

Abstract
Catalytic radical-based domino reactions represent important advances in synthetic organic chemistry. Their development benefits synthesis by providing atom- and step-economical methods to complex molecules. Intricate combinations of radical, cationic, anionic, oxidative/reductive, and transition metal mechanistic steps result in cyclizations, additions, fragmentations, ring-expansions, and rearrangements. This Perspective summarizes recent developments in the field of catalytic domino processes.
Keywords: radical, domino, cascade, tandem, transition metal, photoredox
Introduction
Synthetic organic chemists continuously pursue new reactions and chemoselective transformations under mild conditions. To this end, new methods for the facile and efficient installation of complexity in small molecules are in constant development. The ideal transformation is an efficient single reaction with one set of reagents that yields a desired product. Great efforts have been made to develop efficient single step transformations, which result in the formation and fragmentation of multiple bonds in a step-wise fashion. These reactions have been termed domino, cascade, or tandem processes.1
A domino reaction is a transformation that installs two or more bonds under identical conditions. The advantages of methods that construct complex molecules in a single reaction are self-evident, providing both atom2 and step economy.3 Tietze has described at length the factors that define a domino reaction.1f First, all reactants and reagents must be added at the beginning of the process without further addition. Next, two or more bonds must be formed or, in some cases, broken. Each bond-forming step must happen sequentially, resulting in a “time-resolved succession of steps” that occur linearly from the point of view of a reaction mechanism.1b The steps within the mechanism can be classified as cationic, anionic, pericyclic, photochemical, transition-metal mediated, oxidative or reductive, enzymatic, or radical.
Historically, organic radicals were deemed uncontrollable and unselective except under tightly managed reaction conditions. However, great mechanistic efforts have been applied to gain insight into the reactivity of the free radical in order to understand how best to apply this reactive intermediate. As a result, radical-mediated processes provide nonpolar access to the formation of carbon-carbon and carbon-heteroatom bonds.4 Traditional radical domino reactions are thought of as a series of rapid intramolecular cyclizations of pendant olefins and alkynes. However, radical-based domino processes have emerged that undergo additions, fragmentations, ring-expansions, and rearrangements under a variety of conditions.
Many radical transformations that proceed through atom transfer are initiated by the addition of stoichiometric stannanes, silanes, or peroxides. Additionally, these reactions typically require the use of, albeit a substoichiometric amount, an unstable initiator.5 Redox-mediated transformations usually require one or more equivalents of metal oxidant or reductant.6 Both of these methods are routinely utilized in organic synthesis; however, radical transformations that utilize a catalytic electron transfer agent alleviate the need for stoichiometric metals or harsh atom abstractors. Significant efforts have been focused towards the development of catalytic radical reactions using redox-active transition metals7 and photo-active catalysts.8
This Perspective outlines recent efforts to apply catalytic electron transfer agents towards the development of radical domino reactions. The selection of transformations within highlights the diverse number of methods available to synthetic chemists. Many examples of catalytic radical-mediated processes exist, but we seek to display those that form/break two or more bonds prior to the generation of the final product. These examples were chosen because a radical participates in at least the first bond-forming step and that one or more subsequent steps result from this initial transformation.
Transition metal-mediated domino reactions
The seminal works of Kharash,9 Kochi,10 and Minisci11 clearly demonstrated that carbon radicals could be formed in the presence of a transition metal catalyst. Since their efforts, a great number of transformations have been reported in the literature.7,12 These methods employ copper, manganese, iron, cobalt, titanium, and ruthenium catalysts to perform atom transfer, as well as single electron reductions and oxidations. In recent years, radical domino reactions utilizing transition metal complexes have garnered considerable attention. These systems combine traditional radical transformations with cationic, anionic, oxidative, reductive, or pericyclic steps to provide access to fragmentations, rearrangements, and complex ring systems.
Cu-mediated atom transfer radical cyclizations (ATRC) have been developed extensively over the years.7 In 2007, Quayle and coworkers continued their development of Cu-mediated ATRC reactions with trichloroacetates.13 They observed that microwave irradiation at 200 °C supplemented the ATRC reaction of 1, resulting in a domino benzannulation to form chloronaphthalenes (Figure 1). The authors propose a radical/radical/pericyclic/anionic domino process. Beginning with a Cu(I)-mediated chlorine atom transfer from 11, dichlororadical 4 and Cu(II) complex 5 are formed at the onset. Radical 4 then undergoes a preferred 8-endo-trig cyclization14 to yield radical 6, which is then chlorinated by 5 to form lactone 7. Quayle et al. detected no annulation when 7, easily accessible via conventional heating, was irradiated for 2 h in the absence of other reagents. 2 was only observed in the presence of CuCl and 3. As a result, they proposed that 7 reacts with a second Cu complex to yield radical 8 and chloro-complex 5. This radical then undergoes a reversible 4-exo-trig cyclization, which is facilitated by chlorine atom transfer from 5, to form spirolactone 9. Retro [2+2] cyclization, driven by elimination of CO2, converts 9 to 10, which, upon the elimination of 2 equiv HCl, yields 4-chlorophenanthrene (2).
Figure 1.

