

Abstract
Drug resistance is a principal concern in the treatment of quickly evolving diseases. The viral protease NS3/4A is a primary drug target for the hepatitis C virus (HCV) and is known to evolve resistance mutations in response to drug therapy. At the molecular level, drug resistance reflects a subtle change in the balance of molecular recognition by NS3/4A; the drug resistant protease variants are no longer effectively inhibited by the competitive active site inhibitors but can still process the natural substrates with enough efficiency for viral survival. In previous works we have developed the “substrate envelope” hypothesis, which posits that inhibitors should be less susceptible to drug resistance if they better mimic the natural substrate molecular recognition features. In this work, we perform molecular dynamics simulations on four native substrates bound to NS3/4A and discover a clearly conserved dynamic substrate envelope. We show that the most severe drug resistance mutations in NS3/4A occur at residues that are outside the substrate envelope. Comparative analysis of three NS3/4A inhibitors reveals structural and dynamic characteristics of inhibitors that could lead to resistance. We also suggest inhibitor modifications to improve resistance profiles based on the dynamic substrate envelope. This study provides a general framework for guiding the development of novel inhibitors that will be more robust against resistance by mimicking the static and dynamic binding characteristics of natural substrates.
Introduction
Drug resistance presents a great challenge in treating quickly evolving diseases, including the hepatitis C viral (HCV) infection. Direct-acting anti-viral agents specifically target the viral enzymes, inhibiting viral replication and eventually the disease progress. Two inhibitors targeting HCV NS3/4A protease (telaprevir and boceprevir) are approved by the US Food and Drug Administration (FDA) for treatment of chronic HCV genotype 1 infection in combination with pegylated interferon and ribavirin. Both telaprevir and boceprevir are peptidomimetic small-molecule inhibitors that associate with NS3/4A through a reversible covalent linkage to the catalytic serine (S139) as well as short-range molecular interactions with the binding site. The narrow selectivity of the two FDA-approved drugs to a single genotype renders them susceptible to drug resistance and creates need for inhibitors with broader selectivity profiles.
Several non-covalent NS3/4A inhibitors, including macrocyclic compounds, are at various stages of clinical development. These inhibitors contain a macrocycle connecting either the P1 and P3 groups (ITMN-191; danoprevir1) or alternatively the P2 and P4 groups (MK-51722 and MK-7009; vaniprevir3). These compounds are more potent than telaprevir and boceprevir against wild-type virus. Nevertheless, HCV quickly evolves due to the high replication rate combined with the lack of proofreading in the viral RNA-dependent RNA polymerase. Under the selective pressure of drug therapy, resistant variants are rapidly populated even at early stages of clinical trials, compromising the high efficacy of protease inhibitors and eventually restricting their usage to treatment-naïve patients for a limited period of time.4–6
Ideally, the molecular recognition aspects of drug resistance should be elucidated and incorporated a priori into the structure-based drug design process to develop more robust inhibitors. To achieve this goal, structural requirements for the protease to fulfill its enzymatic function in the viral life cycle should be thoroughly studied, since the biological function imposes evolutionary constraints on the protease under the selective pressure of drug therapy. An important lesson learned from studying human immunodeficiency virus type 1 (HIV-1) protease, also a quickly evolving enzyme, is that designing robust protease inhibitors can be facilitated by an understanding of the key molecular recognition elements of the natural substrates. For example, inhibitors should stay within the substrate recognition regions (i.e. the “substrate envelope”) to minimize the likelihood that the enzyme can mutate to reduce the inhibitor efficacy while still maintaining sufficient enzymatic activity on the substrates.7–11 Resistance to protease inhibitors reflects a subtle change in the balance of molecular recognition events, in favor of substrate processing versus inhibitor binding.
Crystallographic studies from our group have shown that the substrate recognition motif for HIV-1 protease is not a consensus sequence of the substrates but a consensus volume adopted by the non-homologous cleavage site sequences within the binding site, which we term the “substrate envelope”.12 In addition, protein dynamics have been incorporated into the substrate envelope to assess the effects of local conformational fluctuations of the bound substrate.13 The dynamic substrate envelope better captures the specificity determinants of substrate recognition and is preserved even in drug resistant HIV-1 protease variants in the presence of compensatory mutations in the cleavage sites.13, 14 The primary drug resistance mutations in HIV-1 protease occur at residues outside the substrate envelope that are contacted by the inhibitors.15
HCV NS3/4A protease is the N-terminal domain of the viral bifunctional protein NS3, which also has a C-terminal helicase domain. Independently determined crystal structures of the full length NS3/4A versus the individual helicase and protease domains show that the structures of the individual domains are independent of each other.16–20 In addition, the protease inhibitors have similar affinities towards the full-length protein and the protease domain.21 Therefore, the current study is focused on the protease domain only. Efficient proteolytic activity of NS3 requires the cofactor NS4A, a 54-amino acid peptide that tightly associates with the protease.22 The viral genome is translated as a single polyprotein along the endoplasmic reticulum by host cell machinery. NS3/4A hydrolyzes the polyprotein precursor at four known cleavage sites (3-4A in cis, 4A-4B, 4B-5A, and 5A-5B) yielding non-structural proteins essential for viral maturation.23, 24 The four known substrates of HCV protease share little sequence homology except for an Asp/Glu at P6, Cys/Thr at P1, and Ser/Ala at P1′ positions. Our group reported co-crystal structures of the wild-type protease domain bound to substrate cleavage products and inhibitors that included the macrocyclic compounds ITMN-191, MK-5172, and MK-7009, which are used in the current study.20 In the co-crystal structures, the substrate products adopt a consensus volume despite the low sequence homology. Similar to HIV-1 protease, the most severe resistance mutations in NS3/4A correspond to the residues that are contacted by the inhibitors outside the substrate envelope. In a recent study, Xue et al. further investigated the structural basis of drug resistance mutations R155K, A156T, and D168A against MK-5172 and MK-7009.25
In the current work, we present a comprehensive and comparative analysis of substrate versus inhibitor binding and dynamics to reveal a list of recommendations to improve the resistance profiles of the current inhibitors in structure-based drug design. In contrast to the previous works, we explicitly compare the interaction profiles between substrates and inhibitors. In addition to identifying known resistance mutations, this approach facilitates the prediction potential resistance mutations that can arise as a result of differential interaction dynamics between substrates and inhibitors. Three HCV NS3/4A protease inhibitors in the clinic are analyzed in the context of a dynamic substrate envelope obtained from running molecular dynamics on the four viral substrates. The structural and dynamic analyses reveal several features of inhibitors that could render them susceptible to drug resistance. This analysis in particular (1) allows predictions of potential drug resistance mutation sites, at which the enzyme can afford amino acid substitutions with minimal effect on substrate recognition and (2) reveals the untapped regions of the HCV NS3/4A protease binding site that satisfy the substrate envelope and can be explored by new inhibitors to potentially gain affinity without compromising their resistance profiles.
Results and Discussion
Comparative analysis of the binding modes and molecular interactions of substrates versus inhibitors is critical to guide the design of new small-molecule inhibitors that better overlap the natural substrate binding features, minimizing the likelihood of resistance. Toward this goal, the regions of the substrate binding surface that are unexploited by the current inhibitors as well as the regions the inhibitors contact on the protein surface beyond the substrate envelope were revealed using molecular dynamics simulations.
