

Abstract
Objective
To determine whether subjects of Puerto Rican heritage are at increased risk for a specific mutation of the proton-coupled folate transporter (PCFT) causing hereditary folate malabsorption (HFM).
Study design
Three percent of the births in Puerto Rico in 2005, with additional regional oversampling, were screened for the prevalence of the c.1082G>A; p.Y362_G389 del PCFT gene mutation. Six new subjects of Puerto Rican heritage with the clinical diagnosis of HFM were also assessed for this mutation.
Results
Six subjects of Puerto Rican heritage with the clinical diagnosis of HFM were all homozygous for the c.1082G>A; p.Y362_G389 del PCFT mutation. Three heterozygote carriers were identified from the 1582 newborn samples randomly selected from births in Puerto Rico in 2005. The carrier frequency for the mutated allele was 0.2% island-wide and 6.3% in Villalba.
Conclusion
These findings are consistent with a common mutation in the PCFT gene causing HFM that has disseminated to Puerto Ricans who have migrated to mainland United States. Because prompt diagnosis and treatment of infants with HFM can prevent the consequences of this disorder, newborn screening should be considered in high-risk populations and physicians should be aware of its prevalence in infants of Puerto Rican ancestry.
Hereditary folate malabsorption (HFM), a rare autosomal recessive disorder first described in 1961, arises in early infancy and usually presents with failure to thrive and anemia.1 Some subjects have hypogammaglobulinemia with immune deficiency and infections, such as Pneumocystis jirovecii. Developmental delays and a variety of neurologic defects are seen. Seizure disorders, particularly when diagnosis and treatment are delayed, can occur even many years after birth and can be refractory to treatment. 2-5 The major defects in HFM are impaired intestinal folate absorption and impaired transport of folates across the blood/choroid plexus/cerebrospinal fluid barrier. The molecular basis for HFM is loss-of-function mutations in the proton-coupled folate transporter gene (PCFT-SLC46A1), the primary transporter responsible for intestinal folate absorption.3,6 We have reported 10 unrelated families in which there has been at least one member with the clinical diagnosis of HFM and loss-of-function PCFT mutations.3,6-10 Three additional families have been reported by others.4,11,12
All reported mutations associated with HFM have been novel except for two families of Puerto Rican origin, one living in mainland United States.4,6 Both were homozygous for a c.1082G>A; p.Y362_G389del mutation (position 5882, GenBank accession number DQ496103), located in the splice acceptor of intron 2 (intron 2/exon 3 boundary), causing skipping of exon 3 and resulting in a splice variant (Figure 1, A) and a protein with decreased stability and impaired trafficking to the cell membrane.6 In this article we report on the identification of the same mutation in six additional unrelated families of Puerto Rican heritage along with the results of a random screen of 3% of the births on the island in 2005 along with oversampling in specific regions.
Figure 1.
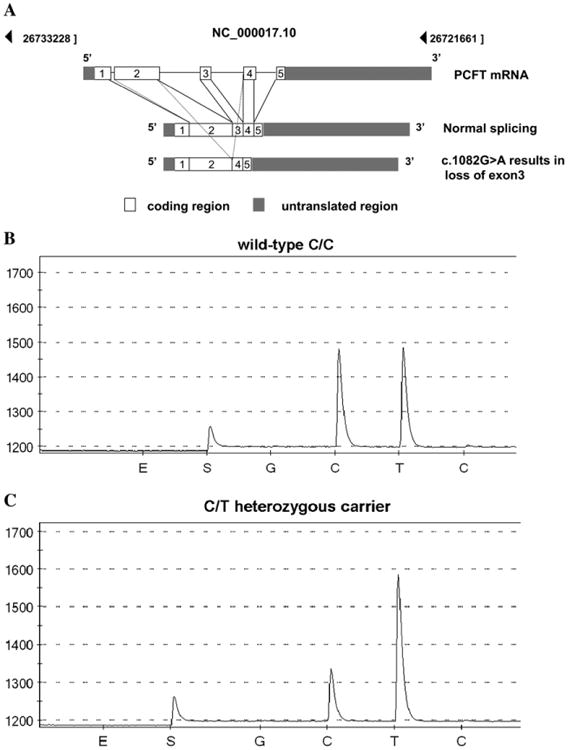
A, The genomic organization and splicing of the PCFT mRNA along with the loss of exon 3 resulting in a truncated PCFT mRNA in patients of Puerto Rican ancestry with HFM. B and C, Representative pyrograms show the wild-type CC sequence and the CT carrier sequence identified from the 1582 newborn samples randomly selected from one year's births (2005) in Puerto Rico. The pyrogram shows a nucleotide sequence around the intron/exon boundary. The y-axis indicates the light emission and the x-axis indicates nucleotide binding. E and S indicate enzyme and substrate, respectively, followed by a negative control base (G), the two peaks of interest (C and T), and a negative control base (C). For the C/T heterozygous mutation, a decrease in the height of the “C” peak and an increase in the height of the “T” peak are compared with the C/C wild-type sequence.