Cu-mediated domino benzannulation of 1 and mechanism.
In 2006, Yang et al. found that ATRC reactions of α,α’-dichloro-β-ketoesters could be facilitated by 30 mol% CuCl in the presence of chiral, bidentate amine ligands (Figure 2).15 However, when the system was extended beyond the formation of a single C-C bond, use of 2,2’-bipyridine (bpy) provided optimal reactivity. The domino bicyclization occurs through a radical/radical process initiated by removal of a chlorine atom from 11 by Cu(I). Radical 13 then undergoes a 6-endo-trig cyclization to form 14, which is then trapped by a rapid 5-exo-trig cyclization to yield primary radical 15. The primary radical then reacts with Cu(II) complex 14 to form 12 in a 61% yield as a 2.3:1 ratio of diastereomers and turnover of the catalyst.
Figure 2.

Cu-mediated ATRC of 11 and mechanism.
Pérez, Belderraín, and Muñoz-Molina developed a diastereoselective Cu-catalyzed ATRC domino reaction (Figure 3).16 Additionally, they found that when Mg was added as a reducing agent, they observed improved yields. This radical/radical process is initiated by abstraction of a chlorine atom from CCl4. The resulting trichloromethyl radical then adds to one of the allyl groups of 17 to form radical 19. This product radical then undergoes a rapid 5-exo-trig cyclization followed by chlorine atom transfer to provide 18 in a 99% yield and turn over the catalyst. Pintauer and coworkers found that the turnover efficiency of this Cu-mediated reaction could be greatly enhanced to where only 0.01 mol% catalyst is required with tris(2-pyridylmethyl)amine as a ligand and substoichiometric amounts of either a diazo initiator17 or ascorbic acid18 as reducing agents.
Figure 3.

Cu-mediated radical addition/cyclization of 17 and mechanism.
In their seminal work, Mori and Ban first demonstrated the ability of low-valent Pd to mediate ATRC processes of α-haloamides.19 In recent years, chemists have catalytic single electron reductions with carbonylations. Ryu, Komatsu, and coworkers found that when 21 was exposed to light under 40 atm of CO and a catalytic amount of Pd(PPh3)4, γ-ketoester 22 was formed (Figure 4).20 They propose a radical/radical/radical/reduction process. Under irradiation, an iodine atom is removed from 21 by Pd(0) to form primary radical 23. This radical adds to CO, forming 24, and then undergoes a rapid 5-exo-trig cyclization, yielding radical 25. The resulting β-keto radical then attacks another equivalent of CO to form 26, which is reduced by Pd(I) to form organopalladium(II) complex 27. Addition of butanol and reductive elimination yields 22 and turns over the catalyst. They later found that when H2O and a boronic acid (28) were substituted for the alcohol, transmetallation and reductive elimination resulted in the formation of diketone 29 (Figure 5).21
Figure 4.

Pd-mediated radical cyclization and carbonylation of 21 and mechanism.
Figure 5.
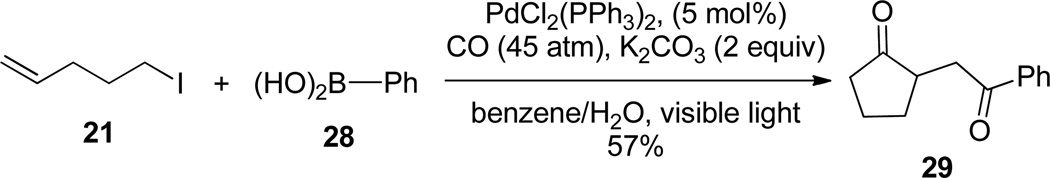
Pd-mediated radical cyclization, carbonylation, and arylation of 21.
Continuing their work on radical carbonylations, Ryu and coworkers designed a method for the formation of lactones (32) from their conditions (Figure 6).22 When irradiated with light, this radical/radical/reduction process begins with the abstraction of an iodine atom from 30 to form radical 33. This primary radical then adds to olefin 31 to form 34, which can add to an equivalent of CO to form keto-radical 35. Reduction of this intermediate results in the formation of organopalladium(II) complex 36. Addition of the pendant hydroxyl group and reductive elimination regenerates the catalyst and forms lactone 32 in a 77% yield.
Figure 6.

Pd-mediated radical addition and carbonylation of 30.
Similarly, Alexanian and Bloom found that an analogous transformation could be mediated by low-valent palladium in the absence of light.23 Alkyl iodide 37 participates in a carbonylative-Heck reaction at elevated temperatures in toluene to give spiro-cyclopentanone 38 (Figure 7). This radical/radical/oxidative process begins with removal of an iodine atom from 37 to form primary radical 39. Similar to the mechanism in Figure 4, radical addition to CO provides access to a rapid 5-exo-trig cyclization, yielding tertiary radical 41. Single electron oxidation by Pd(I) yields tertiary cation 42, which can undergo facile deprotonation at the newly formed α-position of 38.
Figure 7.

