

Abstract
(R,R′)-4′-Methoxy-1-naphthylfenoterol (MNF) promotes growth inhibition and apoptosis of human HepG2 hepatocarcinoma cells via cannabinoid receptor (CBR) activation. The synthetic CB1R inverse agonist, AM251, has been shown to block the anti-mitogenic effect of MNF in these cells; however, AM251 is also an agonist of the recently deorphanized, lipid-sensing receptor, GPR55, whose upregulation contributes to carcinogenesis. Here, we investigated the role of MNF in GPR55 signaling in human HepG2 and PANC-1 cancer cell lines in culture by focusing first on internalization of the fluorescent ligand Tocrifluor 1117 (T1117). Initial results indicated that cell pretreatment with GPR55 agonists, including the atypical cannabinoid O-1602 and L-α-lysophosphatidylinositol, dose-dependently reduced the rate of cellular T1117 uptake, a process that was sensitive to MNF inhibition. GPR55 internalization and signaling mediated by O-1602 was blocked by MNF in GPR55-expressing HEK293 cells. Pretreatment of HepG2 and PANC-1 cells with MNF significantly abrogated the induction of ERK1/2 phosphorylation in response to AM251 and O-1602. Moreover, MNF exerted a coordinated negative regulation of AM251 and O-1602 inducible processes, including changes in cellular morphology and cell migration using scratch wound healing assay. This study shows for the first time that MNF impairs GPR55-mediated signaling and, therefore, may have therapeutic potential in the management of cancer.
Keywords: G-protein coupled receptor, GPR55, ligand internalization, cellular morphology, cell motility
1. Introduction
(R,R′)-4′-methoxy-1-naphthylfenoterol (MNF) is an analog of (R,R′)-fenoterol with a 573-fold greater selectivity for β2-AR than β1-AR [1, 2]. It enhances cAMP accumulation with EC50 value of 3.90 nM in human β2-AR-overexpressing cells [2] and attenuates proliferation of 1321N1 astrocytoma cells with IC50 value of 3.98 nM [3]. In contrast to (R,R′)-fenoterol, MNF activates both Gαs and Gαi proteins and potently stimulates cardiomyocyte contractility, consistent with its role as a β2-AR agonist [2]. However, we have recently reported that MNF treatment of the human-derived HepG2 hepatocarcinoma cell line causes growth arrest and apoptosis via a β2-AR-independent route. The MNF response was found to be insensitive to the β2-AR antagonist ICI 118,551, and U87MG cells, which lack β2-AR binding activity, were responsive to the antimitogenic effects of MNF [4]. The presence of the naphthyl moiety in MNF led us to speculate that it may share structural similarities with other ligands and, therefore, behave as a dually acting compound with unique affinity and selectivity profile.
Cannabinoid receptors (CBRs) are often co-expressed with β2-AR in many tissues and various cell types, and their propensity to heterodimerize demonstrates the potential for crosstalk between the two receptors [5, 6]. In fact, CBRs can modulate β2-AR activity [7]. The engagement of CBRs by endogenous and synthetic cannabinoid ligands results in the regulation of proliferation and apoptosis of cancer cells [8, 9], including HepG2 cells [4, 10]. It is interesting that treatment with selective pharmacological inverse agonists of CBRs blocks the antiproliferative actions of MNF in HepG2 cells, consistent with the potential role of CBRs in MNF signaling [4]. Even though AM251 and its clinical analog, rimonabant, interact with CB1R as inverse agonists, there is growing body of evidence to suggest that they also act as agonists for the recently deorphanized GPR55 [11, 12]. GPR55 is a G protein-coupled receptor with lipid-sensing properties whose upregulation contributes to the aggressive behavior of various cancer types [13–16]. A role for ERK/MAP kinase signaling during microglial activation and the promotion of cancer cell proliferation by GPR55 has been proposed [14, 17]. AM251 also promotes neutrophil chemotaxis by acting as a GPR55 agonist [18]. Thus, the actions of AM251 and rimonabant, which have been widely interpreted as being mediated by CB1R, may, in fact, include off-target cannabinoid effects, some of which mediated by GPR55 activation.
The current study was designed to investigate the contribution of GPR55 in MNF actions in two human cancer cell lines in culture, HepG2 and PANC-1 cells. Pharmacodynamic studies were first performed to determine whether MNF significantly affected the internalization and/or recycling of GPR55. In order to achieve this aim, we used Tocrifluor 1117 (T1117), a fluorescent ligand that binds to endogenous GPR55 with low affinity for CB1R [19]. Then, the impact of MNF treatment on ligand-mediated activation of signaling pathways downstream of GPR55 was explored. Further studies will be required to determine if administration of MNF leads to antitumor activity in vivo.
2. Materials and Methods
2.1 Reagents
(R,R′)-4′-methoxy-1-naphthylfenoterol (MNF) was synthesized as described previously [1]. Dulbecco’s modified Eagle’s medium (DMEM), Eagle’s minimum essential medium, trypsin solution, phosphate-buffered saline (PBS), fetal bovine serum (FBS), 100× solutions of sodium pyruvate (100 mM), L-glutamine (200 mM), and penicillin/streptomycin (a mixture of 10,000 units/ml penicillin and 10,000 μg/ml streptomycin) were obtained from Quality Biological (Gaithersburg, MD). Phenol red-free DMEM and sodium bicarbonate (7.5% solution) were purchased from Life Technologies (Grand Island, NY). WIN 55,212-2, AM251, and AM630 were purchased from Cayman Chemical (Ann Arbor, MI), whereas rimonabant (SR 141716A), CP 55,940, O-1602 and Tocrifluor T1117 were from Tocris Bioscience (Ellisville, MO). L-α-lysophosphatidylinositol (LPI) and G418 was purchased from Sigma-Aldrich (St-Louis, MO). The structures of all ligands used in this study, their known specificity for β2-AR, CBRs and GPR55, and relevant references are found in Table 1.
Table 1.
List of structures of select ligands and their known specificity to the various receptors.
| Structure | Name | Chemical name | Properties | References |
|---|---|---|---|---|
|
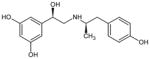
(R,R′)-Fenoterol (Fen) | 5-[(1R)-1-hydroxy-2-{[(2R)-1-(4-hydroxyphenyl) propan-2-yl]amino}ethyl] benzene-1,3-diol |
|
[1] |
|
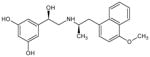
(R,R′)-4-methoxy-1-naphthylfenoterol (MNF) | 5-[(1R)-1-hydroxy-2-{[(2R)-1-(4-methoxynaphthalen-1-yl)propan-2-yl]amino} ethyl]benzene-1,3-diol |
|
[2] |
|
[4] | |||
|
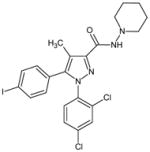
AM251 | N-(Piperidin-1-yl)-5-(4-iodophenyl)--1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide |
|
[20] |
|
[21] | |||
|
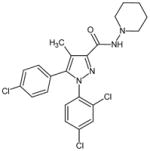
rimonabant, SR 141716 | N-(Piperidin-1-yl)-5-(4-chloropheny-l)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide |
|
[22] |
|
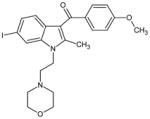
AM630 | 6-Iodo-2-methyl-1-[2-(4-morpholinyl-)ethyl]-1H-indol-3-yl](4-methoxy phenyl)methanone |
|
[23] |
|
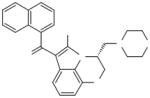
WIN 55,212-2 | (R)-(+)-[2,3-Dihydro-5-methyl-3-(4--morpholinylmethyl) pyrrolo[1,2,3-de]-1,4-benzoxazin--6-yl]-1-naphthalenylmethanone |
|
[24] |
|
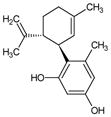
O-1602 | 5-Methyl-4-[(1R,6R)-3-methyl-6-(1-c-yclohexen-1-yl]-1,3-benzenediol |
|
[21] |
|
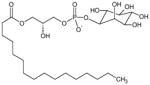
L-α-Lysophosphatidyl inositol (LPI) | 1-Acyl-sn-glycero-3-phospho-(1-D-myo-inositol) |
|
[25] |
|
CP 55,940 | (−)-cis-3-[2-Hydroxy-4-(1,1-dimethy-lheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol |
|
[26] |
|
[11] |
2.2 Maintenance and Treatment of Cell Lines
Human HepG2 hepatocarcinoma cells and human PANC-1 cells were purchased from ATCC (Manassas, VA). HepG2 cells were maintained in Eagle’s minimum essential medium supplemented with 1% L-glutamine, 1% sodium pyruvate, 1% penicillin/streptomycin, and 10% FBS (Hyclone, Logan, UT). PANC-1 cells were cultured in phenol red-free DMEM supplemented with 4.5 g/L glucose and 1.5 g/L sodium bicarbonate together with glutamine, pyruvate, penicillin/streptomycin and 10% FBS. HEK293 cells stably expressing the HA-tagged human GPR55 (3xHA-tagged hGPR55-HEK293) were a generous gift of Maria Waldhoer (Medical University of Graz, Graz, Austria) [27]. The cells were maintained in DMEM with 4.5 g/L glucose supplemented with 10% FBS, 0.2 mg/ml G418, and penicillin/streptomycin. All cell lines were maintained in culture at 37 °C in 5% CO2, and the medium was replaced every 2–3 days.