Three macrocyclic NS3/4A protease inhibitors (ITMN-191, MK-5172, and MK-7009, Figure 1A) were compared to the natural substrates (3-4A, 4A-4B, 4B-5A, and 5A-5B) in terms of dynamics and molecular interactions in the NS3/4A protease complex (Table 1). For substrates, each of the six amino acid residues defines a site (P6 to P1, Figure 1B); the inhibitor functional groups were assigned positions by superimposing the crystal structures of the inhibitor complexes onto the crystal structures of the substrate complexes and assigning inhibitor groups based on the overlap with substrate residues. Because none of the inhibitors span the P6-P5 region, only four chemical groups (P4 to P1, Figure 1C) and one group outside the envelope (P1′) were defined for the inhibitors.
Figure 1.
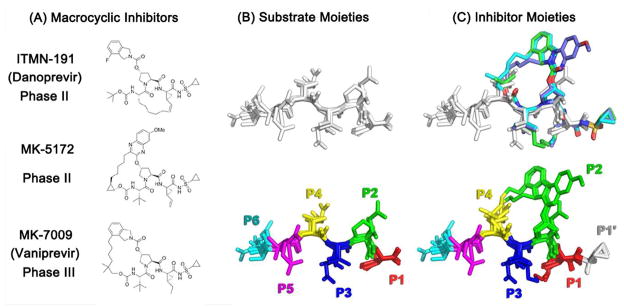
HCV protease substrate and inhibitor structures. (A) The 2D chemical structures of the inhibitors (ITMN-191, MK-5172, and MK-7009) (B) The overlaid substrate cleavage products (3-4A, 4A-4B, 4B-5A, and 5A-5B) are displayed in sticks with the substrate sites indicated by distinct colors. The amino acid sequences are tabulated (Table 1) (C) The inhibitors superimposed onto the substrates, inhibitor chemical moieties colored in the same scheme as the substrates (additionally P1′: white). The substrates do not have a P1′ position in the co-crystal structures due to the residual activity of the S139A protease construct used in crystallization.
Table 1.
The cleavage site products and macrocyclic small-molecule inhibitors of HCV NS3/4A Protease
| Substrates
|
Starting structure PDB Code |
# of atoms | Vout (Å3) | Vtot (Å3) | vdW kcal/mol | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P6 | P5 | P4 | P3 | P2 | P1 | ||||||
| 3-4A | D | L | E | V | V | T | 1CU1 | 47 | 341 ± 31 | 1042±14 | −26.9±2.8 |
| 4A-4B | D | E | M | E | E | C | 3M5M | 50 | 431 ± 40 | 1097±15 | −31.9±2.5 |
| 4B-5A | E | C | T | T | P | C | 3M5N | 43 | 300 ± 26 | 950±17 | −27.7±2.7 |
| 5A-5B | E | D | V | V | C | C | 3M5O | 44 | 318 ± 24 | 993±14 | −27.9±2.2 |
| Inhibitors | PDB Code | # of atoms | Vout (Å3) | Vtot (Å3) | vdW kcal/mol |
|---|---|---|---|---|---|
| ITMN-191 (Danoprevir) | 3M5L | 51 | 567±18 | 1082±15 | −36.5±2.0 |
| MK-5172 | 3SUD | 54 | 633±20 | 1165±16 | −35.0±2.8 |
| MK-7009 (Vaniprevir) | 3SU3 | 53 | 606±19 | 1152±14 | −36.6±1.8 |
Vout: Volume of a cleavage product/inhibitor outside the dynamic substrate envelope
Vtot: Total vdW volume of a cleavage product/inhibitor
vdW: Total vdW contact energy of a cleavage product/inhibitor with the protease
Dynamic Substrate Envelope and the Molecular Shape of Inhibitors
Four substrates in complex with NS3/4A protease adopt a similar overall shape in each of their crystal structures and the overlapping consensus volume defines the static substrate envelope (Figure S1). A continuous representation of the dynamic substrate envelope was defined from 100 ns of production molecular dynamics (MD) simulations and mapping van der Waals (vdW) volumes of P6 to P1 residues of substrate conformers onto a three-dimensional grid, similar to our previous work with HIV-1 protease.13, 14 The dynamic substrate envelope, therefore, is a probability distribution of vdW occupancies based on the conformations of the substrates throughout the MD trajectory, which should provide a more relevant comparison between substrate and inhibitor complexes.
To assess the macrocyclic compounds in terms of their overlap with the substrate envelope, the probabilistic volume occupied by each inhibitor during 100 ns MD simulations was compared with the dynamic substrate envelope. Due to the macrocycle linkage the three inhibitors are not as conformationally flexible as the substrates, sampling a relatively restricted conformational space and resulting in a more homogeneous volume distribution (more red regions for inhibitors in Figure 2). As such, the inhibitors are expected to be affected more by mutations in the binding site compared to the flexible substrates. In fact, the recently published co-crystal structures of these inhibitors with the wild type and three of the most severe drug resistant protease variants (R155K, A156T, and D168A) revealed a complete shift in the binding mode of the inhibitor, likely because the rigid nature of these inhibitors does not allow for minor conformational adjustments that the substrates could undergo to avoid clashes with the mutation.20
Figure 2.

Macrocyclic inhibitors are more rigid than the substrates, resulting in a more homogeneous dynamic volume. The four superimposed substrates, 3-4A, 4A-4B, 4B-5A, 5A-5B, are shown as sticks in red, green, blue, yellow, respectively. P1 and P6 sites are labeled to indicate the directionality of the substrate envelope. The volume distribution of the substrate envelope and each inhibitor is represented with a spectrum from red to blue, reflecting respectively more and less likely occupied regions.
The most well defined region of the dynamic substrate envelope corresponds to the P4 to P1 segment (red region of Figure 2A), which is seen by the low computed volume of substrate atoms outside of the substrate envelope (Figure 3). The 4A-4B cleavage site is an exception, with larger Vout values, especially at P2 position. Two factors contribute to this: first, 4A-4B has a bulky glutamic acid at P2 with a higher degree of side-chain conformational freedom; and second, 4A-4B appears to be inherently more flexible than the other substrates as suggested by alpha carbon root-mean-squared fluctuations (RMSF) of 1.6 Å from the MD simulations, which is significantly larger than the other substrates (1.0–1.2 Å; Figure S2) resulting in a smaller dynamic volume for this substrate than other substrates (Figure S1). With 4A-4B being the exception, the P4-P1 region is the key conserved volume within which the inhibitors should be designed to fit with minimal protrusion to avoid resistance mutations.
Figure 3.

The HCV NS3/4A dynamic substrate envelope is more conserved in the P4 to P1 region. The volume outside the dynamic substrate envelope for each substrate and inhibitor moiety, Vout, is colored in a spectrum of blue to red. Warmer and cooler colors indicate a higher and lower level of protrusion beyond the dynamic substrate envelope, respectively. The macrocyclic inhibitors protrude from the substrate envelope mainly at P2 and P1′ positions.
