

Abstract
Vitiligo is an autoimmune disease of the skin in which melanocytes are destroyed by antigen-specific T cells, resulting in patchy depigmentation. While adaptive immunity plays a clear role in disease progression, initiating factors are largely unknown. Many studies report that cellular stress pathways are dysregulated in melanocytes from vitiligo patients, suggesting that melanocyte-intrinsic defects participate in disease pathogenesis. Recent studies reveal that melanocyte stress generates damage-associated molecular patterns that activate innate immunity, thus connecting stress to organ-specific inflammation. Genetic studies in vitiligo support a role for stress, innate immunity, and adaptive mechanisms. Here, we discuss advances in the field that highlight how cellular stress, endogenous danger signals, and innate immune activation promote the onset of vitiligo.
Introduction
The white, patchy depigmentation that is characteristic of vitiligo is disfiguring (Fig 1), and is often psychologically devastating for patients [1]. Approximately 0.5–2% of the world population is afflicted [2]. Factors involved in the initiation of vitiligo are unknown, although both genetic and environmental factors have been implicated [3]. Due to its location within the skin, vitiligo provides an opportunity to directly observe the course of disease, isolate the target tissue for research studies, and culture primary melanocytes, the target cells. Therefore, vitiligo is an excellent disease in which to use translational research strategies to study the pathogenesis of organ-specific autoimmunity. Research into the pathogenesis of human vitiligo over the past 30 years has sparked controversy, as evidence for both autoimmune-mediated destruction of melanocytes and melanocyte-intrinsic abnormalities appeared to be at odds [4]. However recent advances in the field support both hypotheses, and evidence suggests that each is linked to the other through innate immune mechanisms. Several damage-associated molecular patterns (DAMPs) have been associated with cellular stress, and act as ligands for innate pattern recognition receptors (PRRs). It is likely that in vitiligo, stressed melanocytes activate the innate immune system through the generation and release of DAMPs, which provide the initiating danger signal. The inflammation that ensues ultimately leads to activation of the adaptive immune system, thereby facilitating autoimmune destruction and vitiligo progression.
Figure 1.
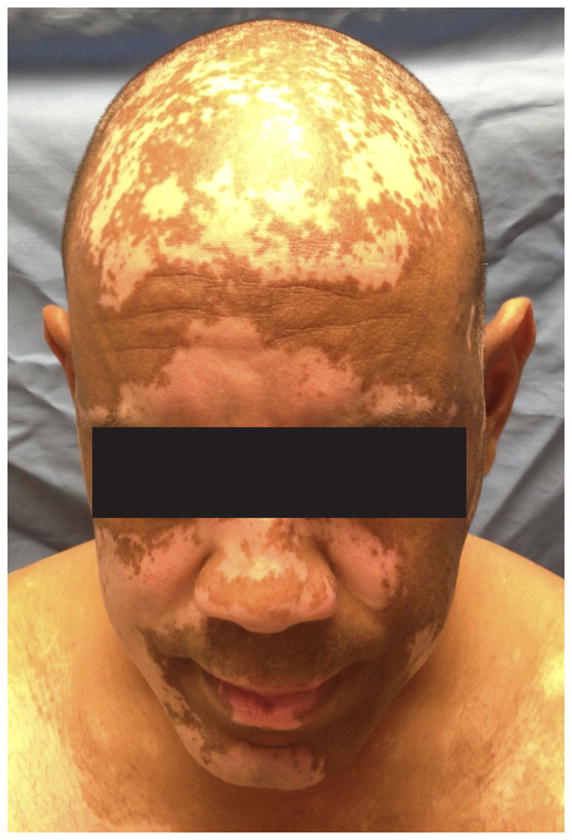
Vitiligo is characterized by disfiguring white patches on the skin due to the loss of melanocytes.