Upon receipt of the HepG2 and PANC-1 cell lines from ATCC, cells were expanded for a few passages to enable the generation of new frozen stocks. Cells were resuscitated as needed and used for no more than 10–12 passages. Cells were never passaged more than 8–10 weeks after resuscitation. HepG2 and PANC-1 cells were authenticated by ATCC using short tandem repeat (STR) analysis.
2.3 Synthesis of 5′-TAMRA-3-phenylpropan-1-amine
Ten μmoles of the NHS ester of 5′-TAMRA (Sigma-Aldrich) was incubated with 20 μmoles of 3-phenylpropan-1-amine (Sigma-Aldrich) in 1 ml of 0.1M PBS, pH 8.0 for 4 h. The solution was stream dried under nitrogen and reconstituted in 500 μl of a 1:1 solution of 10 mM Tris-HCl, pH 8.0, in ethanol. The formation of 5′-TAMRA-3-phenylpropan-1-amine (TAMRA-PPA) and absence of unconjugated NHS ester of 5′-TAMRA was confirmed by mass spectrometry (Fig. 1A, B). A stock solution of 10 mM of TAMRA-PPA was prepared, aliquoted and stored at −20°C.
Fig. 1. Characterization of TAMRA-PPA structure and lack of cellular internalization in HepG2 cells.

A, Structure of 5′-TAMRA-3-phenylpropan-1-amine (TAMRA-PPA) and T1117. B, Mass spectrum of TAMPRA-PPA ion, m/z equals 548.0. C, Serum-depleted HepG2 cells were maintained on a confocal microscope stage equipped with a temperature-controlled, humidified CO2 chamber at 37°C, and rates of cellular accumulation of T1117 (10 nM) vs. TAMRA-PPA (20 nM) were determined in ring-shaped regions of interest (ROI) in the cytoplasmic compartments. Plots of signal intensity vs. time were generated from defined ROIs. Note the absence of TAMRA-PPA incorporation in cells.
2.4 Cellular Uptake of TocriFluor T1117, a Fluorescently Labeled GPR55 Agonist
Two distinct approached were used to measure cellular accumulation of T1117. In the first method, cells were grown in 35-mm glass bottom culture dishes (MatTek Corp., Ashland, MA) for 48 h until reaching ~70% confluence. Serum-depleted cells were incubated either with DMSO (vehicle, 0.1%), MNF (1 μM), or synthetic cannabinoid compounds (AM630, AM251, O-1602, CP 55,940) for 30 min prior to the addition of the novel fluorescent diarylpyrazole cannabinoid ligand, Tocrifluor T1117 (10–100 nM). Cells were imaged with a Zeiss LSM 710 confocal microscope (Thornwood, NY) equipped with a temperature-controlled and humidified CO2 chamber and a definite focus system. A 561 nm DPSS laser was used for the excitation of the 5-TAMRA moiety. The time-series function of the Zeiss Zen software was used to collect images with a 40× 1.3 NA objective every 30 s for up to one hour, with all confocal settings remaining the same throughout the experiments. Still images, movies and fluorescent intensity quantitation were obtained from these series using the Zeiss Zen software. Experiments were repeated at least two to three times. The same procedure was followed for cell incubation with TAMRA-PPA.
In the second method, HepG2 cells were seeded at 1 × 104 cells/well in poly-D-lysine (Sigma-Aldrich) coated 96-well black/clear bottom plates (BD Biosciences, San Jose, CA). The next day, cells were stained with 1 μg/ml of DNA-specific fluorescent dye Hoechst 33342 (Life Technologies) and washed once with serum-free medium. Subsequently, cells were treated with O-1602, AM251 or LPI for 30 min followed by the addition of 100 nM T1117. After another 30 min of incubation cells were washed once with PBS and fixed with 4% (v/v) paraformaldehyde in PBS. For the assessment of T1117 incorporation, cells were imaged on the BD Pathway 855 Bioimager workstation (BD Biosciences) using 20× NA 0.75 dry objective (Olympus, Tokyo, Japan); 380/535 nm and 548/570 nm excitation/emission filter sets were used for acquisition of Hoechst 33342 and T1117 signals, respectively. AttoVision v1.7 software (BD Biosciences) was applied to analyze T1117 fluorescence within cytoplasmic compartments defined as ring-shaped regions of interest (ROIs) established around Hoechst-stained nuclei. Numerical data were generated with BD Image Data Explorer (BD Biosciences) and plotted using Graph Pad Prism 5.03 (GraphPad Software, CA, USA).
2.5 RNA Interference Experiments
HepG2 and PANC-1 cells were transfected with siRNA oligos (1.25 μg) against GPR55 or a non-silencing siRNA control for 48 h using 10 μl of siRNA Transfection Reagent (Santa Cruz Biotechnology, Santa Cruz, CA) following the manufacturer’s protocol. GPR55 siRNA was a pool of 3 target-specific 20–25 nt siRNAs (cat. sc-75183; Santa Cruz Biotechnology) designed to knock down gene expression. Following 48 h of siRNA treatment, cells were washed with PBS, and maintained in serum-free medium before initiating the indicated experiments.
In another series of experiment, HepG2 cells were transfected with siRNA oligos (1.25 μg) against CB1R, CB2R or a non-silencing siRNA control for 48 h as indicated above. Each siRNA was a pool of 3 target-specific 20–25 nt siRNAs (CB1R, sc-39910; CB2R, sc-39912; Santa Cruz Biotechnology).
2.6 RNA Extraction, cDNA Synthesis, and Reverse Transcription-PCR Analysis
Total RNA was isolated from HepG2 and PANC-1 cells by using the RNeasy Mini kit (QIAGEN, Valencia, CA). The RNA preparation included a DNase digestion step. RNA concentration and quality was measured by using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). To obtain cDNA, 1 μg of total RNA was reverse-transcribed by using the Promega reverse transcription kit (Promega, Madison, WI). Semiquantitative RT-PCRs were performed to determine the expression of GPR55 and β2-AR mRNAs by using glyceraldehyde-3-phosphate dehydrogenase as an internal control. The oligonucleotide primer sequences are found in Table 2 whereas cycle number and the thermal cycle profiles are found in Table 3.
Table 2.
Sequences of oligonucleotide primers.
| Gene of interest | Primer sequences |
|---|---|
| β2-AR | F: 5′-CATGTCTCTCATCGTCCTGGCCA-3′ R: 5′-CACGATGGAAGAGGCAATGGCA-3′ |
| GPR55 | F: 5′-GTCCCCCTTCCCGTCCCTGTG-3′ R: 5′-GCTGGCTGCGATGCTGTAGATGC-3′ |
| GAPDH | F: 5′-ACCACAGTCCATGCCATC-3′ R: 5′-TCCACCACCCTGTTGCTG-3′ |
Table 3.