P5 and P6 have higher mobility during the MD simulations (Figure S2). The P6 position of all four substrates has relatively high Vout values compared to the other positions, with 23% to 40% of the total Vout coming from this position. Similarly, at the P5 position the substrates have a lower consensus volume. In general, the lower computed density at P6 and P5 is consistent with the higher temperature factors of P6 and P5 in the crystal structures.18 Indeed, telaprevir, an FDA-approved non-macrocyclic ketoamide inhibitor, has a P5 moiety that does not improve its binding compared to the macrocyclic compounds without a P5 moiety. The Ki for telaprevir is 34.4±3.0 nM against the wild-type enzyme, while macrocyclic compounds without a P5 moiety are more potent (Ki’s, ITMN-191: 1.0±0.1 nM, MK-5172: 0.74±0.07 nM, and MK-7009: 0.14±0.02 nM).20 However, the P6 substrate residue makes extensive electrostatic interactions with the positively charged protease binding site residues R119, R123, R161, or K165, as discussed below, consistent with the strong conservation of P6 acidic side chain for optimal substrate processing.26
According to the substrate envelope hypothesis, the inhibitor moieties that extend beyond the substrate envelope are most susceptible to resistant mutations in the active site. The portion of each inhibitor’s volume that falls outside the dynamic substrate envelope (Vout) is shown in Table 1 and the Vout contribution of each moiety (P4 to P1′) is shown in Figure 3. The larger P2 group, a characteristic feature for the non-covalent inhibitors, extensively protrudes beyond the substrate envelope. This group makes up, on average, 33% of an inhibitor’s total volume and the favorable P2 interactions contribute to the high affinity.27 However, the susceptibility of an inhibitor with a large P2 group to resistance mutations depends on the residues contacted by the P2 group. For example, the quinoxaline of MK-5172 protrudes 361±25 Å3 beyond the substrate envelope and interacts with the invariant catalytic residue H57. While the isoindoline groups in ITMN-191 and MK-7009 protrude slightly less (289±14 Å3 and 299±21 Å3, respectively) but contact the resistance mutation site R155 (Figure 6). Therefore, assessing the degree to which an inhibitor is prone to resistance mutations depends both on the extent of protrusion beyond the substrate envelope and on the specific interactions with the binding site residues. Protruding beyond the substrate envelop may not be detrimental to avoiding resistance if the interactions are primarily with residues of the enzyme that are evolutionarily constrained.
Figure 6.

Differences between the binding surfaces of inhibitors versus substrates provide opportunity for resistance mutations. The protease residues that contact (A) the substrates, (B) ITMN-191, (C) MK-5172, and (D) MK-7009 are colored with respect to their vdW contact potential energies. The catalytic triad residues, H57, D81 and S139, are indicated by underlined labels in italics. The substrate residues P6 and P1 are labeled to show the orientation of the cleavage products while all five moieties (P4 to P1′) are labeled for inhibitors. (E) vdW contact potentials for protease residues. The black and red stars correspond to the natural polymorphic and drug resistance mutation sites, respectively.
In addition to minimizing protrusions from the substrate envelope, a robust inhibitor ideally fills the substrate envelope efficiently to gain maximum potency. This was assessed for the three inhibitors by calculating the average volume of the dynamic substrate envelope that is not being occupied by inhibitor atoms (Vremaining; Figure 4). The volume of the substrate envelope that is not occupied by any inhibitor atom (Vremaining values are 506±15 Å3, 489±16 Å3, and 474±16 Å3; Figure 4) is comparable in magnitude to the volume outside the substrate envelope (Vout values are 567±18 Å3, 633±20 Å3, and 606±19 Å3; Table 1). Therefore, in principle, a compound with the same total volume as the existing inhibitors can be designed to fit fully within the envelope by removing the parts outside the substrate envelope and adding atoms to the regions of the substrate envelope currently not filled. The challenging goal of developing robust inhibitors that avoid drug resistance mutations may be achieved by optimizing the compounds to minimize Vout and Vremaning metrics while retaining potency.
Figure 4.

Macrocyclic inhibitors do not fully occupy the dynamic substrate envelope. The portion of the dynamic substrate envelope that is not used by the inhibitor molecules, Vremaning, is shown on the structures of (A) ITMN-191, (B) MK-5172, and (C) MK-7009. Stereo images were prepared with PyMOL.46
Conformational Flexibility
Conformational flexibility is important for the substrates to be recognized and to adapt to resistance mutations in the protease.14 Previous studies showed that an inhibitor should have the optimal flexibility to adapt to genetic diversity, resistance mutations, and binding site dynamics.28 The optimal inhibitor flexibility should be achieved by mimicking the dynamic aspects of the natural substrates to achieve a flat resistance profile. For this reason, the dynamic features of substrates were characterized to compare with those of the inhibitors by calculating root-mean-squared-fluctuations (RMSF) of alpha carbon atoms in MD simulations and the intramolecular cooperativity of these fluctuations in the bound state. Interdependence of the dynamics of substrate residues plays a role in maintaining the molecular shape necessary for substrate recognition.13, 14 This can be assessed as a time-dependent correlation of atomic fluctuations among the substrate residues (Figure 5). In the correlation plots, residues that fluctuate in a cooperative manner are dynamically coupled and are indicated with deep red color, whereas regions that are not coupled are in white. Strikingly, positive correlations within the P4-P1 region are more pronounced than the correlations across the P6-P5 and P4-P1 regions for all four substrates (Figure S3). The positive correlation within the P4-P1 region is likely to aid in maintaining the consensus volume, which is conserved across the four substrates and could contribute to substrate recognition.
Figure 5.

Correlation of substrate atomic fluctuations within P4-P1 region survives longer than those between P6-P5 and P4-P1. Inhibitor atomic fluctuations are highly correlated. Substrate residues and inhibitor moieties are separated by lines. The P6-P5 and P4-P1 regions of 3-4A are separated by a thicker black line. The macrocycle linker atoms in MK-5172 are indicated by darker lines.
Substrates exhibit subtle variations in the positional fluctuations and their relative time-evolved interdependence. For 3-4A and 4A-4B, atomic fluctuations of the P6 residue are negatively correlated to those of the P3-P1 region. The cooperativity within the 4A-4B substrate is considerably higher than the other substrates (Figure S3). This higher cooperativity may correlate with the number of charged residues and the higher net charge in 4A-4B, as electrostatic interactions are long range and movements in one charged residue can significantly influence a distal charged residue. For example, movement of Asp-P6 is inversely correlated to Glu-P3 and Glu-P2, since their movement in opposite directions would maximize their separation. This kind of cooperativity may result in interdependence within the substrate sequence. Because 4A-4B protrudes beyond the substrate envelope, especially at P2 site, more than the other substrates (Figure 3), the 4A-4B cleavage site is expected to be more susceptible to co-evolution with drug resistant protease variants. The cooperativity within the 4A-4B sequence suggests that a compensatory mutation in the cleavage site may occur at a position other than P2 that would allow the substrate to adapt to drug resistance mutations in the protease.
Similar to the substrates, the conformational flexibility of the inhibitors is dictated by a combination of structural features. Location of a macrocycle linkage (whether the linkage connects the P1 group to P3 or P2 to P4) and the particular atom involved with the linking may also impact the flexibility. The P2-P4 moieties in NS3/4A inhibitors are highly solvent-exposed, hence they are relatively free to move, while the P1-P3 moieties are fully buried within the binding site and therefore more conformationally restricted. As a result, a macrocycle connecting the more flexible P2 and P4 moieties may be more effective in constraining the small molecule inhibitor in the bound state than a linker connecting P1 and P3 moieties that are already relatively rigidified via interactions with the protease. However, the packing of the flexible P2 group may also affect the flexibility of an inhibitor in the binding site. Very tight interactions may lock the inhibitor in a single conformation.
Both MK-5172 and MK-7009 have a P2-P4 linker; however their dynamic envelopes suggest that more local conformational states are accessible to MK-5172 than MK-7009, especially for the P2 group and the linker (Figure 2). MK-5172 has an ether-linked quinoxaline P2 group and a linker to C2 of quinoxaline, while MK-7009 has a carbamate-linked isoindoline P2 group and a linker to the C4 of the isoindoline. In MK-5172, the quinoxaline group is pointing away from the flexible C2-linker and the local fluctuations of this group propagate to the linker. In contrast, the isoindoline in MK-7009 is linked at the C4 position, which is located at the tip of the P2 group and renders the constraint more effective. Thus, the C4 connection in MK-7009 is more conformational restrictive than the C2 connection in MK-5172.