Pd-catalyzed radical cyclization and carbonylation of 37 and mechanism.
Oshima and coworkers developed a Co-mediated process that is initiated by atom transfer in 2003.24 They observed that when 43 and 44 were combined in the presence of trimethylsilylmethylmagnesium chloride and a catalytic amount of CoCl2 ligated by 1,6-bis(diphenylphosphino)hexane (dpph), silane 45 was produced in excellent yield (Figure 8). This transformation provides insight into the mechanism. Furan 48 was produced via a radical/radical/reduction/Tsuji-Trost allylation process. A bromine atom is removed from 43 by Co(0) to form primary radical 46 and Co(I) complex 47. This primary radical undergoes rapid 5-exo-trig cyclization to form 48, which then adds to butadiene 44. Allylic radical 49 is then reduced by 47 to form complex 50. The metal center of 50 is then alkylated by the Grignard reagent to form 51, which undergoes reductive elimination to form furan 45 and regenerate the catalyst.
Figure 8.

Co-catalyzed radical cyclization-Tsuji-Trost allylation of 43 and mechanism.
Fu and Cardenas independently developed Ni-catalyzed cross-coupling methods initiated by atom transfer. Initially, Fu et al. demonstrated that both Ni-catalyzed Stille cross-couplings25 and Suzuki cross-couplings26 of secondary alkyl halides began via low-valent Ni-mediated radical formation (Figure 9). Later, Cardenas et al. demonstrated the same radical process for Ni-catalyzed Negishi cross-coupling of secondary alkyl halides.27 These three methods proceed through similar radical/reduction/anionic domino sequences. Initially, a halogen atom is removed from alkyl halide 59 to form secondary radical 60 and Ni(I) complex 61. The secondary radical undergoes rapid 5-exo-trig cyclization to form primary radical 62, which is then reduced by 61 to form organonickel 63. This complex undergoes transmetallation with agent 64 to form organonickel 65. Upon reductive elimination, Ni(0) is regenerated, yielding 66.
Figure 9.

Ni-catalyzed radical cyclization-Stille coupling of 52, Ni-catalyzed radical cyclization-Suzuki coupling of 52, Ni-catalyzed radical cyclization-Negishi coupling of 56 and their mechanism.
In addition to atom transfer reactions, radical-mediated domino reactions can be initiated with single electron reductions. In 2012, Barrero, Moral, and coworkers applied a radical/radical/oxidation domino process to the total synthesis of (+)-seco-C-oleanane (68A) (Figure 10).28 Most notable about this synthesis is that performing this reaction with a substoichiometric amount of Cp2TiCl2 improved the efficiency of the desired 6-endo-trig/6-endo-trig bicyclization process over the use of stoichiometric Ti reductant. The authors propose that this process begins with single electron reduction and ring-opening of epoxide 67 to give tertiary radical 69. This radical then undergoes two consecutive 6-endo-trig radical cyclizations to form radical 71. Hydrogen atom abstraction of hydrogen atoms A or B (Figure 10) followed by TMS-protection yields TMS protected TMS-68A as well as TMS protected β-seco-amyrin (TMS-68B).
Figure 10.

Ti-catalyzed reductive polyene cyclization of 67 and mechanism.
Similar to the reductive capabilities of Ti(III), Aubé et al. utilized the reducing power of Cu(I) to ring-open chiral oxaziridines to form nitrogen-centered radicals (Figure 11).29 They observed that when oxaziridine 72 was refluxed in THF in the presence of [Cu(PPh3)Cl]4, pyrroline 73 was formed in 66% yield with greater than 95% ee. This radical rearrangement potentially occurs through a radical/radical/oxidation/pericyclic domino process. At the onset, Cu(I) reduces oxaziridine 72, which ring-opens to form nitrogen-centered radical 74. Rapid 5-exo-trig cyclization of 74 results in pyrrolidine radical 75. The authors then propose that radical 76 is formed when 75 undergoes 1,4-aryl migration via ipso attack on the aryl ring. Finally, Cu(I) and acetaldehyde are eliminated to form pyrroline 73. While not proposed by the authors, this rearrangement/elimination may be initiated by single electron oxidation of the alkoxide to form an oxygen-centered radical and regenerate the catalyst. Intramolecular radical coupling results in the formation of 77, which can undergo a retro [2+2] cyclization to form pyrroline 73.
Figure 11.

Cu-catalyzed redox neutral rearrangement of 72 and mechanism.
Conversely, when the oxaziridine diastereomer 78 was exposed to the same conditions, a radical/radical domino process is observed that results in rearrangement to aziridine 79 (Figure 12), initiated by a Cu-mediated ring-opening of 78. An analogous 5-exo-trig cyclization of 80 leads to pyrroline radical 81. However, they propose that the transition state that would result in phenyl transfer is disfavored due to the steric constraints of this diastereomer. As a result, intramolecular radical addition to the nitrogen simultaneously forms an aziridine moiety and ring-opens the pyrrolidine, resulting in benzylic radical 82. Single electron oxidation by Cu(II) regenerates the catalyst and yields 79.
Figure 12.