Methods
This study was approved by the Albert Einstein College of Medicine's Clinical Committee of Investigation and the University of Puerto Rico Medical Sciences Campus Institutional Review Board, in accordance with the Declaration of Helsinki. Informed consent was obtained as necessary from patients with the clinical diagnosis of HFM and their family members. Separate approval was obtained for the newborn screening component. These protocols have been approved yearly.
Identification of the PCFT Mutation in Subjects with a Clinical Diagnosis of HFM
Peripheral blood samples were collected, and genomic DNA was extracted with the Gentra Systems DNA purification kit (Gentra Systems, Inc, Minneapolis, Minnesota). The primers and conditions for polymerase chain reaction (PCR) were reported previously.6 PCFT genomic fragments that contain exons and flanking introns were purified on agarose gels and sequenced on an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, California) in the Albert Einstein Cancer Center Genomics Shared Resource.
Population Screening
Filter paper blood spots were received from the newborn screening program of the Puerto Rico Department of Public Health. Because the Puerto Rican patients reported previously were from the municipality of Villalba and a sector of the metropolitan San Juan area, an initial screening was performed of randomly selected regional samples that consisted of 12% of births in 2005 for the municipalities of Villalba (n = 48), Juncos (n = 66), Humacao (n = 105), and the Santurce sector of San Juan (n = 141). The two towns in Eastern Puerto Rico (Juncos and Humacao) were chosen for comparison, expecting those samples to be negative for the mutation if the allele frequency was highest in the regions where the patients were located (Villalba and Santurce). However, a carrier in the Juncos samples was also detected, thus a larger prevalence study was performed to determine the island-wide allele frequency and whether there were municipalities with a high allele frequency. For the island-wide study, random samples were collected consisting of 3% of the total births on the island in 2005 (n = 1582).
To obtain DNA from filter paper blood spots, sterile distilled water (100 μL) was added to each sample followed by heating to 95° C for 15 to 30 minutes; samples were vortexed every 2 minutes. The eluates were then spun in a centrifuge briefly at 13 200 g. PCR was performed with 5 μL of DNA template, AmpliTaqGold with GeneAmp (Applied Biosytems, Branchburg, New Jersey), and primers (biotinylated forward primer purified by high-performance liquid chromatography: [BioTEG]CCAGCCCCATTTTCCTGAT and reverse primer ACCAGCTTGGAGAGTTTAGCC). PCR conditions were as follows: cycle 1, 95° C for 5 minutes; cycle 2 (repeated 45 times), 95° C for 15 seconds, annealing temperature 55° C for 30 seconds, elongation temperature 72° C for 15 seconds; and cycle 3, 72° C for 5 minutes. PCR products were analyzed on a 2% agarose gel to confirm that a clear strong product (106 bp) was obtained. PCR products were processed at the Albert Einstein Cancer Center Genomics Shared Resource for analysis on the HS 96 Pyrosequencer (Qiagen, Valencia, California) (with pyrosequencing primer GAAAAGCAACCCATATC). All positive samples and a subset of negative samples were sent for validation and confirmation to the CLIA-licensed laboratory at the Division of Genetics, Newborn Screening Program, Biggs Laboratory, of the New York State Department of Health.
Validation by the Laboratory of Human Genetics, Newborn Screening Program, Genetic Testing Section, New York State Department of Health
Samples were amplified by PCR with intronic primers for exon 3, reported previously, with the addition of M13 sequencing tags.6 PCR was performed in a total reaction volume of 25 μL, containing 4 μL of DNA template from blood spot extractions or 2 ng of purified genomic DNA, 0.5 μmol of each forward and reverse primer, 1.5 μmol MgCl2, 0.2 μL Taq antibody (Clonetech 639251) and 2.5 μL Roche Hybridization 10× PCR Reaction Mix (Roche 2158825). PCR conditions were as follow: cycle 1 at 95° C for 5 minutes; cycle 2 (repeated 35 times), 95° C for 30 seconds, annealing temperature 63° C for 30 seconds, elongation temperature 72° C for 30 seconds; and cycle 3 at 72° C for 5 minutes. PCR products were separated by agarose gel electrophoresis to confirm amplification and purified with the ExoSap-IT reagent (USB 78205). Bidirectional sequencing was performed with the BigDye Terminator v3.0 cycle sequencing kit (Applied Biosystems), on an ABI 3130xl instrument. Sequence results were analyzed with SeqScape software (Applied Biosystems).