ITMN-191 entirely lacks this linker, which makes the isoindoline more flexible than MK-7009 and comparable with the C2-linked quinoxaline in MK-5172. Thus, a macrocycle does not guarantee a significant dynamic constraint in a compound; the connection point of the linker also has a role in restricting the extent of the flexibility. All three effects combined together (location of the macrocycle, the connection points of the linker, and the identity of the P2 moiety connected by the linker) are responsible for the differential dynamics observed in the bound state (Figure 2 and Figure S5).
The backbone of the inhibitors is always relatively rigid throughout the simulations, consistent with the substrate P4-P1 backbone stability (Figure S5). P1-P3 moieties of all three inhibitors have very homogeneous volume distributions due to their rigidity in the bound state (Figure 2). The interplay between the rigidity of the P1-P3 groups and the stability of the favorable interactions with the corresponding binding subsite contributes to the tight binding characteristics to these inhibitors. The structural rigidity of these very tight binding compounds relative to natural substrates gives rise to high orientational correlations of motion within a compound (Figure 5). The effect of a movement in one moiety is propagated to the rest of the molecule because of this rigidity introduced by the linkers. The cross-correlations of atomic fluctuations within the P2-P4 macrocyclic compounds (MK-5172 and MK-7009) are much higher and last longer than those within the P1-P3 linked compound (ITMN-191; Figure S4). The high cooperativity may prevent the inhibitors from local conformational readjusting to adapt to resistance mutations in the protease. For example, based on the recently published resistance data, the P2-P4 linked inhibitors are more susceptible to A156T mutation than the P1-P3 linked compound.20 The fold-change in Ki relative to wild type is 4429 and 1295 for MK-5172 and MK-7009, respectively, but 45 for ITMN-191,20 possibly because these rigid macrocyclic compounds cannot conformationally relax to avoid resistance mutations. To overcome this opportunity for resistance, robust inhibitors to NS3/4A protease should be designed to have conformational flexibility consistent with natural substrates.
Intermolecular Contacts
Van der Waals (vdW) interaction energies represent the local packing and can be used to understand the degree of complementarity between a ligand and receptor. A comparative view of vdW energies from the MD simulations reveals the regions of substrate that interact favorable with HCV protease but are unexploited by the inhibitors. Conversely, there are regions of HCV protease that should be avoided in designing new inhibitors to minimize the likelihood of drug resistance (Figure 6). The protease residues that are contacted by substrates and the extent of these interactions are conserved across the four substrates. The vdW interaction energy of each protease residue was averaged over four substrates and mapped onto the binding surface (Figure 6A). The interactions with the catalytic H57 appear to be critical for all substrates, as well as K136, A156, A157, V158, and S159. A comparison of the natural polymorphisms and drug resistance mutation sites and the contact potential of these sites with the natural substrates reveals that the drug resistance mutations have been selected at sites that are not major contact points for substrates, namely Q41, T42, F53, K80, S138, and D168 (Figure 6E). However, R155, A156, and V158 mutate to confer drug resistance although they have extensive, stable interactions with the natural substrates. Based on this structural observation, the viral variants carrying the mutations at these three sites are expected to pay a fitness cost because of reduced substrate binding, yet the selective pressure of the drug therapy populates the viral variants carrying these mutations.
The total vdW interaction energy of the three inhibitors is comparable (−36.5±2.0, −35.0±2.8, and −36.6±1.8 kcal/mol for ITMN-191, MK-5172, and MK-7009, respectively). The catalytic histidine (H57) is an important residue for all inhibitors but especially critical for MK-5172. The extensive interactions with H57 may contribute to the broader activity of MK-5172 across different genotypes and resistant variants.29 The residues H57, S139, R155, and D168 have more favorable interactions with the inhibitors than substrates, while the residues Q41, T42, F43, Y56, G58, V78, D79, K80, and D81 do not contact the substrates at all but interact with the inhibitors. These residues have relatively low vdW interaction energies (in the range of 0 to −1 kcal/mol) but may have a collective impact on the binding affinity. Taken together, targeting the protease residues that are major contact points for natural substrates, including but not limited to the catalytic triad (H57, D81, S139), and minimizing interactions with the variable sites is a promising strategy for avoiding resistance without compromising selectivity.
A large portion of substrate vdW energy with the protease (~40%) involves interactions with the P1 position (Figure 7). Indeed, the Cys in 3-4A and Thr in the other substrates at P1 position are nicely packed in this small cavity compared to the other substrate residues, which contact the relatively shallower parts of the binding surface. The macrocyclic inhibitors do not have P5 and P6 moieties. Instead, at P2 position, they make more favorable vdW contacts than the substrates due to the bulky isoindoline (ITMN-191/MK-7009) or quinoxaline (MK-5172) groups. While the P2 interactions contribute to the high affinity to the wild-type protease, most of them are outside the substrate envelope (Figure 3) and therefore render the inhibitors susceptible to resistance mutations.
Figure 7.

Differential interaction profiles of inhibitors versus substrates suggest potential improvement at P1 moiety of inhibitors. The vdW contact potential of (A) substrate residues P6 to P1, and (B) inhibitor moieties P4 to P1′.
Electrostatic Interactions
Electrostatic interactions are longer range than vdW interactions and can contribute significantly to binding selectivity. All of the protease-substrate hydrogen bonds, except for the one formed by R123, are conserved across the substrates in the crystal structures.18 A majority of these hydrogen bonds persist throughout the MD simulations (Table 2), especially the ones involving backbone atoms of A157 and C/S159. The hydrogen bonds formed by the substrate backbone are more stable, with all except one (between the ε2 oxygen of the Glu-P4 of 3-4A substrate) existing for at least 50% of the simulated time. Although the hydrogen bonds are conserved across the substrates, the percentage of time that these interactions exist throughout the simulations vary between the complexes. Substrate 4B-5A preserves the complete hydrogen-bonding network with the protease in the simulation while 4A-4B preserves half of the hydrogen bonds, consistent with the higher flexibility of this substrate. All P1 terminal carboxyl groups sit in the oxyanion hole, hydrogen bonding with the ε2 nitrogen of the catalytic histidine (H57), which exists 97% of the time in 4B-5A complex while in other substrates this hydrogen bond exists ~70% of the time. Similarly, the backbone hydrogen bond between P1 and G137 is present 89% of the time in the 4B-5A complex whereas the same bond exists 59% of the time or less for other substrates. These variations likely arise from other differences in molecular interactions with the enzyme and inherent differences in substrate flexibilities and contribute to the substrate specificity.
Table 2.
The percentage of time that the protease-substrate hydrogen bonds existed during the MD simulations.
| Protease | Substrate
|
||||
|---|---|---|---|---|---|
| 3-4A | 4A-4B | 4B-5A | 5A-5B | ||
| H57 – NE2 | P1 – O | 72 | 71 | 97 | 69 |
| G137 – N | P1 - O | 55 | <50 | 89 | 59 |
| S139 – OG | P1 - O | 53 | <50 | 58 | <50 |
| R155 – O | P1 -N | <50 | <50 | 56 | <50 |
| A157 – N | P3 – O | 69 | 85 | 76 | 80 |
| A157 – O | P3 – N | 87 | 82 | 72 | 84 |
| R123 – NH2 | P4 – OE2 | 51 | <50 | <50 | <50 |
| C/S159* – N | P5 - O | 78 | 63 | 57 | 81 |
| S159 – O | P5 - N | <50 | <50 | <50 | 59 |
All but one of the protease-inhibitor hydrogen bonds are formed by the backbone of protease binding site residues (the γ oxygen of the catalytic serine S139 being the exception). Making backbone hydrogen bonds should in theory be preferred when designing new drugs to avoid losing a specific hydrogen bond donor/acceptor with a side chain upon an amino acid mutation. However, a drug resistance mutation can also cause backbone structural changes, which may or may not affect the distance/angle between the donor and the acceptor of a hydrogen bond. Such changes may affect the interactions with substrates, which would then have to adapt to the new protease conformation. This reemphasizes the importance of designing inhibitors that not only mimic the substrate envelope but also mimic the dynamic characteristics of the substrates.