Cu-catalyzed redox neutral rearrangement of 78 and mechanism.
Ag(II) is a powerful oxidant that is easily generated by the combination of Ag(I) and persulfate, which has been utilized by chemists for decades.30 A considerable wealth of mechanistic and kinetic information has been compiled over the years, and chemists use these data to develop new transformations. In 2006, Narasaka and coworkers applied this oxidative system to the single electron-mediated ring-opening of cyclopropanols (Figure 13).31 Notably, this method provided diminished yields when performed in the absence of pyridine. The improvement is attributed to coordination of pyridine to Ag(I), activating the metal towards oxidation by persulfate.32 Attachment of a pendant olefin to the cyclopropanol provided access to 84. This reaction occurs via a radical/radical domino process. Ag complex 85 is oxidized by persulfate to Ag(II) complex 86, which is capable of oxidizing cyclopropanol 83 to an alkoxy radical. This radical then undergoes a retro 3-exo-trig cyclization to form heptanone radical 88. A 5-exo-trig cyclization forms primary radical 89, which abstracts a hydrogen atom from 1,4-cyclohexadiene to form 84.
Figure 13.

Ag-catalyzed oxidative fragmentation/cyclization of 83 and mechanism.
Yao, Hu, Zhang, and coworkers utilized the Minisci reaction33 to generate fused pentacycles (92) from indolylpropanoic acids (90) and quinones (91) (Figure 14).34 These carbazoles are formed via a radical/radical/radical/oxidation/anionic/oxidation domino process. This transformation begins by oxidation of Ag(I) to Ag(II), which then oxidizes 90 to a carboxy radical (93). Elimination of CO2 leads to primary radical 94 that adds to quinone 91. Radical 95 then undergoes a 6-endo-trig cyclization with the pendant indole to complete the pentacyclic framework. Next, tertiary radical 96 is converted to 97 via one of three pathways: 1) hydrogen atom abstraction, 2) oxidation to the cation and then deprotonation, 3) deprotonation to the radical anion followed by oxidation. The central ring of the pentacycle is then aromatized via elimination of HBr and oxidation to yield 92.
Figure 14.

Ag-catalyzed Minisci/cyclization of 90 and 91 and mechanism.
In 2010, Baran and coworkers developed an intermolecular Minisci reaction for boronic acids.35 A year later, they demonstrated an intramolecular variant that could utilize either boronic acids or trifluoroborates (Figure 15).36 Utilizing quinones as radicophiles, the Baran group were able to perform a radical/radical domino reaction to yield 100. In this process, Ag(II) is generated via a persulfate-mediated single electron oxidation. Then, Ag(II) oxidizes quaternized 9837 to form aryl radical 101, which cyclizes to secondary radical 102. Quinone 99 then adds to the radical and forms 100 after hydrogen atom abstraction.
Figure 15.

Ag-catalyzed oxidative cyclization of 98 and addition to 99 and mechanism.
MacMillan et al. devised a series of domino reactions by combining the properties of the SOMO-activated addition of styrene38 and the intramolecular arylation.39 In the original styrene approach, the product was formed by internal ligand transfer of a nitrate from ceric ammonium nitrate to a benzylic cation. MacMillan and coworkers utilized this electrophilic intermediate for follow-up nucleophilic attacks from aromatic ring and pendant, protected amines.40 Application of these nucleophiles provides access to multicyclic ring systems containing substituted benzenes such as 105 (Figure 16) or pyrrolidines. These cyclizations proceed through a radical/oxidation/cationic domino mechanism. First, condensation of aldehyde 103 and catalyst 106 forms electron-rich enamine 107, which is then oxidized by Fe(III) to yield radical cation 108. Addition of radicophile 104 to 108 yields product radical cation 109. A secondary Fe(III)-mediated oxidation forms dication 110, which then undergoes an intramolecular Friedel-Crafts alkylation, proton transfer, and hydrolysis to form 105 and regenerate the catalyst.
Figure 16.

Fe(III)-mediated organo-SOMO cycloaddition of 103 and 104 with mechanism.
The MacMillan group further extended the reactivity of the radical intermediate through unsaturations in polyenes to form multiple fused six-membered rings in one reaction with high enantioselectivity (Figure 17).41 Reaction of 113 in the presence of catalyst 115 and Cu(II) as an oxidant formed hexacyclic 114 in 63% yield and 93% ee. This domino reaction undergoes four consecutive 6-endo-trig cyclizations. The mechanism is initiated by condensation of 113 with 115 to form an enamine, which is oxidized by Cu(II) to form distonic radical cation 116. This radical cation then undergoes rapid and successive 6-endo-trig cyclization, terminating in the formation of conjugated radical 117. Oxidation and elimination of a proton yield iminium 118, which, upon hydrolysis, releases catalyst 115 and forms 114.
Figure 17.

