

Abstract
Kidney uncoupling protein 2 (UCP-2) increases in streptozotocin-induced diabetes, resulting in mitochondria uncoupling, i.e., increased oxygen consumption unrelated to active transport. The present study aimed to investigate the role of UCP-2 for normal and diabetic kidney function utilizing small interference RNA (siRNA) to reduce protein expression. Diabetic animals had increased glomerular filtration rate and kidney oxygen consumption, resulting in decreased oxygen tension and transported sodium per consumed oxygen. UCP-2 protein levels decreased 2 and 50% after UCP-2 siRNA administration in control and diabetic animals respectively. Kidney function was unaffected by in vivo siRNA-mediated gene silencing of UCP-2. The reason for the lack of effect of reducing UCP-2 is presently unknown but may involve compensatory mitochondrial uncoupling by the adenosine nucleotide transporter.
Keywords: Diabetes, Kidney, Mitochondria, siRNA, Uncoupling protein 2
1 Introduction
Diabetic nephropathy is a common complication in diabetes mellitus and often results in the need for dialysis and/or kidney transplantation. In the year of 2030 the projected prevalence of diabetes mellitus worldwide is 366 million people [1] with 37% expected to develop kidney damage [2].
The mechanisms underlying diabetic nephropathy are presently unclear, but there is a connection between kidney dysfunction and alterations in the energy metabolism, increased oxidative stress, mitochondria dysfunction and increased oxygen consumption (QO2) [3–5]. The increased oxidative stress directly contributes to increased QO2 resulting in decreased oxygen tension (pO2) [4]. A major source of reactive oxygen species (ROS) is the mitochondria electron transport chain and normally 0.2% of total QO2 is due to production of superoxide radicals (O2.−) [6] but this increases in diabetes [ 7]. Cultured renal proximal tubular cells exposed to hyperglycemia display increased membrane potential together with increased production of O2.−. After the initial response, a decrease in the mitochondrial membrane potential, increased QO2 and decreased O2.− production has been reported [8], indicating mitochondria uncoupling. Uncoupling proteins (UCP-2) releases protons independently of ATP-production and therefore decreases membrane potential and O2.− production [9, 10]. In diabetes, up-regulation of kidney UCP-2 results in mitochondria uncoupling which may contribute to increased QO2 [5, 11]. Therefore, we investigated the role of kidney UCP-2 in diabetes by utilizing in vivo gene silencing of UCP-2.
2 Materials and Methods
All animal procedures were approved by the Animal Care and Use Committee. Diabetes was induced in male Sprague Dawley rats (250–300 g, streptozotocin 65 mg/kg bw, tail vein). Blood glucose was measured using a reagent test strip (MediSense, Bedford, MA, USA) in a blood sample obtained from the cut tip of the tail and considered diabetic if it increased ≥15 mM within 24 h of injection and remained elevated. For administration of siRNA, a polyethylene catheter was placed in the carotid artery under isoflurane anesthesia (2% in 40% O2). A nonfunctional scrambled (scrl siRNA) or a siRNA directed against UCP-2 (id no. 50931, Ambion, Austin, TX, USA) was administered in a volume of 6 ml of 37 °C sterile saline during 6 s via a catheter in the carotid artery which was subsequently ligated. siRNA was administered at day 5 of diabetes and all experiments carried out 2 days thereafter.
Kidney cortexes were homogenized in RIPA buffer (1% tergitol type NP40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM NaF, 80 mM Tris, pH 7.5). Enzyme inhibitors (phosphatase inhibitor cocktail-2; 10 μl/ml, Sigma–Aldrich, St. Louis, MO, USA and Complete Mini; one tablet/1.5 ml; Roche Diagnostics, Mannheim, Germany) were added before each experiment. Molecular weight separation was performed on 12.5% Tris–HCl gels with Tris/glycine/SDS buffer, the proteins transferred to nitrocellulose membranes and detected with goat anti-rat UCP-2 (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and HRP-conjugated rabbit anti-goat, (1:10,000; Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA). β-actin was detected with mouse anti-rat β-actin antibody (1:10,000, Sigma–Aldrich, St Louis, MO, USA) and secondary HRP-conjugated goat-anti mouse antibody (1:60,000; Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA).