The P6 residue, despite not being an essential part of the dynamic substrate envelope, is conserved (Asp or Glu). Mutational studies show little tolerability of amino acid substitutions at this position for optimal cleavage, suggesting functional importance.26 Crystal structures suggest that this arises from the potential electrostatic interactions of P6 acid with K165 and possibly in long-range substrate recognition, and thus P6 is included in the definition of the dynamic substrate envelope. In all crystal structures except for the product complex 4B-5A, K165 forms a salt bridge with the P6 acids.18 In the MD trajectories, the P6 acid-K165 salt bridge criterion is satisfied for 12%, 26%, 35%, and 57% of the simulated time for 3-4A, 4A-4B, 4B-5A, and 5A-5B, respectively. The percentages are relatively low for the first three complexes, however the disrupted salt bridge re-forms in a recurring manner. In addition, the percentage of time the P6 acid forms a salt bridge with any one of the four binding site basic residues (R113, R119, R161, and K165) is 53%, 68%, 50%, and 83% for 3-4A, 4A-4B, 4B-5A, and 5A-5B, respectively (Figure S6). These numbers suggest that the P6 acid, in solution, interacts with at least three more basic residues on the binding surface in a dynamic manner in addition to K165 observed in the crystal structures. The P6 position, while forming salt bridges with a series of binding site residues, does not adopt a conserved consensus volume. The highly dynamic interaction pattern of the P6 acid with the binding surface rationalizes the strong conservation of Asp or Glu at this position as well as the dramatic reduction in cleavage efficiencies of substrates with positively charged P6 substitutions. To mimic the salt bridges between the protease and the P6 acid, inhibitors would require a negatively charged P6 and/or P5 moiety that is flexible enough to form interchanging salt bridges with a series of binding site residues. The probabilistic character of the dynamic substrate envelope provides a basis for building the P5 and/or P6 moieties with the relevant charge and flexibility requirements, preventing potential pitfalls and misleading results in case the static P6 crystallographic volume were to be satisfied.
The electrostatic network along one surface of the bound cleavage products between protease residues R123, D168, R155, and the catalytic D81 has been proposed to increase the stability of the bound state and therefore contribute to substrate recognition.30 In MD simulations, however, the salt bridges D81-R155 and R155-D168 in the substrate complexes appear to be mutually exclusive, suggesting that R155 is shared between D81 and D168 (Figure 8). This exchange of salt bridges along the binding surface does not occur in inhibitor complexes, as the rigid tight binding inhibitors lock the conformation of the side chains at the binding site stabilizing individual salt bridges. In the inhibitor-bound complexes, the R155-D168 salt bridge is extremely stable. The D81-H57 salt bridge exists throughout the inhibitor simulations, as R155 is not available for salt bridging to D81. The D168-R123 salt bridge is an exception, which appears to be transient in both substrate and inhibitor MD simulations (Figure S7–S8). The enhanced flexibility towards the P6 site in the substrates may have given added conformational freedom to the R123 side chain while the inhibitors have minimal interactions with this residue due to the lack of P6-P5 moieties. Two of the most severe drug resistance mutations, R155K and D168A, could disrupt this salt bridge network causing an energetically unfavorable perturbation in the inhibitor complexes. The maintenance of this electrostatic network within the protease residues should also be assessed in inhibitor design.
Figure 8.

R155 is shared by D81 and D168 in a salt bridge network along the binding surface of substrate-bound complex while in the inhibitor complexes, the R155-D168 salt bridge is stabilized by the formation of the D81-H57 salt bridge. Salt bridges were defined as an interaction between a any side-chain oxygen atom of Asp or Glu within 4.0 Å of any nitrogen atom of Arg or Lys.
Conclusions
In this paper, we propose a dynamic substrate envelope guided drug design strategy that also takes conformational flexibility and intramolecular interactions into account. This strategy is retrospectively validated by the differential sensitivity of the inhibitors to the previously known mutations included here and our group has ongoing efforts to design, synthesize and test new compounds using the dynamic substrate envelope based drug design strategy aiming for inhibitors with improved resistance profiles.
Understanding differential interaction patterns between substrates and inhibitors can help guide the design of new inhibitors that better target resistant protease variants. Comparison of a series of structural and dynamic features of inhibitors and substrates reveals key insights for structure-based design of inhibitors that are robust against resistance mutations. One aspect of substrate specificity that can be used to optimize inhibitors is the dynamic substrate envelope. This is a two-sided optimization problem: (1) the volume of the compound that stays outside the envelope (Vout) should be minimized to avoid contacts with protease residues that are not important for substrate recognition and at the same time (2) the portion of the substrate envelope that is not being filled by the inhibitor (Vremaining) should be minimized to efficiently use the substrate envelope when growing the compounds. These need to be achieved while maintaining the potency of the inhibitors.
Considering the effect of protein dynamics in substrate binding modes and defining the substrate envelope as a probability distribution is especially critical for the HCV NS3/4A protease. The high solvent exposure of the NS3/4A binding site allows the substrates to be highly flexible. Accessibility of more conformational states to the substrates means they are less constrained within the consensus volume defined by the crystal structure conformations; thus, the increased space explored by the substrates results in a smaller consensus volume. The dynamic substrate envelope, defined as a probability distribution of the substrate volume in the binding site, better captures the substrate binding features and allows for a more accurate assessment of the inhibitors during drug design. This probabilistic dynamic substrate envelope allows for a detailed investigation of the individual moieties on an inhibitor and an accurate evaluation of the similarities/differences between substrate and inhibitor complexes.
The most conserved substrate volume when protein dynamics is taken into account is defined by the P1 to P4 residues, while P5 and P6 have more conformational freedom. P6 is able to serve as a hub in an intermolecular electrostatic network involving the protease residues R119, R123, R161, or K165, dynamically flipping between these basic residues. This suggests that an inhibitor can be designed to have a charged P6 moiety with molecular flexibility consistent with the natural substrates to facilitate these electrostatic interactions with the protease. Including the P6 residues of the substrates in the dynamic substrate envelope will provide a useful basis to guide the design of these charged compounds. Based on this profile, inhibitors should be designed such that their regions spanning P1-P4 portion of the substrate envelope are more restrained while the moieties spanning P5-P6 sites are more flexible. Ideal inhibitors of NS3/4A protease should be designed to have the intrinsic flexibility consistent with the natural substrates.