Cu(II)-mediated organo-SOMO polyene cyclization of 113 and mechanism.
Domino reactions mediated by photoredox catalysis
Visible light photoredox catalysis has established itself as a powerful technique for enacting free radical transformations.8,42 Typical photocatalysts are transition metal complexes or organic dyes that, upon photoexcitation by visible light to an excited triplet state, can proceed through oxidative or reductive quenching pathways to enact single electron transfers (Figure 18). It is emerging as a versatile and advantageous technique for mediating free radical reactions for several reasons. First, photoredox catalysts are activated by visible light, and unlike a traditional photochemical apparatus, reactions are conducted in typical borosilicate glassware with simple light fixtures. Second, the stoichiometric electron carrier is typically an inexpensive amine as opposed to stannanes or silanes found in traditional radical processes. Finally, reactions are very robust with typical transformations proceeding efficiently at low catalyst loadings.
Figure 18.

General photoredox paradigm.
In 2013, Zhu and coworkers developed a visible light-mediated, room temperature, decarboxylation/aromatic substitution of aniline 119 to oxindole 120 (Figure 19) via a radical/radical/cation domino process.43 In this transformation, the excited state of Ir(ppy)3 is oxidatively quenched by iodosobenzene diacetate (PIDA). The PIDA radical then fragments into iodobenzene, CO2 and a methyl radical, which adds to 119. Intramolecular cyclization to the arene installs the quaternary center in 122. Thereafter, oxidation to the resonance stabilized cation followed by deprotonation leads to aromatization of the 3-disubstituted product.
Figure 19.

Visible light-mediated methyl transfer to 119 and cyclization and mechanism.
In 2012, König and coworkers developed an eosin Y-catalyzed synthesis of benzothiophene derivatives (126) (Figure 20).44 This method utilizes a radical/radical/oxidative/cation domino reaction to access these heteroarenes. Starting from the o-methylthio-benzenediazonium salt 124, visible light irradiation excites the photocatalyst to the triplet state, which is oxidatively quenched by the diazonium salt to release N2 gas and aryl radical 127. In the presence of p-nitro phenyl acetylene (125), radical addition yields vinyl in the radical 128. Addition to sulfur followed by oxidation forms thionium 130. Methyl transfer results in formation of 126.
Figure 20.

Visible light-mediated reduction/addition/cyclization of 124 and 125 and mechanism.
In a recent report by Zhou and coworkers, eosin Y induced a [4+2] benzannulation of biaryldiazonium salts with alkynes (Figure 21).45 This radical/radical/cation domino reaction produced a variety of 9- and 9,10-disubstituted phenanthrenes. Irradiation with visible light excites eosin Y from the ground state. A single electron transfer from the excited photocatalyst to aryl diazonium 131 results in the radical cation of the catalyst and aryl radical 134. Radical addition to alkyne 132 affords vinyl radical 135. Intramolecular cyclization followed by single electron oxidation yields the carbocation 137 and the regenerated catalyst. Finally, deprotonation of 137 results in desired phenanthrene 133.
Figure 21.

Visible light-mediated reductive benzannulation of 131 and 132 and mechanism.
The Stephenson group reported a tin-free reductive dehalogenation reaction utilizing a visible light activated photocatalyst in 2009.46 This method, which formed a radical intermediate, was applied to the functionalization of indoles and pyrroles (Figure 22).47 Utilizing the radical generated from C-Br cleavage, a radical/radical/oxidation process yielded tetracyclic indole 139. The proposed mechanism begins with irradiation of the photocatalyst with visible light to afford an electron accepting excited state that is reductively quenched by triethylamine to form a triethylamine radical cation and Ru(I). Reduction of the C-Br bond of 138 affords tertiary radical 140, which undergoes successive 5-exo-trig and 6-exo-trig cyclizations to form radical 142. The indole radical is then rearomatized, affording the tetracyclic product 139.
Figure 22.

Visible light-mediated ATRC of 138 and mechanism.
Inspired by the work of Curran48 and Tanabe,49 Stephenson and coworkers continued to develop radical/radical domino reactions onto unactivated π-systems.50 The reaction led to a variety of products including 5- and 6-membered rings as well as fused and spiro products (Figure 23). When applied to this domino system, photoexcitation of the metal catalyst and reductive quenching affords the active single electron reductant Ru(I). The electron rich catalyst then reduces the C-Br bond of 144 to yield the regenerated Ru(II) and radical 146. This radical then undergoes two successive 5-exo-trig cyclizations to form vinyl radical 148. Finally, the carbon radical abstracts a hydrogen atom to form tricyclic product 145 in a 69% yield.
Figure 23.