Animals were sedated with sodium thiobutabarbital (Inactin, 120 mg/kg bw non-diabetic animals, 80 mg/kg bw diabetic animals, i.p) and were placed on a heating pad that was servo-controlled at 37 °C. Tracheotomy was performed and polyethylene catheters were placed in the femoral artery and vein to allow monitoring of blood pressure (Statham P23dB, Statham Laboratories, Los Angeles, CA, USA), blood sampling and infusion of saline (5 ml/kg bw/h nondiabetic animals, 10 ml/kg bw/h diabetic animals). The left kidney was exposed by a subcostal flank incision and immobilized. The left ureter and bladder were catheterized to allow for timed urine sampling and urinary drainage, respectively. After surgery, the animal was allowed to recover for 40 min. Kidney pO2 was measured using Clark-type oxygen electrodes (Unisense, Aarhus, Denmark). Glomerular filtration rate (GFR) and renal blood flow (RBF) were measured by clearance of 14C-inulin and 3H-para-aminohippuric acid (185 kBq bolus followed by 185 kBq/kg bw/h, American Radiolabelled Chemicals, St. Louis, MO, USA). RBF was calculated assuming an extraction of 70%. GFR was calculated as inulin clearance = ([inulin]urine*urine flow)/[inulin]plasma and RBF with PAH-clearance adjusted for the hematocrit. Total kidney QO2 (μmol/min) was estimated from the arteriovenous difference in O2 con+ tent (O2ct = ([Hb]*oxygen saturation*1.34 + pO2*0.003)) *total RBF. Tubular Na+ transport (TNa+, μmol/min) was calculated as follows: TNa+ = [PNa+]*GFR-UNaV, where [PNa+] is plasma Na concentration and UNaV is the urinary Na + excretion. TNa+ per QO2 was calculated as TNa+/QO2. Statistical comparisons were performed using one-way analysis of variance with Bonferroni post hoc test were appropriate. p < 0.05 was considered significant and all values are mean ± SEM.
3 Results
Increased blood glucose and decreased weight were observed with diabetes. Weight loss was also observed after administration of siRNA (Table 30.1). UCP-2 was increased in diabetic rats compared to controls but decreased in both groups by UCP-2 siRNA (Fig. 30.1). Diabetics had increased GFR which was maintained after UCP-2 siRNA (Fig. 30.2). No differences in RBF were detected between any of the groups (data not shown). QO2 increased and TNa+/QO2 decreased in diabetic animals compared to controls (Figs. 30.3 and 30.4). Diabetic animals had decreased kidney pO2 compared to control animals which was unaffected by UCP-2 siRNA (Fig. 30.5). All parameters investigated were unaffected by scrambled siRNA.
Table 30.1.
Body weight and blood glucose
| Bw (g) | BG (mM) | |
|---|---|---|
| Control | 349 ± 7 | 4.8 ± 0.15 |
| Control + scrambled siRNA | 272 ± 7* | 4.9±0.35 |
| Control + UCP-2 siRNA | 303 ± 6* | 5.65±0.47 |
| Diabetes | 278±8* | 19.3±0.52* |
| Diabetes + scrambled siRNA | 296 ± 9* | 18.2±1.1* |
| Diabetes + UCP-2 siRNA | 308 ± 13* | 18.2±0.7* |
Denotes p < 0.05 vs. untreated control animals
Fig. 30.1.