PCR conditions.
| Steps | Temperature (°C) | Duration (min) | Cycle number | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Initial | 94 | 94 | 94 | 6 | 4 | 4 | 1× | 1× | 1× | |
| denaturation | ||||||||||
| Denaturation | 94 | 94 | 94 | 1 | 0.5 | 1 | ||||
| Amplification | Annealing | 58 | 68 | 53 | 0.5 | 0.5 | 1 | 40× | 38× | 35× |
| Extension | 72 | 72 | 72 | 1 | 1 | 1 | ||||
| Final extension | 72 | 72 | 72 | 5 | 5 | 10 | 1× | 1× | 1× | |
| Hold | 4 | 4 | 4 | ∞ | ∞ | ∞ | 1× | 1× | 1× | |
The conditions were for β2-AR (black font), GPR55 (red font), and GAPDH (green font).
2.7 GPR55 Internalization Assay
Endocytosis of GPR55 was observed following a previously described protocol with minor modifications [27]. Briefly, 3xHA-tagged hGPR55-HEK293 cells were grown on Lab-Tek II CC2 chamber slides (Thermo Scientific Nunc, Rochester, NY) for 48 h in regular medium and then were serum-starved for 1 h. A pre-incubation with 1:1000 rabbit HA antibody (Covance, MD) was performed in the presence of vehicle (0.1% DMSO) or 1 μM MNF in serum-free medium for 45 min at 37°C in a CO2 incubator. Cells were then washed extensively with PBS and treated with 5 μM O-1602 in serum-free medium for 20 min at 37°C in the CO2 incubator. Subsequently, cells were washed three times, fixed in fresh 3.7% paraformaldehyde in PBS (10 min), and incubated with anti-rabbit Alexa Fluor 488 antibody (Molecular Probes, Eugene, OR; 1:1000, 30 min). Cells were washed and fixed for a second time prior to permeabilization with 0.2% Triton X-100 (5 min). Incubation with anti-rabbit Alexa Fluor 568 antibody (Molecular Probes; 1:1000, 30 min) was carried out to determine the extent of internalized 3xHA-tagged GPR55•anti-HA antibody complexes. After a washing cycle with PBS, nuclear counterstaining was performed with DAPI (4′,6-diamidino-2-phenylindole) added to the Prolong Gold antifade mounting medium (Invitrogen, Carlsbad, CA). Slides were cured for 24 h at room temperature in the dark, and then images were acquired with a Zeiss LSM 710 confocal microscope using Carl Zeiss LSM software.
2.8 Scratch Assays
These assays were carried out as previously described with slight modifications [28]. In brief, cells were seeded in 12-well nontreated polystyrene cell culture plates with flat bottom (Greiner Bio-One, Monroe, NC). Once the cells became confluent, a scratch wound was made with a pipette tip and pictured immediately (time 0). Cells were pretreated either with vehicle (DMSO, 0.1%) or the synthetic GPR55 ligands AM251 (1 μM) or O-1602 (1 μM) for 30 min followed by the addition of MNF (1 μM) where indicated. Cell migration was examined at 12, 24, 36, 48 h and 12, 18, 24, 48 h after scratch for the HepG2 and PANC-1 cells, respectively. In preliminary experiments, images of the same field were taken every 3 h to determine the rate of cell migration. Images were captured on an Axiovert 200 inverted microscope (Carl Zeiss) mounted with an AxioCam HRc digital camera (Carl Zeiss) and the measurement of scratch area was performed with ImageJ 1.46s software (National Institutes of Health, Bethesda, MD). Each experiment was performed in duplicate dishes and repeated at least twice.
2.9 Western Blot Analysis
For detection of intracellular signaling proteins, cells were lysed in radioimmunoprecipitation buffer containing EGTA and EDTA (Boston BioProducts, Ashland, MA). The lysis buffer contained a protease inhibitor cocktail (cat. #P8340; Sigma-Aldrich) and phosphatase inhibitor cocktail sets I and II (cat. #524624 and 524625; Calbiochem, San Diego, CA). Equivalent amount of proteins (14 and 54 μg/well for PANC-1 and HepG2 cells, respectively) were separated on 4–12% precast gels (Invitrogen) using SDS-polyacrylamide gel electrophoresis under reducing conditions and then electrophoretically transferred onto polyvinylidene fluoride membrane (Invitrogen). Western blots were performed according to standard methods, which involved blocking in 5% non-fat milk and incubated with the antibody of interest, followed by incubation with a secondary antibody conjugated with the enzyme horseradish perodixase. The detection of immunoreactive bands was performed by chemiluminescence using the ECL Plus Western Blotting Detection System (GE Healthcare, Piscataway, NJ). The quantification of bands was done by volume densitometry by using ImageJ software. The rabbit polyclonal antibodies against EGF receptor (EGFR) and GPR55 were obtained from Cell Signaling Technology, Inc. (Beverly, MA) and Cayman Chemical Co. (Ann Arbor, MI), respectively. The monoclonal anti-Hsp90 was purchased from Santa Cruz Biotechnology.
2.10 Effect of MNF on O-1602-mediated Increase in Cell Signaling
Serum-starved cells were pretreated in the absence or presence of 1 μM MNF for 10 min followed by a 10–min incubation with vehicle, 2.5 and 10 μM O-1602. The levels of total ERK2 and phosphorylated forms of ERK1/2 (pErk1/2, Thr202/Tyr204) were then determined by Western blotting technique. The two primary antibodies were purchased from Cell Signaling Technology and used at a dilution recommended by the manufacturer.
2.11 Measurement of Phospho-active ERK Using alphaScreen
3xHA-tagged hGPR55-HEK293 cells were plated at 1 × 104/well in 100 μl DMEM supplemented with 10% FBS and 0.2 mg/ml G418 per well in a 96-well plate and grown at 37 °C for 24 h. Medium was replaced with 100 μl serum-free DMEM medium with G418 (0.2 mg/ml) and incubated for 48 h. Medium was removed and replaced with 50 μl serum-free DMEM and incubated at 37 °C for 2 h (on day-4). Then 25 μl of vehicle (1% DMSO) or pre-warmed 4× MNF solution was added to the assay plate and incubated for 15 min at 37 °C followed by addition of 25 μl of 4× agonist solution composed of LPI + rimonabant. Cells were further incubated for 10 min at 37 °C followed by lysis and detection as per the instructions provided by the vendor (Perkin Elmer Alphascreen Surefire ERK1/2 assay kit, cat# TGRES500). The plate was read with the Envision plate reader (Perkin Elmer) using standard Alphascreen settings.
2.12 Statistical Analysis
Prism 4 (GraphPad Software, Inc., La Jolla, CA) running on a personal computer was used to perform all statistical data analysis. Effects of different doses of GPR55 agonists on T1117 incorporation in HepG2 cells were statistically evaluated using one-way analysis of variance (ANOVA) followed by Dunnett’s test. The same statistical approach was utilized to evaluate the changes in T1117 internalization in HepG2 cells caused by a panel of siRNAs designed to silence the expression of CB1R, CB2R and GPR55. Expression levels of GPR55 and T1117 incorporation patterns in PANC-1 cells evoked by non-silencing control siRNA and GPR55 siRNAs were compared using unpaired Student’s t-test. Effects of 1 μM MNF on T1117 uptake in HepG2 and PANC-1 cells was also evaluated using Student’s t-test. Relative ERK1/2 phosphorylation levels in GPR55-expressing HEK293 cells were compared using one-way ANOVA and Tukey’s post-hoc test. The same method was used to evaluate the effects of GPR55 siRNA on O-1602-dependent increase of ERK 1/2 phosphorylation in PANC-1 cells as well as to compare the effects of MNF and AM251 on changes in morphology and migration rate of HepG2 and PANC-1 cells. Unless otherwise indicated, error bars represent standard error of the mean (SEM).