Visible light-mediated radical cyclization of 144 and mechanism.
Following the development of a general cyclization strategy, the Stephenson group applied this concept to the formation of tricyclic pyrrolidinones (Figure 24).51 The system proceeds through either a radical/radical/pericyclic or a radical/radical/radical domino mechanism. The reaction, outlined in Figure 24, begins with the reductive quenching of photoexcited Ir(III) by Et3N to form an Ir(II) complex. Single electron transfer to 149 provides α-radical 151, which quickly undergoes a 5-exo-dig cyclization with the pendant alkyne to form lactam 152. At this point, the mechanism was less clear. In path A, 152 can undergo hydrogen atom abstraction followed by a thermal sigmatropic rearrangement that is driven by strain release of the cyclopropyl ring to yield 150. Alternatively, radical 152 can undergo an additional cyclization onto the aromatic ring, followed by β-scission to form radical 154B (path B). Then, isomerization and hydrogen atom abstraction may lead to product 150. Interestingly, trace quantities of 153A were isolated from the reaction mixture. To provide further mechanistic insight, intermediate 153A was heated at 40 °C in DMF in the absence of other redox conditions. The desired fused pyrrolidinone (150) was the only observed product, suggesting that path A is the likely route.
Figure 24.

Visible light-mediated radical cyclization/fragmentation of 149 and mechanism.
In 2008, the Yoon group developed a photocatalytic [2+2] reductive cyclization of a (bis)enone bearing a three-carbon tether.52 Later, the authors observed that when the bis(enone) tether was expanded by the addition of a methylene unit under the same conditions, the expected products were not formed and the observed products included dihydropyran 157 (Figure 25).53 Intrigued by this observation, the authors optimized a photocatalytic radical/radical reaction to access dihydropyrans. The proposed mechanism involves reductive quenching of the excited state of the ruthenium catalyst, followed by single electron reduction of Li-activated (bis)enone (LA-156) to form radical 158. This allylic radical then undergoes a 6-exo-trig cyclization to form α-radical 159. Enolate attack on the α-radical leads to ketyl 160, which is then oxidized to form desired tetrahydropyran 157 in high yield (86%).
Figure 25.

Visible light-mediated redox neutral bicyclization of 156 and mechanism.
Continuing their efforts to combine Lewis acid activation with photoredox catalysis, the Yoon group reported a photocatalytic [3+2] cycloaddition of activated cyclopropanes with olefins (Figure 26).54 This radical/radical/radical domino process results in the formation of fused cyclopentane rings. Single electron reduction of LA-activated 161 by the photocatalyst results in ketyl radical anion formation (163). The subsequent retro 3-exo-trig cyclization drives the reaction forward to allyl radical 164. Facile 5-exo-trig cyclization onto the pendant olefin forms tertiary radical intermediate 165. Finally, ring closure via another 5-exo-trig cyclization and single electron oxidation affords fused [3.3.0] product 162 in a 6:1 diastereomeric ratio.
Figure 26.

Visible light-mediated redox neutral fragmentation/bicyclization of 161 and mechanism.
In 2012, Zhao and coworkers showcased a visible light-initiated reductive coupling/aldol cyclization of chalcones (Figure 27).55 During preliminary studies, the expected [2+2] cyclobutane product was not observed. Rather, exposure of chalcone 167 to photoredox conditions resulted in a complex mixture of products, which included cyclic dimer 168 in 24% yield. A Lewis acid screen demonstrated Sm(OTf)3 as an effective additive to activate the carbonyl of 167. Single electron reduction of 167 coordinated to Sm(III) generates reactive β-radical intermediate 169, which adds to another equivalent of 167 to form 170. Hydrogen atom abstraction forms enolate 171 that is subsequently protonated and undergoes an intramolecular aldol addition to form 168 in a 62% yield.
Figure 27.