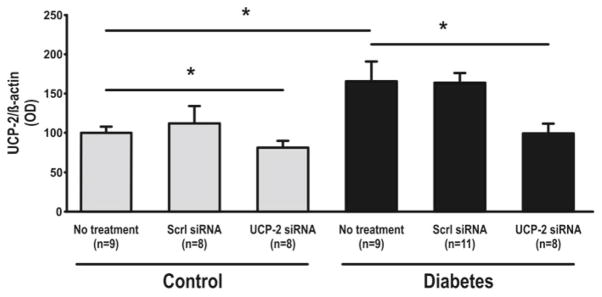
UCP-2 protein expression in kidney cortex. Scrl scrambled, asterisk denotes p<0.05
Fig. 30.2.
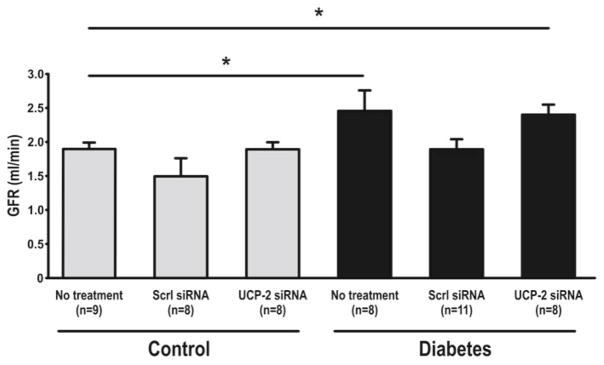
Glomerular filtration. Scrl scrambled, asterisk denotes p<0.05
Fig. 30.3.
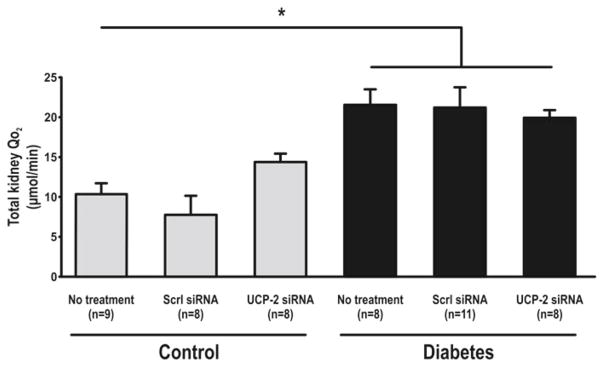
Total kidney oxygen consumption. Scrl scrambled, asterisk denotes p<0.05
Fig. 30.4.
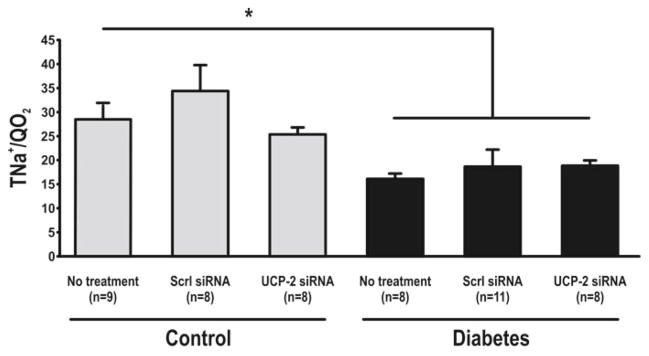
Transported sodium per consumed oxygen. Scrl scrambled, asterisk denotes p<0.05
Fig. 30.5.