3. Results
3.1 A Role for the Deorphanized GPR55 in the Cellular Incorporation of T1117
To establish whether the transport and cellular incorporation of T1117 required the presence of the AM251 moiety, HepG2 cells were incubated for up to 1 h with equimolar amounts of either T1117 or the fluorophore alone, 5′-TAMRA-(3-phenylpropan-1-amine) (TAMRA-PPA). No significant incorporation of fluorescence was observed upon cell incubation with TAMRA-PPA (Fig. 1C).
Initial T1117 uptake rates increased in a dose-dependent manner in HepG2 cells (Fig. 2A). The areas under the curve (AUC) were calculated and represented as bar graphs (Fig. 2B). The results clearly showed that maximal T1117-AUC was achieved at 100 nM with half-maximal signal at ~8 nM. Dropping the temperature to 10 °C reduced the rate of T1117 uptake (data not shown), whereas the simultaneous addition of a 100× molar excess of unlabeled AM251 caused an 18-min delay in the cellular accumulation of T1117 (Fig. 2C, D) likely due to a phenomenon of competition for the receptor.
Fig. 2. Rapid and saturable incorporation of T1117 in HepG2 cells.

Cellular entry of T1117 was measured on a Zeiss 710 confocal microscope with thermoregulated chamber system for live cell imaging. A, Serum-depleted HepG2 cells were incubated in the presence of increasing concentrations of T1117 (2.5–100 nM). Plots of signal intensity vs. time were generated from defined ROIs. Results are from 2–3 independent experiments. B, The area under the curve (AUC) in a plot of T1117 internalization against time was obtained and plotted as bar graphs. Relative AUC data vs. T1117 concentrations is shown, with the T1117-AUC value at 100 nM set at 1. C, The cellular incorporation of T1117 (100 nM) was carried out in the presence of a 100× molar excess of unlabeled AM251. Bars indicate mean ± S.D. (n=3 ROIs) from a single experiment, which was repeated twice with comparable results. D, Representative images at t = 15 min are shown. Bar, 30 μm. DIC, Differential Interference Contrast.
To confirm the role of GPR55 as cell surface receptors responsible for the transport and cellular accumulation of T1117, HepG2 cells were pretreated with various concentrations of the GPR55 agonist O-1602 for 30 min prior to the addition of T1117 (Fig. 3A). The calculation of T1117-AUC showed a significant dose-dependent reduction in the rate of cellular T1117 accumulation in response to O-1602 (Fig. 3B), consistent with ligand-mediated GPR55 occupancy and subsequent lowering in T1117 uptake through competition. We adapted this functional assay to a 96-well format and showed that pretreatment of HepG2 cells with two GPR55 ligands, O-1602 and LPI, dose-dependently reduced subsequent cellular accumulation of T1117 (Fig. 3C and D). The potent synthetic cannabinoid agonist CP 55,940 has been reported to block GPR55 internalization in a heterologous expression system [11]. Here, a 30-min pretreatment with CP 55,940 (0.25 μM) delayed the onset of T1117 uptake by 16 min, which was followed by subsequent decrease in T1117 internalization rate (Fig. 4A). It would appear that blocking GPR55 internalization impairs cellular entry of T1117. As shown previously, inhibition of the internalization process reduced intracellular accumulation of β2-AR complexed with a fluorescently labeled ligand in live cells, following agonist binding [29]. The potency of AM630, a CB2R inverse agonist, was compared to that of the CB1R inverse agonist AM251 (Fig. 2C), and the results showed that cell pretreatment with AM630 had no adverse effect on T1117 incorporation while preincubation with WIN 55,212-2, a CBR agonist, led to marked increase in the rate of T1117 accumulation (Fig. 4B).
Fig. 3. A key role for GPR55 in cellular incorporation of T1117.

A, Serum-depleted HepG2 cells were pretreated with increasing concentrations of O-1602 for 30 min followed by the addition of 10 nM T1117. B, Bars represent the average T1117-AUC ± S.D. (n=3 independent experiments). C and D, HepG2 cells seeded in 96-well plates were serum-starved and subjected to O-1602 (C) and LPI (D) treatment at the indicated concentrations for 30 min. Cellular incorporation of T1117 was carried out as indicated in Material and Methods, method 2. Bars indicate mean ± S.D. of averaged T1117 ROI intensities from 8 wells across two independent experiments. **, *** P < 0.01 and 0.001, respectively.
Fig. 4. Key role of GPR55 in cellular incorporation of T1117.

A, Serum-depleted HepG2 cells were treated with vehicle (0.01% DMSO) or CP 55,940 (0.25μM) for 30 min followed by the addition of 10 nM T1117. B, AM630 (1μM), WIN 55,212-2 (1μM). A and B, Bars indicate mean ± S.D. (n=3 ROIs) from single experiment, which was repeated twice with comparable results. C, Expression of GPR55, β2-AR and GAPDH mRNA was determined by semi-quantitative PCR analysis in PANC-1 (lane 1) and HepG2 (lane 2) cells. Water control is shown in lane 3. M, size markers. D, PANC-1 cells were treated with non-silencing control siRNA (crtl) or GPR55 siRNA oligos for 48 h. Levels of GPR55 were determined in cell lysates by Western blotting and normalized to β-actin. Upper panel, representative immunoblot; Lower panel, Bars represent the mean ± SEM from three independent experiments, each performed in duplicate dishes. **, P < 0.05. E, PANC-1 cells transfected for 48 h with control (crtl) and GPR55 siRNAs were serum-starved for 3 h followed by the addition of 10 nM T1117. Plots of signal intensity vs. time were generated from defined ROIs. Bars indicate mean ± S.E.M. of 3 independent experiments, each performed with 3–4 ROIs. F, Relative T1117-AUC data in PANC-1 cells with control siRNA values set at 1. G, HepG2 cells were transfected with siRNA oligos either against CB1R, CB2R or GPR55 or the non-silencing siRNA control for 48 h. Cells were maintained in serum-free medium for 3 h followed by the addition of 10 nM T1117. Plots of signal intensity vs. time were generated from defined ROIs. Bars indicate mean ± S.E.M. of 3 independent experiments, each performed with 3–4 ROIs. H, Relative T1117-AUC data in HepG2 cells with control siRNA values set at 1. *, *** P < 0.05 and 0.001, respectively.
In order to independently validate our results, the following series of experiments were performed in parallel using both the HepG2 cells and the PANC-1 cell line, which was previously shown to be responsive to AM251 treatment [30]. Semiquantitative RT-PCR analysis established that HepG2 and PANC-1 cells actually expressed GPR55 at the mRNA level, with PANC-1 cells expressing the most (Fig. 4C). A similar profile was obtained when looking at β2-AR mRNA levels. Both cell lines were then incubated with siRNA oligos against GPR55 and the non-silencing siRNA control for 48 h to assess their impact on T1117 incorporation. Control experiment established that treatment of PANC-1 cells with GPR55 siRNA led to a ~2-fold reduction in the endogenous protein levels of GPR55 (Fig. 4D). Under these conditions, the silencing of GPR55 caused a significant delay in internalization and the total amount of T1117 incorporated, as evidenced by a 70.4 ± 8.6% reduction in T1117-AUC when compared with PANC-1 cells transfected with control siRNA (Fig. 4E and F). Similarly, more than 90.5 ± 6.3% reduction in T1117-AUC was noted when HepG2 cells were treated with a pool of GPR55 siRNAs (Fig. 4G and H).
In HepG2 cells transfected with CB2R siRNA, the T1117 incorporation rate was comparable to that of cells transfected with a non-silencing siRNA control (Fig. 4G and H), consistent with the data collected with the CB2R inverse agonist, AM630 (Fig. 4B). Moreover, upon CB1R silencing there was a 10–12 min delay before the start of T1117 incorporation, which ultimately resulted in a 31.8 ± 11.2% reduction in T1117-AUC (Fig. 4G and H). It is possible that constitutive CB1R activity participates to some extent in T1117 entry. Because of the poor quality of commercial antibodies raised against CB1R and CB2R, we were unable to demonstrate down-regulation of those endogenously expressed GPCRs upon siRNA transfection.