Visible light-mediated redox neutral dimerization/cyclization of 167 and mechanism.
Conclusion
These transition metal- and visible light-mediated radical processes demonstrate the efforts of the synthetic organic community to develop new and efficient methods for the generation of complex molecules in both an atom- and step-economical fashion. Both transition metal and photoredox domino processes garner access to a wide variety of products. While the mechanisms illustrated here all show clear, succinct catalytic systems, it is also possible for these reactions to operate additionally through short-chain propagation. However, in all of the systems presented, a catalyst is required at a minimum for initiation. Further studies of the discrete interactions of each system might shed insight onto which mechanistic pathway is dominant. These data could provide important details necessary to expand the selection of complex transformations in this area.
ACKNOWLEDGMENT
Financial support from the NIH-NIGMS (R01-GM096129) the Alfred P. Sloan Foundation, the Camille and Henry Dreyfus Foundation, Amgen, Boehringer Ingelheim, Eli Lilly, Novartis and the University of Michigan is gratefully acknowledged. We would like to thank Mr. Joel Beatty, Dr. Milena Czyz, Dr. James Douglas, Mr. Mitch Keylor, Mr. Bryan Matsuura, Mr. John Nguyen, and Dr. Elizabeth Swift for their helpful discussions and suggestions.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.For reviews on these types of reactions, see: Tietze LF, Beifuss U. Angew. Chem. Int. Ed. in Engl. 1993;32:131–163. Tietze LF. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e. Denmark SE, Thorarensen A. Chem. Rev. 1996;96:137–166. doi: 10.1021/cr940277f. Ryu I, Sonoda N, Curran DP. Chem. Rev. 1996;96:177–194. doi: 10.1021/cr9400626. Malacria M. Chem. Rev. 1996;96:289–306. doi: 10.1021/cr9500186. Tietze LF, Brasche G, Gericke KM. Domino Reactions in Organic Synthesis. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2006. Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem. Int. Ed. in Engl. 2006;45:7134–7186. doi: 10.1002/anie.200601872.
- 2.Trost B. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 3.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 4.Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2001. [Google Scholar]
- 5.(a) Khattab MA, Elgamal MA, El-Batouti M. Fire Mater. 1996;20:253–259. [Google Scholar]; (b) O’Mahony G. Synlett. 2004:572–573. [Google Scholar]
- 6.(a) Molander GA. Chem. Rev. 1992;92:29–68. [Google Scholar]; (b) Snider BB. Chem. Rev. 1996;96:339–364. doi: 10.1021/cr950026m. [DOI] [PubMed] [Google Scholar]
- 7.(a) Iqbal J, Bhatia B, Nayyar NK. Chem. Rev. 1994;94:519–564. [Google Scholar]; (b) Eckenhoff WT, Pintauer T. Cat. Rev. - Sci. Eng. 2010;52:1–59. [Google Scholar]; (c) Pintauer T. Eur. J. Inorg. Chem. 2010;2010:2449–2460. [Google Scholar]; (d) Muñoz-Molina JM, Belderrain TR, Pérez PJ. Eur. J. Inorg. Chem. 2011;2011:3155–3164. and references therein. [Google Scholar]
- 8.Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kharasch MS, Arimoto FS, Nudenberg W. J. Org. Chem. 1951;16:1556–1565. [Google Scholar]
- 10.Kochi JK. Acc. Chem. Res. 1974;7:351–360. [Google Scholar]
- 11.(a) Minisci F. Acc. Chem. Res. 1975;8:165–171. [Google Scholar]; (b) Minisci F, Citterio A, Giordano C. Acc. Chem. Res. 1983;16:27–32. [Google Scholar]
- 12.Ford L, Jahn U. Angew. Chem. Int. Ed. in Engl. 2009;48:6386–6389. doi: 10.1002/anie.200901761. [DOI] [PubMed] [Google Scholar]
- 13.Bull JA, Hutchings MG, Quayle P. Angew. Chem. Int. Ed. in Engl. 2007;46:1869–1872. doi: 10.1002/anie.200603416. [DOI] [PubMed] [Google Scholar]
- 14.Beckwith ALJ, Schiesser CH. Tetrahedron. 1985;41:3925–3941. [Google Scholar]
- 15.Yang D, Yan Y-L, Zheng B-F, Gao Q, Zhu N-Y. Org. Lett. 2006;8:5757–5760. doi: 10.1021/ol0623264. [DOI] [PubMed] [Google Scholar]
- 16.Muñoz-Molina JM, Belderrain TR, Pérez PJ. Adv. Synth. Catal. 2008;350:2365–2372. [Google Scholar]
- 17.Ricardo C, Pintauer T. Chem. Commun. 2009:3029–3031. doi: 10.1039/b905839g. [DOI] [PubMed] [Google Scholar]
- 18.Taylor MJW, Eckenhoff WT, Pintauer T. Dalton Trans. 2010;39:11475–11482. doi: 10.1039/c0dt01157f. [DOI] [PubMed] [Google Scholar]
- 19.Mori M, Oda I, Ban Y. Tetrahedron Lett. 1982;23:5315–5318. [Google Scholar]
- 20.Ryu I, Kreimerman S, Araki F, Nishitani S, Oderaotoshi Y, Minakata S, Komatsu M. J. Am. Chem. Soc. 2002;124:3812–3813. doi: 10.1021/ja017315e. [DOI] [PubMed] [Google Scholar]
- 21.Sumino S, Ui T, Ryu I. Org. Lett. 2013;15:3142–3145. doi: 10.1021/ol401363t. [DOI] [PubMed] [Google Scholar]