To minimize the susceptibility of an inhibitor to drug resistance mutations, interactions with functional residues, which include the catalytic triad and also the residues that play a key role in substrate recognition in HCV protease, should be enhanced. In contrast, the interactions with known drug resistance mutation sites or polymorphic sites should be minimized. For example, R155 (one of the most severe drug resistance mutation sites) is in extensive contact with the P2 groups of all three inhibitors. During the optimization of new compounds, favoring the P2 conformations that interact with D81 and H57 while avoiding R155, as in MK-5172, is expected to be useful for better performance against the R155K variant. In fact, the fold-change in the inhibition constant Ki against the R155K variant relative to wild type is 162, 6, and 749 for ITMN-191, MK-5172, and MK-7009, respectively.20 In addition, the P2-P4 macrocycle enhances the protease interactions through D168 and V158. Breaking this macrocycle could reduce the loss of potency to the D168A variant. In fact, fold changes in Ki values for ITMN-191 and MK-7009 are 208 and 3561, respectively, against the D168A variant.20 Finally, the P1-P3 macrocycle appears to hinder P1 to fill the S1 pocket completely. P1 may be modified to explore the possibility of enhancing the interactions with F154 in the S1 pocket, consistent with the substrates’ highly favorable interactions at P1 site.
Another binding interaction to optimize is the hydrogen bonding. When placing the hydrogen bond donors/acceptors in the compounds, the protease backbone donors/acceptors could be targeted at polymorphic sites. While a conformational readjustment is possible with an amino acid substitution at these sites, a hydrogen bond with the side chain is more likely to be lost upon a mutation. In addition, the substrate P6 residues salt bridge with a series of protease binding site residues. Maintaining this electrostatic network within the protease could also be taken into account during inhibitors design.
In addition to the electrostatic interactions within the protease substrate or inhibitor complexes, recent work also suggests that water molecules can play an important role in molecular recognition.31–33 While the substrates do not have conserved water-mediated hydrogen bonds with the protease in the crystal structures, the inhibitors ITMN-191 and MK-7009 have eight crystallographic water molecules that are within 4 Å of both protease and inhibitor. The solvent exposure of the binding site combined with the effects of crystal contacts may present challenges in accurately assessing the relative importance of these water molecules.
In conclusion, the binding volume of an inhibitor that is robust to resistance mutations should ideally not fall outside of the dynamic substrate envelope to achieve a better resistance profile. When searching the chemical space in the design process, ranking the candidates based on their Vout and Vremaining in combination with vdW contacts and the electrostatic interactions may predict which compounds will be more susceptible to resistance. To maintain efficacy against the extremely shallow binding surface of NS3/4A, staying completely within the substrate envelope may not always be practical. In such cases, the exact location where the inhibitor protrudes beyond the substrate envelope is critical. Some regions of the dynamic substrate envelope are more heterogeneous, therefore the compound could have more freedom at those regions. Relative importance of subpockets should be explored for each compound in development. Protruding beyond the substrate envelope might be tolerable to pick up interactions with the catalytic triad since the catalytic residues are evolutionarily conserved and highly unlikely to mutate to confer drug resistance. In addition, modifications to ligands may be needed to alter properties other than target affinity and selectivity (i.e. solubility, metabolism, permeability, or efflux).34 Extending beyond the substrate envelope in regions that do not contact the enzyme could be used to tune such properties and modifications in these regions are likely to be better tolerated than those that contact the enzyme. Finally, designing inhibitors to have flexibility consistent with substrates should improve the chances of avoiding resistance mutations.
Methods
Protease-substrate/inhibitor complex structures
Structural and dynamic properties of four substrates and three small-molecule inhibitors in complex with the NS3/4A serine protease were investigated (Table 1). Crystal structures of the three protease-substrate complexes, which correspond to the cleavage sites 4A-4B, 4B-5A, and 5A-5B on the viral polyprotein, were available (PDB ID: 3M5M, 3M5N, 3M5N).18 The structure of the fourth cleavage site, 3-4A, was modeled based on the full-length helicase/protease structure19 (PDB ID: 1CU1) by deleting the helicase domain but keeping the C-terminal eight residues in the enzyme binding site. This C-terminal region corresponds to the product of the cleavage site 3-4A. In addition, the co-crystal structures of three macrocyclic inhibitors ITMN-191, MK-5172, and MK-7009 (PDB ID: 3SU0, 3SUD, 3SU3) were used.20
Structure preparation
For the co-crystal structures in which more than one molecule exists in the asymmetric unit, all of the molecules were inspected for major structural changes in the backbone, average temperature factors, the number of missing side chains, and the ambiguity of the electron density for the substrate or inhibitor in the binding site. The complex molecules with protease chain IDs A, B, D, and A were chosen for the simulations of 3-4A, 4A-4B, 4B-5A, and 5A-5B, respectively. The crystallographic waters within 4.0 Å of any protein or ligand atom were kept, however all the buffer salts were removed from the coordinate files.
The protease in the substrate co-crystal structures, except for the 3-4A complex, has the S139A substitution in the active site, which was originally thought to inactivate the enzyme, prevent hydrolysis, and allow capturing the intact substrates in the active site. To get a more realistic model of the active enzyme interactions with the substrates, the A139S back-mutation was introduced in silico to the crystal structures by deleting the Ala side chain and predicting the conformation of the Ser side chain using the software Prime.35 The N-terminal residues in the NS3/4A construct, GSHMASMKKK, were extremely flexible in the first set of trial simulations and did not form stable secondary structure. In addition, these residues did not interact with either protease or substrate. Therefore, this region was deleted in subsequent simulations to reduce the computational cost of the simulations.
As the x-ray structures are not suitable for immediate use in MD simulations,36 the above described structures were prepared using the Protein Preparation Wizard from Schrödinger.37, 38 The process adds hydrogen atoms, builds side chains with missing atoms, and determines the optimal protonation states for ionizable side chains and ligand groups. In addition, the hydrogen bonding network is optimized by flipping the terminal chi angle of Asn, Gln, and His residues and sampling hydroxyl/thiol polar hydrogens. The exhaustive sampling option with the inclusion of water orientational sampling was used. Following this step, the structure was minimized in vacuum with restraints on heavy atoms using the Impact Refinement module with the OPLS2005 force field and terminated when the root-mean square deviation (RMSD) reached a maximum cutoff of 0.3 Å. This step allows hydrogen atoms to be freely minimized, while allowing heavy-atom movement to relax strained bonds, angles, and clashes.
Molecular dynamics simulations
Desmond39, 40 with OPLS2005 force field41, 42 was used in all simulations. The prepared systems were solvated in an orthorhombic solvent box with the SPC water model extending 10 Å beyond the protein in all directions using the System Builder utility. The overall charge of the system was neutralized by adding the appropriate number of counterions (Na+ or Cl−).
Each system was relaxed using a protocol consisting of an initial minimization restraining the solute heavy atoms with a force constant of 1000 kcal mol−1 Å−2 for 10 steps with steepest descent and with LBFSG method up to 2000 total steps with a convergence criterion of 50.0 kcal mol−1 Å−2. The system was further minimized by restraining only the backbone and allowing the free motion of the side chains. At this stage, the restraint on the backbone was gradually reduced from 1000 to 1.0 kcal mol−1 Å−2 with 5000 steps (250 steepest descent plus 4750 LBFSG) for each value of force constant (1000, 500, 250, 100, 50, 10, 1.0 kcal mol−1 Å−2) and finally an unrestrained energy minimization was performed.