Results
Prevalence of the c.1082G>A; p.Y362_G389del Mutation among Patients with a Clinical Diagnosis of Hereditary Folate Malabsorption
The Table (available at www.jpeds.com) lists all published PCFT gene mutations along with their geographic areas of origin and location at the time of the current study. The Table includes two families of Puerto Rican heritage previously reported, along with six new unrelated cases confirmed in this report. Of 19 known affected families in which there is one member with both alleles of the PCFT gene mutated, eight are of Puerto Rican heritage; all are homozygous for the c.1082G>A; p.Y362_G389del mutation, which leads to the loss of exon 3 resulting in a truncated PCFT mRNA and a non functional protein (Figure 1, A).6
Table. Summary of published PCFT mutations in subjects with the clinical diagnosis of hereditary folate malabsorption.
| DNA Nucleotide Change | Protein amino acid change | Ethnicity | Location time of study |
|---|---|---|---|
| c.1082-1G>A6 | p.Tyr362_Gly389del | Puerto Rican | Puerto Rico |
| c.194delG3 | p.Gly65AlafsX25 | African American | Pennsylvania |
| c.337C>A3 | p.Arg113Ser | Turkish | Europe |
| c.439G>C3 | p.Gly147Arg | Caucasian | Europe |
| c.1126C>T | p.Arg376Trp | Mexican | Texas |
| c.954C>G3 | p.Ser318Arg | ||
| c.1274C>G3 | p.Pro425Arg | Arab | Israel |
| c.337C>T11 | p.Arg113Cys | Arab | Israel |
| c.197_198delGCinsAA (c.197GC>AA)7 | p.Cys66X | Portugese | New Jersey |
| c.1082-1G>A4 | p.Tyr362_Gly389del | Puerto Rican | Boston, Massachusetts |
| c.194dupG12 | p.Cys66LeufsX99 | Pakistani | London |
| c.1127G>A8 | p.Arg376Gln | Chinese | Australia |
| c.204 del CC9 | p. Asp68LysfsX94 | Turkish | Turkey |
| c.466G>T10 | p.Asp156Tyr | Pakistani | Canada |
| c.1082-1G>A | p.Tyr362_Gly389del | Puerto Rican | Puerto Rico |
| c.1082-1G>A | p.Tyr362_Gly389del | Puerto Rican | Rochester, New York |
| c.1082-1G>A | p.Tyr362_Gly389del | Puerto Rican | Worcester, Massachusetts |
| c.1082-1G>A | p.Tyr362_Gly389del | Puerto Rican | Puerto Rico |
| c.1082-1G>A | p.Tyr362_Gly389del | Puerto Rican | Puerto Rico |
| c.1082-1G>A | p.Tyr362_Gly389del | Puerto Rican | Puerto Rico |
Prevalence of the c.1082G>A; p.Y362_G389del Mutation
The initial regional oversampling (12% of births in 2005) revealed a carrier frequency of 6.3% in Villalba and 1.5% in Juncos; no carriers from Humacao or Santurce were identified. Three heterozygote carriers were identified from the 1582 newborn samples randomly selected from one year's births (2005) in Puerto Rico. Figure 1, B and C, shows a representative pyrosequencing assay. No homozygotes were identified. The carrier frequency for the mutated allele island-wide was 0.2%. The identified carriers were born in the towns of Ponce, Bayamon, and Villalba. Hence, although the highest allele frequency was found in the central municipality of Villalba, carriers were detected in municipalities in other parts of the island (Figure 2).
Figure 2.