- 22.Fusano A, Sumino S, Nishitani S, Inouye T, Morimoto K, Fukuyama T, Ryu I. Chemistry - A European Journal. 2012;18:9415–9422. doi: 10.1002/chem.201200752. [DOI] [PubMed] [Google Scholar]
- 23.Bloome KS, Alexanian EJ. J. Am. Chem. Soc. 2010;132:12823–12825. doi: 10.1021/ja1053913. [DOI] [PubMed] [Google Scholar]
- 24.Mizutani K, Shinokubo H, Oshima K. Org. Lett. 2003;5:3959–3961. doi: 10.1021/ol0356643. [DOI] [PubMed] [Google Scholar]
- 25.Powell DA, Maki T, Fu GC. J. Am. Chem. Soc. 2005;127:510–511. doi: 10.1021/ja0436300. [DOI] [PubMed] [Google Scholar]
- 26.González-Bobes F, Fu GC. J. Am. Chem. Soc. 2006;128:5360–5361. doi: 10.1021/ja0613761. [DOI] [PubMed] [Google Scholar]
- 27.Phapale VB, Buñuel E, García-Iglesias M, Cardenas DJ. Angew. Chem. Int. Ed. in Engl. 2007;46:8790–8795. doi: 10.1002/anie.200702528. [DOI] [PubMed] [Google Scholar]
- 28.Domingo V, Arteaga JF, López Pérez JL, Peláez R, Quílez del Moral JF, Barrero AF. J. Org. Chem. 2012;77:341–350. doi: 10.1021/jo201968t. [DOI] [PubMed] [Google Scholar]
- 29.Aube J, Peng X, Wang Y, Takusagawa F. J. Am. Chem. Soc. 1992;114:5466–5467. [Google Scholar]
- 30.House DA. Chem. Rev. 1962;62:185–203. [Google Scholar]
- 31.Chiba S, Cao Z, Bialy, El SA, Narasaka K. Chem. Lett. 2006;35:18–19. [Google Scholar]
- 32.(a) Alexiev A, Bontchev PR. Mikrochim Acta. 1970;58:13–19. [Google Scholar]; (b) Bonchev PR, Aleksiev AA. Theor Exp Chem. 1975;9:144–147. [Google Scholar]
- 33.Minisci F, Bernardi R, Bertini F, Galli R, Perchinummo M. Tetrahedron. 1971;27:3575–3579. [Google Scholar]
- 34.Ding C, Tu S, Yao Q, Li F, Wang Y, Hu W, Zhang A. Adv. Synth. Catal. 2010;352:847–853. [Google Scholar]
- 35.(a) Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J. Am. Chem. Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patel NR, Flowers RA., II J. Am. Chem. Soc. 2013;135:4672–4675. doi: 10.1021/ja400712g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lockner JW, Dixon DD, Risgaard R, Baran PS. Org. Lett. 2011;13:5628–5631. doi: 10.1021/ol2023505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shundrin LA, Bardin VV, Frohn H-J. Z. anorg. allg. Chem. 2004;630:1253–1257. [Google Scholar]
- 38.Graham TH, Jones CM, Jui NT, MacMillan DWC. J. Am. Chem. Soc. 2008;130:16494–16495. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J. Am. Chem. Soc. 2009;131:11640–11641. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Jui NT, Lee ECY, MacMillan DWC. J. Am. Chem. Soc. 2010;132:10015–10017. doi: 10.1021/ja104313x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jui NT, Garber JAO, Finelli FG, MacMillan DWC. J. Am. Chem. Soc. 2012;134:11400–11403. doi: 10.1021/ja305076b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rendler S, MacMillan DWC. J. Am. Chem. Soc. 2010;132:5027–5029. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.For additional reviews on visible light photoredox catalysis, see: Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2010;40:102. doi: 10.1039/b913880n. Teplý F. Collect. Czech. Chem. Commun. 2011;76:859–917. Tucker JW, Stephenson CRJ. J. Org. Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x. Yoon TP. ACS Catal. 2013;3:895–902. doi: 10.1021/cs400088e.
- 43.Zhu C, Xie J, Xu P, Li H, Xue Q, Jin H, Yixiang C. Chem. Commun. 2013;49:5672–5674. doi: 10.1039/c3cc42672f. [DOI] [PubMed] [Google Scholar]
- 44.Hari DP, Hering T, König B. Org. Lett. 2012;14:5334–5337. doi: 10.1021/ol302517n. [DOI] [PubMed] [Google Scholar]
- 45.Xiao T, Dong X, Tang Y, Zhou L. Adv. Synth. Catal. 2012;354:3195–3199. [Google Scholar]
- 46.Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- 47.Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org. Lett. 2010;12:368–371. doi: 10.1021/ol902703k. [DOI] [PubMed] [Google Scholar]
- 48.Curran DP, Rakiewicz DM. J. Am. Chem. Soc. 1985;107:1448–1449. [Google Scholar]
- 49.Tanabe Y, Nishii Y, Wakimura K-I. Chem. Lett. 1994:1757–1760. [Google Scholar]
- 50.Tucker JW, Nguyen JD, Narayanam JMR, Krabbe SW, Stephenson CRJ. Chem. Commun. 2010;46:4985. doi: 10.1039/c0cc00981d. [DOI] [PubMed] [Google Scholar]
- 51.Tucker JW, Stephenson CRJ. Org. Lett. 2011;13:5468–5471. doi: 10.1021/ol202178t. [DOI] [PubMed] [Google Scholar]
- 52.Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 53.Hurtley AE, Cismesia MA, Ischay MA, Yoon TP. Tetrahedron. 2011;67:4442–4448. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu Z, Shen M, Yoon TP. J. Am. Chem. Soc. 2011;133:1162–1164. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao G, Yang C, Guo L, Sun H, Lin R, Xia W. J. Org. Chem. 2012;77:6302–6306. doi: 10.1021/jo300796j. [DOI] [PubMed] [Google Scholar]