3.2 MNF Inhibits Cellular Incorporation of T1117
A dose-response study was carried out in serum-depleted HepG2 cells to define the working effective range of MNF (10 nM – 10 μM) on T1117 internalization. MNF inhibited T1117 incorporation with an IC50 of 0.51 μM (Fig. 5A) and required short-term pretreatment as opposed to the 16-h period needed to promote apoptosis in these cells [4]. From these results, the concentration of 1 μM MNF was chosen for the next series of studies aimed at comparing the effects of MNF in HepG2 and PANC-1 cells. As anticipated, MNF was equipotent at inhibiting T1117 incorporation in both cell lines (Fig. 5B–D).
Fig. 5. Effect of MNF on cellular uptake of T1117.

A, Dose-response study of MNF (10nM–10μM) was carried out in serum-depleted HepG2 cells incubated with 10 nM T1117. Each datapoint represents the mean ± S.E.M. of T1117-AUC data normalized to DMSO controls (n=3 experiments). B–D, Serum-depleted HepG2 (B) and PANC-1 (C) cells were pretreated with vehicle or MNF (1 μM) for 30 min followed by the addition of 10 nM T1117. Plots of signal intensity vs. time were generated from defined ROIs. D, Bars represent the mean ± S.E.M. of T1117-AUC data normalized to the DMSO controls (n=3). ***, P < 0.001.
3.3 Effect of MNF on GPR55 Internalization and Downstream Signaling
The effect of MNF on ligand-induced GPR55 internalization and signaling was performed in HEK293 cells stably expressing 3xHA-tagged GPR55. Using confocal laser scanning microscopy, GPR55 was found to be located largely at the plasma membrane of unstimulated cells (Fig. 6A). Addition of O-1602 for 20 min led to marked endocytosis of 3xHA-tagged GPR55, which was blocked by pretreatment with MNF (Fig. 6B). The determination of pixel count distribution was carried out on the merged images depicted in Fig. 6B, and the results showed cytoplasmic localization of the receptor in O-1602-treated cells and its accumulation with the plasma membrane in response to MNF (Fig. 6C).
Fig. 6. MNF impairs ligand-induced GPR55 internalization and signaling.

HEK293 cells stably transfected with 3xHA-tagged hGPR55 vector (panel A) were serum-starved, and then incubated with anti-HA antibody in the absence or presence of MNF (1 μM) for 45 min at 37 °C. After extensive washing, O-1602 (5 μM) was added to the cells for 20 min at 37 °C to promote GPR55 internalization. Intact cells were fixed and then incubated with anti-rabbit Alexa Fluor 488 antibody (green) to label cell surface GPR55. After a permeabilization step, anti-rabbit Alexa Fluor 568 antibody (red) was added to detect intracellular GPR55. Nuclei were counterstained with DAPI (blue). Yellow indicates signal coalescence in the merged images. Scale bar = 20 μm. C, Merged images with pixel intensities for cell surface GPR55 (green), internalized GPR55 (red) and nuclei (blue) are shown. D, GPR55-expressing HEK293 cells were incubated in the absence or presence of 100 nM MNF for 15 min followed by the addition of LPI (1 μM) + rimonabant (10 μM) for 10 min. The rationale of having used two GPR55 agonists together stemmed from the recent observation that AM251 and rimonabant are allosteric ligands of GPR55 in addition to their capacity to modulate the LPI (ligand)-binding site through different pharmacophores [46]. The detection of phosphorylated ERK was carried out with a Perkin Elmer Alphascreen Surefire kit and the signal normalized to total ERK levels. Rimonabant alone had no effect on basal ERK phosphorylation levels, although the LPI response was enhanced. ***, P < 0.001 (n=3 independent experiments).
Additional events downstream of GPR55 internalization may be impaired upon MNF treatment, as the redistribution of ligand-bound receptors from the cell surface to endosomal compartment differentially regulates various signaling pathways and their associated biological outcomes. Indeed, spatio-temporal activation of extracellular signal-regulated kinase (ERK)-MAP kinase plays an important role in the dynamic control of complex cellular functions [31]. Here, agonist stimulation of 3xHA-tagged GPR55-expressing HEK293 cells led to significant increase in ERK phosphorylation levels, which was abrogated by MNF pretreatment (Fig. 6D). Similarly, exposure of HepG2 cells to O-1602 dose-dependently increased ERK phosphorylation and MNF pretreatment blunted O-1602 responsiveness (Fig. 7A and B). As anticipated, PANC-1 cells exhibited the same behavior as HepG2 cells and displayed comparable sensitivity to MNF (Fig. 7C and D). The requirement of GPR55 in O-1602 signaling was assessed by performing GPR55 siRNA knockdown experiments. The results indicated significant reduction in O-1602-mediated ERK phosphorylation in PANC-1 cells after GPR55-specific gene silencing (Fig. 7E and F).
Fig. 7. Impairment in GPR55 downstream signaling by MNF.

Serum-depleted HepG2 (A, B) and PANC-1 (C, D) cells were pretreated or not in the presence of MNF (1 μM) for 10 min followed by the addition of vehicle, O-1602 (2.5 and 10 μM), or 10% FBS for an additional 10 min. Cell lysates were prepared, separated by reducing SDS-PAGE gel electrophoresis and immunoblotted for total and phosphoactive forms of ERK. A and C, Representative immunoblots; B and D, Phospho-ERK1/2 bands were normalized to total ERK2, and the O-1602-10μM values were set at 1. Data are means of two independent dishes ± range. The migration of molecular-mass markers (values in kilodaltons) is shown on the left of immunoblots. E and F, PANC-1 cells were transfected with control (crtl) or GPR55 siRNA for 48 h, and then were serum-starved for 3 h. ERK1/2 phosphorylation was monitored in cell lysates from vehicle or O-1602 (10 μM)-treated cells. E, Representative immunoblots; F, Signals associated with phospho-active ERK1/2 was normalized to β-actin. Bars represent the mean ± SEM from three independent experiments. *, P < 0.05.
3.4 A Role of MNF in the Morphology and Motility of Tumor cells
The role of MNF in GPR55-mediated responses was investigated further in HepG2 and PANC-1 cells. Cells with irregular appearance, long filipodia and lamellipodia were observed in response to AM251 and O-1602 stimulation (Fig. 8A and C, white arrows), and pretreatment with MNF rendered both cell lines refractory to these changes in morphology (Fig. 8A–D).
Fig. 8. MNF interferes with inducible changes in cell morphology and expression of EGFR.

Serum-starved HepG2 (A) and PANC-1 (C) cells were pre-incubated in the presence of DMSO (0.1%) or MNF (1 μM) for 30 min followed by the addition of AM251 (5 μM) or O-1602 (5 μM) for 16 h. Unstimulated PANC-1 cells displayed cuboidal morphology with and without MNF. White arrows show individual cells with filopodia. B and D, Bars represent the average number of cells with filipodia per ‘frame’ ± SEM (n=16). Each frame contained an average of 50 and 15 cells for HepG2 and PANC-1 cells, respectively. **, *** P < 0.01 and 0.001. E, Cell lysates were prepared from similar experiments and immunoblotted for EGFR. The membranes were reprobed for Hsp90, which served as a loading control.
The presence of epidermal growth factor receptor (EGFR) on filopodia has been proposed to play a significant role in the ‘remote’ sensing of growth factors required for the regulation and coordination of cell motility [32]. Of significance, AM251-induced increase in EGFR expression has been implicated in greater motility and invasiveness of PANC-1 cells [30]. As shown in Fig. 8E, treatment of HepG2 and PANC-1 cells with O-1602 led to higher EGFR levels when compared to vehicle-treated cells, and MNF blocked this effect (Fig. 8E, lane 4 vs. 3) and that of AM251 (data not shown).
A wound-healing assay in vitro was then performed to investigate the effects of MNF on cell motility, a well-known readout of GPR55 signaling [13, 33]. MNF (1 μM) had minimal effect on the motility of HepG2 cells under basal conditions, a result that contrasted with its significant inhibitory effect toward AM251-mediated increase in cell motility (Fig. 9A and B). Depicted in Fig. 9C is the comparison of the relative reduction of the wound surface area at the 24-h time-point between the treatment groups. Similar to its effects in HepG2 cells, MNF also produced significant decrease in AM251-induced motility of PANC-1 cells without impacting on their basal migration rate (Fig. 9D–F). The ability of MNF to inhibit the wound closure evoked by O-1602 was also observed in PANC-1 cells (data not shown).