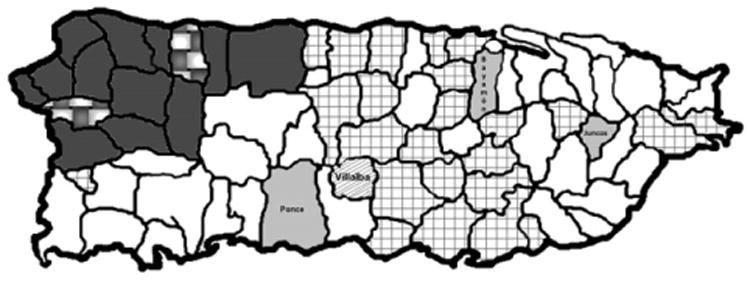
Map of the island of Puerto Rico indicating the regional distribution of common mutations that can cause rare autosomal recessive disorders. Municipalities of high carrier frequency (1.7% or higher) for the HPS3 3,904 bp gene deletion mutation are indicated with a grid pattern. Municipalities where the HPS1 gene 16 bp duplication is prevalent are indicated in dark grey; towns where the carrier frequencies for both the HPS1 and HPS3 founder mutations are high are indicated in a checkered pattern. Municipalities where carriers of the PCFT gene c.1082G>A mutation were detected are shown in light grey; the town of Villalba, where carriers of both the HPS3 gene deletion and the PCFT gene mutation have been identified, is indicated by the diagonal points.
Discussion
Among the earliest reports of HFM were children of Puerto Rican heritage and the molecular basis for HFM was established in a Puerto Rican family.2,6,13 The fact that all affected Puerto Rican families are homozygous for the same mutation is consistent with a common mutation on the island, with highest allele frequency in the municipality of Villalba. Additional genetic epidemiological studies will be necessary to confirm more definitively the site(s) of highest prevalence on the island. It is clear that this allele has disseminated and is now present in individuals of Puerto Rican heritage living in the United States.
Although c.1082G>A; p.Y362_G389del is the first mutation found in multiple subjects, in three instances two mutations have been detected at the same residues, as indicated in the Table. In a Turkish subject, a Ser was substituted for Arg1133; in an Israeli Arab, there was a Cys substitution at this residue.11 These mutations resulted in a loss of function of the carrier.3,11 In a Mexican subject with heterozygous mutations, Trp was substituted for Arg376 in one allele and, although expression of the protein at the cell surface was unchanged, there was a complete loss of function.3 On the other hand, in a Chinese subject, glutamine was substituted at this residue in both alleles. This resulted in only a modest reduction in protein expression at the cell surface and some retention of function that was substrate-dependent, but insufficient to maintain adequate folate absorption or folate transport across the choroid plexus.8 There have been multiple mutations within the first intron involving substitutions and deletions. Mutations at Cys66 resulted in a frame-shift9 and stop codon.7 This highly GC-rich region of the first exon (the GC content of residues 63 to 70 is 75%) is also the site of two other mutations, frame-shifts at Gly653 and Cys66.12
Other prevalent mutations associated with rare diseases have been identified among the population of Puerto Rico. Two separate founder mutations in the HPS1 (10q23) and HPS3 (3q24) genes associated with the Hermansky-Pudlak syndrome have been identified on the island.14-16 The p.G47D (11q14) mutation associated with Type I-A oculocutaneous albinism in Puerto Rico appeared to have originated from a common founder.17 In addition, the p.R457Q (11p11) substitution is a common mutation in Puerto Rico and is linked to the otherwise extremely rare genetic prothrombin deficiency disorder.18
On the basis of findings in this article, a case can be made that screening for the c.1082G>A mutation should be considered for high-risk regions of Puerto Rico, and infants of parents from this region living elsewhere, especially because diagnosis and treatment shortly after birth can prevent all the adverse consequences of this disorder. Of particular importance is the prevention of seizures, that can be very refractory to treatment, that occur when diagnosis and treatment are delayed.2,5
Acknowledgments
We thank Michele Caggana, ScD (Laboratory of Human Genetics, New York State Department), for supervision of the confirmatory analytical studies of blood specimens obtained in the newborn screen.
Funded by the St. Baldrick's Foundation, which had no role in the design and conduct of the study, in the collection, management, analysis, and interpretation of the data, nor in the preparation, review, or approval of the manuscript.
Glossary
- HFM
Hereditary folate malabsorption
- PCFT
Proton-Coupled Folate Transporter
- PCR
Polymerase chain reaction
Footnotes
The authors declare no conflict of interest.