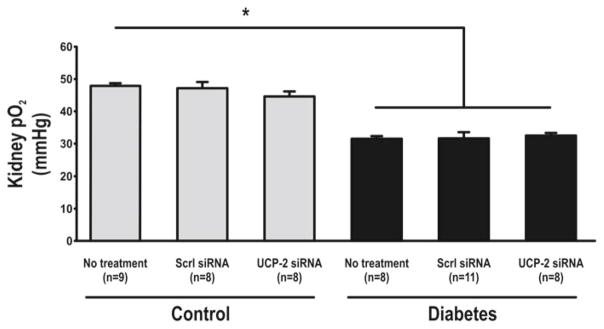
Oxygen tension in kidney cortex. Scrl scrambled, asterisk denotes p<0.05
4 Discussion
The present study demonstrates increased protein levels of UCP-2 in the diabetic rat kidney concomitant with increased total kidney QO2 and decreased TNa+/QO2. This isconsistent with the hypothesis that defective renal oxygenation in diabetes is mediated by enhanced expression of UCP-2. However, neither GFR, RBF, pO2, QO2 nor TNa+/QO2 were affected by selective siRNA-mediated gene silencing of UCP-2. There are several possible explanations. First, we only achieved a partial knockdown in UCP-2. It is possible that the residual UCP-2, possibly through increased activity, prevented a significant effect on in vivo kidney function. Second, it can be argued that diabetic nephropathy is a disease evolving during several years and 48 h knockdown of UCP-2 might not be enough. Third, possible compensatory up-regulation of other mitochondrial uncoupling mechanisms that were not investigated in the present study, but it is also possible the increased compensatory mitochondria uncoupling may have obscured an effect of reduced UCP-2 expression. Specifically, mitochondrial uncoupling can also be mediated by the adenosine nucleotide transporter.
5 Conclusion
The diabetic kidney had defective oxygenation and enhanced expression of UCP-2. siRNA-mediated knockdown of UCP-2 for 48 h in the diabetic kidney did not reverse that defect on kidney function in vivo. The role of UCP-2 and mitochondria uncoupling in diabetes needs to be further evaluated, preferentially using genetically modified mice.
Acknowledgments
MPF and FP were supported by a grant from the Swedish Research Council and the Swedish Diabetes Foundation whereas WJW and CSW were supported by a grant from the NIH (DK 49870; DK 36079; HL 68686).
Contributor Information
Malou Friederich Persson, Email: Malou.Friederich@mcb.uu.se, Division of Integrative Physiology, Department of Medical Cell Biology, Biomedical Center, Uppsala University, Box-571 75123 Uppsala, Sweden.
William J. Welch, Division of Nephrology and Hypertension, Department of Medicine, Medical Center, Georgetown University, Washington, DC, USA
Christopher S. Wilcox, Division of Nephrology and Hypertension, Department of Medicine, Medical Center, Georgetown University, Washington, DC, USA
Fredrik Palm, Division of Integrative Physiology, Department of Medical Cell Biology, Biomedical Center, Uppsala University, Box-571 75123 Uppsala, Sweden. Division of Nephrology and Hypertension, Department of Medicine, Medical Center, Georgetown University, Washington, DC, USA. Department of Medicine and Health Sciences, Linköping University, Linköping, Sweden.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Ostchega Y, Yoon S, Hughes J, Louis T. Hypertension awareness, treatment and control – continued disparities in adults: United States, 2005–2006. NCHS DATA Brief. 2008 http://www.cdc.gov/nchs/data/databriefs/db03. [PubMed]
- 3.Palm F. Intrarenal oxygen in diabetes and a possible link to diabetic nephropathy. Clin Exp Pharmacol Physiol. 2006;33:997–1001. doi: 10.1111/j.1440-1681.2006.04473.x. [DOI] [PubMed] [Google Scholar]
- 4.Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46:1153–1160. doi: 10.1007/s00125-003-1155-z. [DOI] [PubMed] [Google Scholar]
- 5.Friederich M, Fasching A, Hansell P, Nordquist L, Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. 2008;1777:935–940. doi: 10.1016/j.bbabio.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 6.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 7.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial super-oxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 8.Munusamy S, MacMillan-Crow LA. Mitochondrial superoxide plays a crucial role in the development of mitochondrial dysfunction during high glucose exposure in rat renal proximal tubular cells. Free Radic Biol Med. 2009;46:1149–1157. doi: 10.1016/j.freeradbiomed.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 10.Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. Faseb J. 1997;11:809–815. [PubMed] [Google Scholar]
- 11.Friederich M, Nordquist L, Olerud J, Johansson M, Hansell P, Palm F. Identification and distribution of uncoupling protein isoforms in the normal and diabetic kidney. Adv Exp Med Biol. 2009;645:205–212. doi: 10.1007/978-0-387-85998-9_32. [DOI] [PubMed] [Google Scholar]