Fig. 9. MNF inhibits ligand-induced motility of HepG2 and PANC-1 cells in a wound-healing assay.

Confluent HepG2 (A, B, C) and PANC-1 (D, E, F) cells were subjected to scratch wound as described in Materials and Methods. Cells were incubated in the presence of DMSO (0.1%) or the GPR55 agonist AM251 (1 μM) for 30 min, followed by the addition of MNF (1 μM) where indicated. Images were captured at various time-points. B and E, The relative wound surface area was measured over time and plotted, and values at time 0 were set at 1. C and F, The relative wound surface area of four independent observations at the 24-h time point is plotted. *, *** P < 0.05 and 0.001.
4. Discussion
Engagement of the ‘cannabinoid-like receptor’ GPR55 triggers a number of signaling cascades that promote cell proliferation, migration, survival and oncogenesis (reviewed in [34]). MNF displays a number of characteristics associated with selective attenuation in GPR55 signaling, including 1) delayed cellular entry of a fluorescent GPR55 ligand, 2) inhibition of the internalization of the ligand-occupied GPR55, and 3) a significant reduction in GPR55 agonist efficacy with regard to a number of biological readouts (Fig. 10).
Fig. 10. Schematic diagram of the modulation of GPR55 signaling.
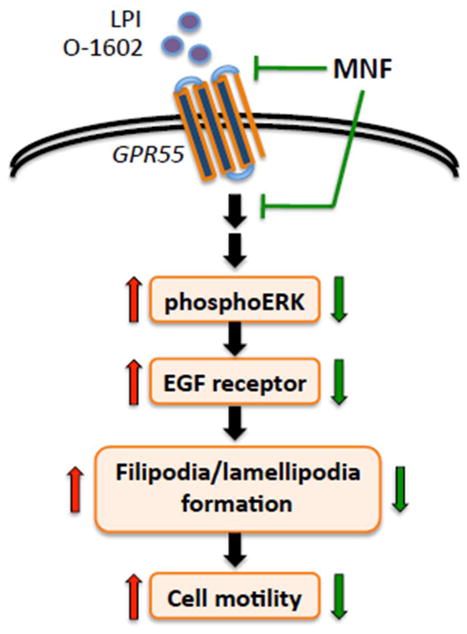
Binding of agonists, such as LPI and O-1602, elicits the activation of GPR55 and its downstream signaling cascade ultimately resulting in increased cancer cell motility. Cell treatment with (R,R′)-MNF has the capacity to inhibit the pro-oncogenic activity of GPR55.
In cellular assays, the low level of non-specific uptake of the fluorophore alone (5′-TAMRA-PPA) makes T1117 (5′-TAMRA-PPA conjugate of AM251) suitable for in vivo imaging approaches aimed at assessing occupancy and internalization of GPR55. The compound T1117 has been shown previously to measure the distribution of endogenously expressed GPR55 in small mouse arteries [19]. Here, employing the siRNA-based gene silencing method, we confirmed that GPR55 is a key player in T1117 entry in intact cells. Although CB2R interacts cooperatively with GPR55 to influence inflammatory responses of neutrophils [18] pharmacological inhibition and siRNA-mediated silencing of CB2R did not alter T1117 incorporation in HepG2 cells. However, a CB1R-dependent mechanism appears to have contributed to some extent to T1117 uptake, as the silencing of CB1R by siRNA led to lower cellular incorporation of the GPR55 fluorescent ligand. Both receptors trigger distinct signaling pathways in endothelial cells [35], and our study confirmed their presence in HepG2 and PANC-1 cells. Heterodimerization between CB1R and various GPCRs has functional consequences on receptor trafficking and signaling [6, 36–38]. The recent observation that GPR55 can heterodimerize with CB1R [39] led us to speculate that CB1R/GPR55 physical interaction may have potential functional implications in promoting some of the physiological responses of MNF.
Analysis of the data revealed that MNF significantly delayed the cellular accumulation of T1117 in serum-depleted cells expressing endogenous levels of GPR55, suggestive of a decrease in the binding affinity of T1117 to GPR55 and/or impairment in constitutive cell surface GPR55 internalization and recycling pathways. In this model, O1602-bound GPR55 complexes were internalized and any residual cell surface GPR55 receptors were targeted by MNF, making this GPCR inaccessible for efficient T1117 binding and/or internalization. Similarly, interaction of GPR55 with AM251 may have also contributed to the observed potency in MNF signaling. The ability of CP 55,940 to block cellular entry of T1117 was consistent with its role as a GPR55 antagonist [11].
The stimulation of 3xHA-tagged GPR55-expressing HEK-293 cells with the atypical cannabinoid O-1602 triggered rapid internalization of GPR55 through a MNF-inhibitable mechanism. These and other results illustrate the in vitro potency of MNF in cells that contain endogenous and overexpressed GPR55. GPCR desensitization and internalization requires the participation of β-arrestin translocation to the activated receptor [40, 41]. Using a β-arrestin translocation assay in a transient transfection format, AM251 and its clinical analog rimonabant exhibit potent activity as GPR55 agonists [11, 42] whereas CP 55,940 blocks the formation of β-arrestin•GPR55 complexes [11]. The possibility exists that MNF prevents the recruitment of β-arrestin to the GPR55, thereby providing a negative impact on internalization and recycling of this GPCR after agonist exposure. In addition to its role in the promotion of GPCR internalization, β-arrestin is required for activation of downstream signaling (e.g., ERK activation) [43, 44]. GPR55 is thought to bind predominantly G-protein α13, where it promotes Rho-dependent signaling in endothelial cells [35]. Additional events downstream of GPR55 include activation of ERK and Ca2+ release from internal stores (for review, see [45]). Here, in vitro exposure of HepG2 and PANC-1 cells to AM251 or O-1602 resulted in rapid increase in ERK phosphorylation, a process that was inhibited by cell pretreatment with MNF. The simplest explanation for the significant reduction in agonist-stimulated increase in ERK phosphorylation in response to MNF is that ligand-bound GPR55 stimulates ERK activity once the receptor is internalized. Alternatively, MNF may interact with a putative allosteric binding site on GPR55 [46] and elicit negative modulation of GPR55 function. These allosteric sites, which differ from agonist-binding and channel-permeation sites, provide means to modulate, either positively or negatively, receptor function. It is noteworthy that CB1R is believed to contain extracellular regulatory sites that recognize small molecule ligands that are likely to regulate its cellular activity [47]. However, more work will be required to provide direct experimental evidence of MNF binding at GPR55.
Another striking observation from our study was the cause-effect relationship between the effect of MNF on agonist-induced ERK phosphorylation and on biological readouts, including GPR55-dependent change in cellular morphology and cell motility. ERK has been found to coordinate and regulate cell migration by promoting lamellipodial leading edge movement via phosphorylation of the WAVE2 regulatory complex [48]. Here, treatment of HepG2 and PANC-1 cells with AM251 or O-1602 led to fillipodia extension, which was blocked by MNF pretreatment. Moreover, MNF elicited a significant reduction in the rate of wound closure prompted by GPR55 agonists using a scratch wound-healing assay. Because β-arrestin proteins are signaling scaffolds capable at facilitating cell migration through segregation and reorganization of the actin cytoskeleton (reviewed in [49]), a study to investigate the role of β-arrestin in the MNF control of GPR55 signaling will soon be initiated in our laboratory.