After energy minimization, each system was equilibrated by running a series of short MD steps. First, a 10 ps MD simulation at 10 K was performed with a 50 kcal mol−1 Å−2 restraint on solute heavy atoms and using Berendsen thermostat in the NVT ensemble. MD steps were integrated using a two time-step algorithm, with 1 fs steps for bonded and short-range interactions within the 9 Å cutoff and 3 fs for long-range electrostatic interactions, which were treated with the smooth particle-mesh Ewald (PME) method.43, 44 Time steps were kept shorter at this first MD stage to reduce numerical issues associated with large initial forces before the system equilibrates. This was followed by another restrained MD simulation for 10 ps at 10 K with a 2 fs inner and 6 fs outer time step in NPT ensemble. The temperature of the system was slowly increased from 10 K to 300 K over 50 ps retaining the restraint on the system and 10 ps MD was performed without the harmonic restraints. Production MD simulations were carried out at 300 K and 1 bar for 100 ns using the NPT ensemble, Nose-Hoover thermostat, and Martyna-Tuckerman-Klein barostat. The long-range electrostatic interactions were computed using a smooth particle mesh Ewald (PME)44 approximation with a cutoff radius of 9 Å for the transition between the particle-particle and particle-grid calculations and van der Waals (vdW) interactions were truncated at 9 Å. The coordinates and energies were recorded every 5 ps.
Quantitative Definition of the Dynamic Substrate Envelope
All MD trajectories were aligned onto the 4A-4B co-crystal structure using the alpha carbon of binding site residues 137–139 and 154–160, which is less flexible compared to the rest of the molecule. The effect of structural alignment on the resulting substrate envelope was previously assessed to show that the selection of residues for alignment does not have a significant impact on the final substrate envelope as long as the selected residues are located in a relatively less mobile region of the enzyme.14 The structural alignments were performed using the Visual Molecular Dynamics (VMD) software package.45
Substrate conformers were extracted from the aligned MD trajectories. The frequency of each conformer is dictated by the free energy of the system, since sampling is performed under constant pressure at 300 K (NPT ensemble) with explicit solvent. The vdW volumes of all conformers were mapped onto the three-dimensional grid placed on the binding site of the enzyme and normalized by the total number of conformers to obtain a probability distribution. The vdW radii were taken from OPLS2005 force field. The mathematical details of the grid-based volume calculations can be found elsewhere.13
Estimating the van der Waals Contact Potential
The vdW contact energy between the protease and a substrate/inhibitor was estimated by a simplified Lennard-Jones potential, V(rij), using the relation 4ε[(σ/rij)12−(σ/rij)6], where r is the interatomic distance between the protease atom i and the substrate/inhibitor atom j. The ε and σ are the well depth and hard sphere diameter, respectively, for the ij protease-substrate/inhibitor atom pair. V(rij) for all possible protease-substrate/inhibitor atom pairs was computed within 6 Å, and when the distance between nonbonded pairs are less than that corresponds to ε, V(rij) was equated to ε. The rationale for this modification to the original 6–12 Lennard-Jones potential was previously described in detail.13 Using this simplified potential for each nonbonded protease-substrate/inhibitor pair, ΣV(rij) was then computed for each protease and substrate/inhibitor residue. The vdW parameters, ε and σ, were taken from OPLS2005. For the pairs involving two separate atom types, the vdW parameters were geometrically averaged.
Hydrogen Bonds and Salt Bridges
The percentage of time a hydrogen bond existed between the protease and a substrate/inhibitor was calculated using VMD.45 A hydrogen bond was defined by a distance between the donor and acceptor of less than 3.5 Å and a hydrogen-donor-acceptor angle of less than 30°. Only the hydrogen bonds that existed more than 50% of the time were considered in the analyses. Salt bridges were defined as an interaction between a side-chain oxygen atom of Asp or Glu within 4.0 Å of a nitrogen atom of Arg or Lys.
Root-mean-squared Fluctuations and Time-delayed Correlations
The normalized time-delayed orientational cross-correlations between residue pairs are defined as
where ΔRi(t) is the fluctuation in the position vector Ri of site i at time t around its average position throughout the trajectory and ΔRj(t+τ) is the fluctuation in the position vector Rj of site j at time t+τ. The brackets represent time averages. The cross-correlations vary in the range [−1, 1] with the upper and lower limits indicating fully correlated (moving in the same direction) and fully anti-correlated (moving in the opposite direction) atomic fluctuations, respectively. τ=0 gives the equal-time cross-correlations of atomic fluctuations.
Supplementary Material
Table 3.
The maximum percentage of time that the protease-inhibitor hydrogen bonds existed during the MD simulations.
| Protease | Inhibitor
|
|||
|---|---|---|---|---|
| ITMN-191 | MK-5172 | MK-7009 | ||
| G137 – N | P1′ | 78 | 77 | 74 |
| S139 – N | P1′ | 71 | 67 | 69 |
| G137 – N | P1 | <50 | <50 | <50 |
| S139 – OG | P1 | 95 | 95 | 90 |
|
|
||||
| R155 – O | P1 | 89 | 53 | 93 |
| A157 – N | P3 | 89 | 87 | 89 |
| A157 – O | P3 | 76 | 90 | 88 |
Acknowledgments
Funding Resources
This work was supported by the National Institute of Health grant R01-AI085051.
We thank Nese Kurt Yilmaz for critical reading of the manuscript and further editorial assistance.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Figures showing the individual substrate volumes compared to the dynamic substrate envelope, flexibility of substrate/inhibitor subsites, correlated atomic fluctuations and the dynamic network of salt bridges formed at the binding surface in substrate/inhibitor complexes are provided in the Supporting Information. Supporting figures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Seiwert SD, Kossen K, Pan L, Liu J, Buckman BO. Antiviral Drugs. John Wiley & Sons, Inc; 2011. Discovery and Development of the HCV NS3/4A Protease Inhibitor Danoprevir (ITMN-191/RG7227) pp. 257–271. [Google Scholar]
- 2.Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B, Koch U, Petrocchi A, Holloway MK, Butcher JW, Romano JJ, Bush KJ, Gilbert KF, McIntyre CJ, Nguyen KT, Nizi E, Carroll SS, Ludmerer SW, Burlein C, DiMuzio JM, Graham DJ, McHale CM, Stahlhut MW, Olsen DB, Monteagudo E, Cianetti S, Giuliano C, Pucci V, Trainor N, Fandozzi CM, Rowley M, Coleman PJ, Vacca JP, Summa V, Liverton NJ. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med Chem Lett. 2012;3(4):332–336. doi: 10.1021/ml300017p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCauley JA, McIntyre CJ, Rudd MT, Nguyen KT, Romano JJ, Butcher JW, Gilbert KF, Bush KJ, Holloway MK, Swestock J, Wan BL, Carroll SS, DiMuzio JM, Graham DJ, Ludmerer SW, Mao SS, Stahlhut MW, Fandozzi CM, Trainor N, Olsen DB, Vacca JP, Liverton NJ. Discovery of vaniprevir (MK-7009), a macrocyclic hepatitis C virus NS3/4a protease inhibitor. J Med Chem. 2010;53(6):2443–2463. doi: 10.1021/jm9015526. [DOI] [PubMed] [Google Scholar]
- 4.Qureshi SA. Hepatitis C virus--biology, host evasion strategies, and promising new therapies on the horizon. Med Res Rev. 2007;27(3):353–373. doi: 10.1002/med.20063. [DOI] [PubMed] [Google Scholar]
- 5.Cabot B, Martell M, Esteban JI, Piron M, Otero T, Esteban R, Guardia J, Gomez J. Longitudinal evaluation of the structure of replicating and circulating hepatitis C virus quasispecies in nonprogressive chronic hepatitis C patients. J Virol. 2001;75(24):12005–12013. doi: 10.1128/JVI.75.24.12005-12013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martell M, Esteban JI, Quer J, Genesca J, Weiner A, Esteban R, Guardia J, Gomez J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J Virol. 1992;66(5):3225–3229. doi: 10.1128/jvi.66.5.3225-3229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chellappan S, Kiran Kumar Reddy GS, Ali A, Nalam MN, Anjum SG, Cao H, Kairys V, Fernandes MX, Altman MD, Tidor B, Rana TM, Schiffer CA, Gilson MK. Design of mutation-resistant HIV protease inhibitors with the substrate envelope hypothesis. Chem Biol Drug Des. 2007;69(5):298–313. doi: 10.1111/j.1747-0285.2007.00514.x. [DOI] [PubMed] [Google Scholar]
