. doi: 10.1523/JNEUROSCI.0981-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanelli RJ, Wood TL. Directing traffic in neural cells: determinants of receptor tyrosine kinase localization and cellular responses. J Neurochem. 2008;105:2055–2068. doi: 10.1111/j.1471-4159.2008.05263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Seabra MC. Membrane association and targeting of prenylated Ras-like GTPases. Cell Signal. 1998;10:167–172. doi: 10.1016/S0898-6568(97)00120-4. [DOI] [PubMed] [Google Scholar]
- Shin J, Park HC, Topczewska JM, Mawdsley DJ, Appel B. Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci. 2003;25:7–14. doi: 10.1023/B:MICS.0000006847.09037.3a. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simons M, Krämer EM, Thiele C, Stoffel W, Trotter J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J Cell Biol. 2000;151:143–154. doi: 10.1083/jcb.151.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders I, Smets I, Maier O, vandeVen M, Steels P, Ameloot M. Simvastatin interferes with process outgrowth and branching of oligodendrocytes. J Neurosci Res. 2010;88:3361–3375. doi: 10.1002/jnr.22490. [DOI] [PubMed] [Google Scholar]
- Sperber BR, McMorris FA. Fyn tyrosine kinase regulates oligodendroglial cell development but is not required for morphological differentiation of oligodendrocytes. J Neurosci Res. 2001;63:303–312. doi: 10.1002/1097-4547(20010215)63:4<303::AID-JNR1024>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Thorpe JL, Doitsidou M, Ho SY, Raz E, Farber SA. Germ cell migration in zebrafish is dependent on HMGCoA reductase activity and prenylation. Dev Cell. 2004;6:295–302. doi: 10.1016/S1534-5807(04)00032-2. [DOI] [PubMed] [Google Scholar]
- Tierney E, Bukelis I, Thompson RE, Ahmed K, Aneja A, Kratz L, Kelley RI. Abnormalities of cholesterol metabolism in autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:666–668. doi: 10.1002/ajmg.b.30368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Frost E, To V, Robinson S, Ffrench-Constant C, Geertman R, Ransohoff RM, Miller RH. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110:373–383. doi: 10.1016/S0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- Umemori H, Sato S, Yagi T, Aizawa S, Yamamoto T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature. 1994;367:572–576. doi: 10.1038/367572a0. [DOI] [PubMed] [Google Scholar]
- Van Doren M, Broihier HT, Moore LA, Lehmann R. HMG-CoA reductase guides migrating primordial germ cells. Nature. 1998;396:466–469. doi: 10.1038/24871. [DOI] [PubMed] [Google Scholar]
- Vasudevan A, Qian Y, Vogt A, Blaskovich MA, Ohkanda J, Sebti SM, Hamilton AD. Potent, highly selective, and non-thiol inhibitors of protein geranylgeranyltransferase-I. J Med Chem. 1999;42:1333–1340. doi: 10.1021/jm9900873. [DOI] [PubMed] [Google Scholar]
- Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]