As indicated earlier, MNF is characterized by a ~500-fold greater selectivity for β2-AR than β1-AR, and yet, it exhibits off-target activities towards GPR55. A number of β2-AR ligands have been reported to display off-target effects that cannot be explained solely by their interactions with β2-AR. It can be assumed that characteristics of β-blocker drugs, such as lipophilicity and hydrophilicity, the ratio of antagonist versus (partial) agonist properties, affinity to non-β-AR receptor sites e.g., 5-HT receptors [50], strength of membrane-stabilizing activity [51, 52], stereospecificity, and potency, all come into play when considering nonspecific membrane effects of certain β-blockers, which are thought to derive from the physicochemical interaction of the drugs with the cell membrane [53]. Notably, the presence of specific chemical moieties in certain β-blockers confers unique profile of signaling characteristics with ability to stimulate signaling pathways in a G protein-independent, β-arrestin-dependent fashion [54, 55]. Ligands capable of interacting with two different species of G protein-coupled receptors (GPCRs) have been synthesized [56], giving rise to bivalent ligands of β2-AR and adenosine A1 receptor capable of generating biphasic pattern of cAMP production in cells expressing both receptors [57]. The ability of β2-AR to form functional heterodimeric complex with other GPCRs and elicit transactivation of the heterocomplex can further increase the list of off-target effects evoked by β2-AR ligands. For example, isoproterenol activates the ζ isoform of protein kinase C (PKCζ) only in cells co-expressing oxytocin receptor and the β2-AR [58].
In conclusion, this investigation has provided the first evidence for the modulation of GPR55-mediated signaling events by MNF, which offers the possibility that MNF will facilitate future research on GPR55 with respect to its pleiotropic functions in normal and pathological states.
Acknowledgments
We wish to thank Maria Waldhoer from the Institute of Experimental and Clinical Pharmacology, Medical University of Graz, Graz, Austria for her generous gift of HEK-GPR55 cells and Amina S. Adem from SRI Internationals for performing the Alphacreen Surefire-pERK assays. A.W. received funding from the Foundation for Polish Science for his NIH Visiting Program internship within the grant TEAM 2009-4/5. The BD Pathway 855 Bioimager workstation was purchased within the Operational Program Development of Eastern Poland 2007–2013, Priority Axis I Modern Economy, Operations I.3 Innovation Promotion. This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging. This paper is subject to the NIH Public Access Policy.
Abbreviations
- MNF
(R,R′)-4′-methoxy-1-naphthylfenoterol
- CP55
940, 3-(2-hydroxy-4-(1,1-dimethylheptyl)phenyl)-4-(3-hydroxypropyl)cyclohexanol
- AM251
1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide
- CBR
cannabinoid receptor
- O-1602
[5-methyl-4-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-1,3-benzenediol
- rimonabant
(SR141716A; N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2, 4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride)
- AM630
6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone
- β2-AR
beta2 adrenoreceptor
- T1117
Tocrifluor 1117
- ROI
region of interest
- LPI
L-α-lysophosphatidylinositol
- WIN 55
212-2, (R)-(+)-[2,3-Dihydro-5-methyl-3-(4--morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin--6-yl]-1-naphthalenylmethanone
- EGFR
EGF receptor
- DIC
Differential Interference Contrast
Footnotes
Authorship Contributions
Participated in research design: Paul, Wnorowski, Indig, Gonzalez-Mariscal, Moaddel, Wainer, Bernier
Conducted experiments: Paul, Wnorowski, Indig, Gonzalez-Mariscal, Nayak, Pajak
Contributed new reagents or analytical tools: Moaddel, Pajak
Performed data analysis: Paul, Wnorowski, Indig, Gonzalez-Mariscal, Wainer, Bernier
Wrote or contributed to the writing of the manuscript: Wainer, Bernier
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Rajib K. Paul, Email: paulrk@mail.nih.gov.
Artur Wnorowski, Email: artur.wnorowski@gmail.com.
Isabel Gonzalez-Mariscal, Email: isabel.gonzalezmariscal@nih.gov.
Surendra K. Nayak, Email: surendra.nayak@sri.com.
Karolina Pajak, Email: karolina.pajak@umlub.pl.
Ruin Moaddel, Email: moaddelru@grc.nia.nih.gov.
Fred E. Indig, Email: indigfr@grc.nia.nih.gov.
Michel Bernier, Email: bernierm@mail.nih.gov.
Irving W. Wainer, Email: wainerir@grc.nia.nih.gov.
References
- 1.Jozwiak K, Khalid C, Tanga MJ, Berzetei-Gurske I, Jimenez L, Kozocas JA, et al. Comparative molecular field analysis of the binding of the stereoisomers of fenoterol and fenoterol derivatives to the beta2 adrenergic receptor. J Med Chem. 2007;50:2903–15. doi: 10.1021/jm070030d. [DOI] [PubMed] [Google Scholar]
- 2.Jozwiak K, Woo AY, Tanga MJ, Toll L, Jimenez L, Kozocas JA, et al. Comparative molecular field analysis of fenoterol derivatives: A platform towards highly selective and effective beta(2)-adrenergic receptor agonists. Bioorg Med Chem. 2010;18:728–36. doi: 10.1016/j.bmc.2009.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toll L, Jimenez L, Waleh N, Jozwiak K, Woo AY, Xiao RP, et al. {Beta}2-adrenergic receptor agonists inhibit the proliferation of 1321N1 astrocytoma cells. J Pharmacol Exp Ther. 2011;336:524–32. doi: 10.1124/jpet.110.173971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paul RK, Ramamoorthy A, Scheers J, Wersto RP, Toll L, Jimenez L, et al. Cannabinoid receptor activation correlates with the proapoptotic action of the beta2-adrenergic agonist (R,R′)-4-methoxy-1-naphthylfenoterol in HepG2 hepatocarcinoma cells. J Pharmacol Exp Ther. 2012;343:157–66. doi: 10.1124/jpet.112.195206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hudson BD, Hebert TE, Kelly ME. Physical and functional interaction between CB1 cannabinoid receptors and beta2-adrenoceptors. Br J Pharmacol. 2010;160:627–42. doi: 10.1111/j.1476-5381.2010.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardiner SM, March JE, Kemp PA, Bennett T. Involvement of CB1-receptors and beta-adrenoceptors in the regional hemodynamic responses to lipopolysaccharide infusion in conscious rats. Am J Physiol Heart Circ Physiol. 2005;288:H2280–8. doi: 10.1152/ajpheart.00851.2004. [DOI] [PubMed] [Google Scholar]
- 8.Guindon J, Hohmann AG. The endocannabinoid system and cancer: therapeutic implication. Br J Pharmacol. 2011;163:1447–63. doi: 10.1111/j.1476-5381.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermanson DJ, Marnett LJ. Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev. 2011;30:599–612. doi: 10.1007/s10555-011-9318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu WJ, Yang Q, Cao QF, Zhang YW, Xia YJ, Hu XW, et al. Membrane cholesterol mediates the endocannabinoids-anandamide affection on HepG2 cells. Zhonghua Gan Zang Bing Za Zhi. 2010;18:204–8. doi: 10.3760/cma.j.issn.1007-3418.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 11.Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, et al. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem. 2009;284:29817–27. doi: 10.1074/jbc.M109.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther. 2010;126:301–13. doi: 10.1016/j.pharmthera.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ford LA, Roelofs AJ, Anavi-Goffer S, Mowat L, Simpson DG, Irving AJ, et al. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br J Pharmacol. 2010;160:762–71. doi: 10.1111/j.1476-5381.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andradas C, Caffarel MM, Perez-Gomez E, Salazar M, Lorente M, Velasco G, et al. The orphan G protein-coupled receptor GPR55 promotes cancer cell proliferation via ERK. Oncogene. 2011;30:245–52. doi: 10.1038/onc.2010.402. [DOI] [PubMed] [Google Scholar]
- 15.Pineiro R, Maffucci T, Falasca M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene. 2011;30:142–52. doi: 10.1038/onc.2010.417. [DOI] [PubMed] [Google Scholar]
- 16.Perez-Gomez E, Andradas C, Flores JM, Quintanilla M, Paramio JM, Guzman M, et al. The orphan receptor GPR55 drives skin carcinogenesis and is upregulated in human squamous cell carcinomas. Oncogene. 2013;32:2534–42. doi: 10.1038/onc.2012.278. [DOI] [PubMed] [Google Scholar]