References
- 1.Lubhy AL, Eagle FJ, Roth E, Cooperman JM. Relapsing megaloblastic anemia in an infant due to a specific defect in gastrointestinal absorption of folic acid. Am J Dis Child. 1961;102:482–3. [Google Scholar]
- 2.Geller J, Kronn D, Jayabose S, Sandoval C. Hereditary folate malabsorption: family report and review of the literature. Medicine (Baltimore) 2002;81:51–68. doi: 10.1097/00005792-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Zhao R, Min SH, Qiu A, Sakaris A, Goldberg GL, Sandoval C, et al. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood. 2007;110:1147–52. doi: 10.1182/blood-2007-02-077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borzutzky A, Crompton B, Bergmann AK, Giliani S, Baxi S, Martin M, et al. Reversible severe combined immunodeficiency phenotype secondary to a mutation of the proton-coupled folate transporter. Clin Immunol. 2009;133:287–94. doi: 10.1016/j.clim.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahadeo KM, Min SH, Diop-Bove N, Kronn D, Goldman ID. Hereditary Folate Malabsorption. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet] Seattle, WA: University of Washington, Seattle; 2010. [Google Scholar]
- 6.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917–28. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 7.Min SH, Oh Sy, Karp GI, Poncz M, Zhao R, Goldman ID. The clinical course and genetic defect in the PCFT in a 27-year-old woman with Hereditary folate malabsorption. J Pediatr. 2008;153:435–7. doi: 10.1016/j.jpeds.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahadeo K, Diop-Bove N, Shin D, Unal E, Teo J, Zhao R, et al. Properties of the Arg376 residue of the proton-coupled folate transporter (PCFT-SLC46A1) and a glutamine mutant causing hereditary folate malabsorption. Am J Physiol Cell Physiol. 2010;299:C1153–61. doi: 10.1152/ajpcell.00113.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atabay B, Turker M, Ozer EA, Mahadeo K, Diop-Bove N, Goldman ID. Mutation of the proton-coupled folate transporter gene (PCFT-SLC46A1) in Turkish siblings with hereditary folate malabsorption. Pediatr Hematol Oncol. 2010 Aug 26;27:614–9. doi: 10.3109/08880018.2010.481705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin DS, Min SH, Russell L, Zhao R, Fiser A, Goldman ID. Functional roles of aspartate residues of the proton-coupled folate transporter (PCFT; SLC46A1); a D156Y mutation causing hereditary folate malabsorption. Blood. 2010 Aug 30;116:5162–9. doi: 10.1182/blood-2010-06-291237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lasry I, Berman B, Straussberg R, Sofer Y, Bessler H, Sharkia M, et al. A novel loss of function mutation in the proton-coupled folate transporter from a patient with hereditary folate malabsorption reveals that Arg 113 is crucial for function. Blood. 2008 Jun 17;112:2055–61. doi: 10.1182/blood-2008-04-150276. [DOI] [PubMed] [Google Scholar]
- 12.Meyer E, Kurian MA, Pasha S, Trembath RC, Cole T, Maher ER. A novel PCFT gene mutation (p.Cys66LeufsX99) causing hereditary folate malabsorption. Mol Genet Metab. 2010 Mar;99:325–8. doi: 10.1016/j.ymgme.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santiago-Borrero PJ, Santini R, Jr, Perez-Santiago E, Maldonado N. Congenital isolated defect of folic acid absorption. J Pediatr. 1973 Mar;82:450–5. doi: 10.1016/s0022-3476(73)80119-2. [DOI] [PubMed] [Google Scholar]
- 14.Anikster Y, Huizing M, White J, Shevchenko YO, Fitzpatrick DL, Touchman JW, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet. 2001 Aug;28:376–80. doi: 10.1038/ng576. [DOI] [PubMed] [Google Scholar]
- 15.Oh J, Bailin T, Fukai K, Feng GH, Ho L, Mao JI, et al. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996 Nov;14:300–6. doi: 10.1038/ng1196-300. [DOI] [PubMed] [Google Scholar]
- 16.Torres-Serrant M, Ramirez SI, Cadilla CL, Ramos-Valencia G, Santiago-Borrero PJ. Newborn screening for hermansky-pudlak syndrome type 3 in Puerto Rico. J Pediatr Hematol Oncol. 2010 Aug;32:448–53. doi: 10.1097/MPH.0b013e3181e5e1f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oetting WS, Witkop CJ, Jr, Brown SA, Colomer R, Fryer JP, Bloom KE, et al. A frequent tyrosinase gene mutation associated with type I-A (tyrosinase-negative) oculocutaneous albinism in Puerto Rico. Am J Hum Genet. 1993 Jan;52:17–23. [PMC free article] [PubMed] [Google Scholar]
- 18.Lefkowitz JB, Weller A, Nuss R, Santiago-Borrero PJ, Brown DL, Ortiz IR. A common mutation, Arg457→Gln, links prothrombin deficiencies in the Puerto Rican population. J Thromb Haemost. 2003 Nov;1:2381–8. doi: 10.1046/j.1538-7836.2003.00420.x. [DOI] [PubMed] [Google Scholar]