- 8.Altman MD, Ali A, Reddy GS, Nalam MN, Anjum SG, Cao H, Chellappan S, Kairys V, Fernandes MX, Gilson MK, Schiffer CA, Rana TM, Tidor B. HIV-1 protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. J Am Chem Soc. 2008;130(19):6099–6113. doi: 10.1021/ja076558p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kairys V, Gilson MK, Lather V, Schiffer CA, Fernandes MX. Toward the design of mutation-resistant enzyme inhibitors: further evaluation of the substrate envelope hypothesis. Chem Biol Drug Des. 2009;74(3):234–245. doi: 10.1111/j.1747-0285.2009.00851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altman MD, Nalivaika EA, Prabu-Jeyabalan M, Schiffer CA, Tidor B. Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease. Proteins. 2008;70(3):678–694. doi: 10.1002/prot.21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalam MN, Ali A, Altman MD, Reddy GS, Chellappan S, Kairys V, Ozen A, Cao H, Gilson MK, Tidor B, Rana TM, Schiffer CA. Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol. 2010;84(10):5368–5378. doi: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure. 2002;10(3):369–381. doi: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- 13.Ozen A, Haliloglu T, Schiffer CA. Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J Mol Biol. 2011;410(4):726–744. doi: 10.1016/j.jmb.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozen A, Haliloglu T, Schiffer CA. HIV-1 Protease and Substrate Coevolution Validates the Substrate Envelope as the Recognition Pattern. J Chem Theory Comput. 2012;8:703–714. doi: 10.1021/ct200668a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA. Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem Biol. 2004;11(10):1333–1338. doi: 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 16.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6(1):89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 17.Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4(6):463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 18.Romano KP, Ali A, Royer WE, Schiffer CA. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc Natl Acad Sci USA. 2010;107(49):20986–20991. doi: 10.1073/pnas.1006370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure. 1999;7(11):1353–1363. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 20.Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 2012;8(7):e1002832. doi: 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali A, Aydin C, Gildemeister R, Romano KP, Cao H, Ozen A, Soumana D, Newton A, Petropoulos CJ, Huang W, Schiffer CA. Evaluating the Role of Macrocycles in the Susceptibility of Hepatitis C Virus NS3/4A Protease Inhibitors to Drug Resistance. ACS Chem Biol. 2013;8(7):1469–1478. doi: 10.1021/cb400100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin C, Thomson JA, Rice CM. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J Virol. 1995;69(7):4373–4380. doi: 10.1128/jvi.69.7.4373-4380.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J Virol. 1994;68(8):5045–5055. doi: 10.1128/jvi.68.8.5045-5055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin C, Pragai BM, Grakoui A, Xu J, Rice CM. Hepatitis C virus NS3 serine proteinase: trans-cleavage requirements and processing kinetics. J Virol. 1994;68(12):8147–8157. doi: 10.1128/jvi.68.12.8147-8157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue W, Ban Y, Liu H, Yao X. Computational Study on the Drug Resistance Mechanism against HCV NS3/4A Protease Inhibitors Vaniprevir and MK-5172 by the Combination Use of Molecular Dynamics Simulation, Residue Interaction Network, and Substrate Envelope Analysis. J Chem Inf Model. 2013 doi: 10.1021/ci400060j. [DOI] [PubMed] [Google Scholar]
- 26.Shiryaev SA, Thomsen ER, Cieplak P, Chudin E, Cheltsov AV, Chee MS, Kozlov IA, Strongin AY. New Details of HCV NS3/4A Proteinase Functionality Revealed by a High-Throughput Cleavage Assay. PLoS One. 2012;7(4):e35759. doi: 10.1371/journal.pone.0035759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cummings MD, Lindberg J, Lin TI, de Kock H, Lenz O, Lilja E, Fellander S, Baraznenok V, Nystrom S, Nilsson M, Vrang L, Edlund M, Rosenquist A, Samuelsson B, Raboisson P, Simmen K. Induced-fit binding of the macrocyclic noncovalent inhibitor TMC435 to its HCV NS3/NS4A protease target. Angew Chem, Int Ed. 2010;49(9):1652–1655. doi: 10.1002/anie.200906696. [DOI] [PubMed] [Google Scholar]
- 28.Velazquez-Campoy A, Freire E. Incorporating target heterogeneity in drug design. J Cell Biochem Suppl. 2001;(Suppl 37):82–88. doi: 10.1002/jcb.10068. [DOI] [PubMed] [Google Scholar]
- 29.Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, Dimuzio JM, Ferrara M, Di Filippo M, Gates AT, Graham DJ, Harper S, Hazuda DJ, McHale C, Monteagudo E, Pucci V, Rowley M, Rudd MT, Soriano A, Stahlhut MW, Vacca JP, Olsen DB, Liverton NJ, Carroll SS. MK-5172, a selective inhibitor of Hepatitis C Virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 doi: 10.1128/AAC.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romano KP, Laine JM, Deveau LM, Cao H, Massi F, Schiffer CA. Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J Virol. 2011;85(13):6106–6116. doi: 10.1128/JVI.00377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snyder PW, Mecinovic J, Moustakas DT, Thomas SW, 3rd, Harder M, Mack ET, Lockett MR, Heroux A, Sherman W, Whitesides GM. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc Natl Acad Sci USA. 2011;108(44):17889–17894. doi: 10.1073/pnas.1114107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beuming T, Che Y, Abel R, Kim B, Shanmugasundaram V, Sherman W. Thermodynamic analysis of water molecules at the surface of proteins and applications to binding site prediction and characterization. Proteins. 2012;80(3):871–883. doi: 10.1002/prot.23244. [DOI] [PubMed] [Google Scholar]
- 33.Robinson DD, Sherman W, Farid R. Understanding kinase selectivity through energetic analysis of binding site waters. Chem Med Chem. 2010;5(4):618–627. doi: 10.1002/cmdc.200900501. [DOI] [PubMed] [Google Scholar]
- 34.Huggins DJ, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design. J Med Chem. 2012;55(4):1424–1444. doi: 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suite 2011: Prime, Version 3.0. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
- 36.Davis AM, Teague SJ, Kleywegt GJ. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew Chem, Int Ed. 2003;42(24):2718–2736. doi: 10.1002/anie.200200539. [DOI] [PubMed] [Google Scholar]
- 37.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput-Aided Mol Des. 2013 doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 38.Suite 2011: Maestro, Version 9.2. Schrödinger, LLC; Portland, OR: 2011. [Google Scholar]
- 39.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida. 2006. [Google Scholar]
- 40.Suite 2011: Desmond Molecular Dynamics System, version 3.0. D. E. Shaw Research; New York, NY: 2011. [Google Scholar]; Maestro-Desmond Interoperability Tools, version 3.0. Schrödinger; New York, NY: 2011. [Google Scholar]
- 41.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 42.Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J Chem Theory Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 43.Darden T, Darrin Y, Pedersen L. Particle mesh Ewald: An N–log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10093. [Google Scholar]
- 44.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 45.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 46.The PyMOL Molecular Graphics System. Schrödinger, LLC; New York, NY: 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.