- 17.Pietr M, Kozela E, Levy R, Rimmerman N, Lin YH, Stella N, et al. Differential changes in GPR55 during microglial cell activation. FEBS Lett. 2009;583:2071–6. doi: 10.1016/j.febslet.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 18.Balenga NA, Aflaki E, Kargl J, Platzer W, Schroder R, Blattermann S, et al. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011;21:1452–69. doi: 10.1038/cr.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daly CJ, Ross RA, Whyte J, Henstridge CM, Irving AJ, McGrath JC. Fluorescent ligand binding reveals heterogeneous distribution of adrenoceptors and ‘cannabinoid-like’ receptors in small arteries. Br J Pharmacol. 2010;159:787–96. doi: 10.1111/j.1476-5381.2009.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, et al. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42:769–76. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- 21.Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinaldi-Carmona M, Barth F, Heaulme M, Alonso R, Shire D, Congy C, et al. Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci. 1995;56:1941–7. doi: 10.1016/0024-3205(95)00174-5. [DOI] [PubMed] [Google Scholar]
- 23.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol. 1999;126:665–72. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–50. [PubMed] [Google Scholar]
- 25.Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362:928–34. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- 26.Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278:989–99. [PubMed] [Google Scholar]
- 27.Henstridge CM, Balenga NA, Schroder R, Kargl JK, Platzer W, Martini L, et al. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br J Pharmacol. 2010;160:604–14. doi: 10.1111/j.1476-5381.2009.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiori JL, Zhu TN, O’Connell MP, Hoek KS, Indig FE, Frank BP, et al. Filamin A modulates kinase activation and intracellular trafficking of epidermal growth factor receptors in human melanoma cells. Endocrinology. 2009;150:2551–60. doi: 10.1210/en.2008-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hegener O, Prenner L, Runkel F, Baader SL, Kappler J, Haberlein H. Dynamics of beta2-adrenergic receptor-ligand complexes on living cells. Biochemistry. 2004;43:6190–9. doi: 10.1021/bi035928t. [DOI] [PubMed] [Google Scholar]
- 30.Fiori JL, Sanghvi M, O’Connell MP, Krzysik-Walker SM, Moaddel R, Bernier M. The cannabinoid receptor inverse agonist AM251 regulates the expression of the EGF receptor and its ligands via destabilization of oestrogen-related receptor alpha protein. Br J Pharmacol. 2011;164:1026–40. doi: 10.1111/j.1476-5381.2011.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu P, Wee P, Jiang J, Chen X, Wang Z. Differential regulation of transcription factors by location-specific EGF receptor signaling via a spatio-temporal interplay of ERK activation. PLoS One. 2012;7:e41354. doi: 10.1371/journal.pone.0041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lidke DS, Lidke KA, Rieger B, Jovin TM, Arndt-Jovin DJ. Reaching out for signals: filopodia sense EGF and respond by directed retrograde transport of activated receptors. J Cell Biol. 2005;170:619–26. doi: 10.1083/jcb.200503140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Maor Y, Wang JF, Kunos G, Groopman JE. Endocannabinoid-like N-arachidonoyl serine is a novel pro-angiogenic mediator. Br J Pharmacol. 2010;160:1583–94. doi: 10.1111/j.1476-5381.2010.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pineiro R, Falasca M. Lysophosphatidylinositol signalling: new wine from an old bottle. Biochim Biophys Acta. 2012;1821:694–705. doi: 10.1016/j.bbalip.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Waldeck-Weiermair M, Zoratti C, Osibow K, Balenga N, Goessnitzer E, Waldhoer M, et al. Integrin clustering enables anandamide-induced Ca2+ signaling in endothelial cells via GPR55 by protection against CB1-receptor-triggered repression. J Cell Sci. 2008;121:1704–17. doi: 10.1242/jcs.020958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carriba P, Navarro G, Ciruela F, Ferre S, Casado V, Agnati L, et al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods. 2008;5:727–33. doi: 10.1038/nmeth.1229. [DOI] [PubMed] [Google Scholar]
- 37.Hojo M, Sudo Y, Ando Y, Minami K, Takada M, Matsubara T, et al. mu-Opioid receptor forms a functional heterodimer with cannabinoid CB1 receptor: electrophysiological and FRET assay analysis. J Pharmacol Sci. 2008;108:308–19. doi: 10.1254/jphs.08244fp. [DOI] [PubMed] [Google Scholar]
- 38.Callen L, Moreno E, Barroso-Chinea P, Moreno-Delgado D, Cortes A, Mallol J, et al. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J Biol Chem. 2012;287:20851–65. doi: 10.1074/jbc.M111.335273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kargl J, Balenga N, Parzmair GP, Brown AJ, Heinemann A, Waldhoer M. The cannabinoid receptor CB1 modulates the signaling properties of the lysophosphatidylinositol receptor GPR55. J Biol Chem. 2012;287:44234–48. doi: 10.1074/jbc.M112.364109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–65. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 41.Charest PG, Terrillon S, Bouvier M. Monitoring agonist-promoted conformational changes of beta-arrestin in living cells by intramolecular BRET. EMBO Rep. 2005;6:334–40. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J Biol Chem. 2009;284:12328–38. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–61. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 44.Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen B, et al. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. J Biol Chem. 2000;275:2479–85. doi: 10.1074/jbc.275.4.2479. [DOI] [PubMed] [Google Scholar]
- 45.Nevalainen T, Irving AJ. GPR55, a lysophosphatidylinositol receptor with cannabinoid sensitivity? Curr Top Med Chem. 2010;10:799–813. doi: 10.2174/156802610791164229. [DOI] [PubMed] [Google Scholar]
- 46.Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, et al. Modulation of L-alpha-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J Biol Chem. 2012;287:91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–95. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 48.Mendoza MC, Er EE, Zhang W, Ballif BA, Elliott HL, Danuser G, et al. ERK-MAPK drives lamellipodia protrusion by activating the WAVE2 regulatory complex. Mol Cell. 2011;41:661–71. doi: 10.1016/j.molcel.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Min J, Defea K. beta-arrestin-dependent actin reorganization: bringing the right players together at the leading edge. Mol Pharmacol. 2011;80:760–8. doi: 10.1124/mol.111.072470. [DOI] [PubMed] [Google Scholar]
- 50.Castro ME, Harrison PJ, Pazos A, Sharp T. Affinity of (+/−)-pindolol, (−)-penbutolol, and (−)-tertatolol for pre- and postsynaptic serotonin 5-HT(1A) receptors in human and rat brain. J Neurochem. 2000;75:755–62. doi: 10.1046/j.1471-4159.2000.0750755.x. [DOI] [PubMed] [Google Scholar]
- 51.Hachisu M, Koeda T. Study on the pharmacological actions of beta-adrenoceptor blockers with reference to their physico-chemical properties. J Pharmacobiodyn. 1980;3:183–90. doi: 10.1248/bpb1978.3.183. [DOI] [PubMed] [Google Scholar]
- 52.Koella WP. CNS-related (side-)effects of beta-blockers with special reference to mechanisms of action. Eur J Clin Pharmacol. 1985;28 (Suppl):55–63. doi: 10.1007/BF00543711. [DOI] [PubMed] [Google Scholar]
- 53.Imai S. Pharmacologic characterization of beta blockers with special reference to the significance of nonspecific membrane effects. Am J Cardiol. 1991;67:8B–12B. doi: 10.1016/0002-9149(91)90814-2. [DOI] [PubMed] [Google Scholar]
- 54.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104:16657–62. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci U S A. 2008;105:14555–60. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2012;52:153–78. doi: 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- 57.Karellas P, McNaughton M, Baker SP, Scammells PJ. Synthesis of bivalent beta2-adrenergic and adenosine A1 receptor ligands. J Med Chem. 2008;51:6128–37. doi: 10.1021/jm800613s. [DOI] [PubMed] [Google Scholar]
- 58.Wrzal PK, Goupil E, Laporte SA, Hebert TE, Zingg HH. Functional interactions between the oxytocin receptor and the beta2-adrenergic receptor: implications for ERK1/2 activation in human myometrial cells. Cell Signal. 2012;24:333–41. doi: 10.1016/j.cellsig.2011.09.019. [DOI] [PubMed] [Google Scholar]