Adaptive immunity in vitiligo
Multiple studies implicate antigen-specific, CD8+ T cell-mediated destruction of melanocytes in human vitiligo. Early observations reported infiltration of T cells in lesional skin from patients with vitiligo [5], and CD8+ T cells were found adjacent to dying melanocytes in the epidermis [6]. Further, the frequency of melanocyte antigen tetramer-positive CD8+ T cells in the blood of vitiligo patients correlates with disease severity, and these cells are capable of killing melanocytes in vitro [7,8]. Finally, purified CD8+ T cells isolated from lesional skin of vitiligo patients, but not CD8-depleted T cells, infiltrate unaffected skin from the patients ex vivo and induce melanocyte apoptosis in situ, revealing that CD8+ T cells are both necessary and sufficient for melanocyte destruction in human vitiligo [9]. Antigenic proteins in vitiligo have been identified, and include gp100, MART1, tyrosinase, and tyrosinase related proteins 1 and 2 [10–12,7]. Certain mouse models of vitiligo also implicate CD8+ T cells as key effectors in disease, and further identify IFN-γ as a critical cytokine in pathogenesis [13,14]. While CD4+ T cells are present in vitiligo lesions [6], a convincing role for them in pathogenesis has not yet been identified. They are dispensable in a mouse model of vitiligo, and disease is exacerbated in their absence, suggesting a possible role for T regulatory (Treg) suppression [13]. However other mouse models of vitiligo have been developed that are CD4-dependent [15,16]. Tregs are reported to be dysregulated in patients with vitiligo, although there is no clear consensus on specific abnormalities (i.e. decreased numbers, skin homing defects, or functional defects) [17–20].
Cellular stress in melanocytes
The earliest indication that melanocytes from vitiligo patients were intrinsically abnormal was the observation that they were more difficult to culture ex vivo compared to those from healthy controls [21], and were more sensitive when exposed to exogenous stressors [22,23]. Ultrastructural analysis revealed a dilated endoplasmic reticulum suggesting increased cellular stress [24], and elevated levels of H2O2 and oxidative byproducts reflected oxidative stress [25–27]. Further evidence for the role of stress in vitiligo came from studies on monobenzone and other phenols, commonly found in commercial products (including rubber, leather products, cosmetic dyes, etc.), that are well known to both induce and exacerbate vitiligo [28–30]. While very high doses of these chemicals induced melanocyte death in vitro, lower and more likely physiologic doses did not kill the cells but induced reactive oxygen species (ROS) and activated the unfolded protein response (UPR) [31,32]. This effect was dependent on tyrosinase, and occurs through the ability of certain phenols to mimic the chemical structure of the amino acid tyrosine (also a phenol), which is a basic building block of the pigment molecule melanin [31].
In addition to the very specific effect of phenols in melanocytes through interaction with tyrosinase, cellular stress results from oxygen and/or nutrient imbalances, chemical or physical agents, infection, inflammation, and misfolded proteins. Melanocytes are particularly susceptible to stress because they perform melanogenesis, an energy-expensive process by which they produce a large amount of the pigment melanin. Mitochondrial energy metabolism generates ROS, and the production of large quantities of protein increases the risk of protein misfolding in the endoplasmic reticulum, a trigger that activates the UPR. Furthermore, the skin is readily exposed to environmental insults such as UV light, which generates intracellular ROS, hydrogen peroxide, and superoxide anions. Finally, the process of melanogenesis itself liberates hydrogen peroxide, a ROS precursor [33].
Therefore, these data indicate that melanocytes in vitiligo patients are more susceptible to oxidative damage than melanocytes from unaffected individuals due to an inherited inability to manage stressors from normal cellular processes, or exposure to environmental chemicals. However while cellular stress in melanocytes provides a reasonable explanation for how vitiligo is induced, it cannot completely account for the disease, since stressed melanocytes remain viable. Bridging the gap between cellular stress and adaptive immunity in vitiligo requires a better understanding of how stress signals are communicated, recognized, and translated into proinflammatory signals. Studies of gene expression profiles in a chicken model of spontaneous vitiligo revealed links between oxidative stress and innate immune activation [34], supporting a role for innate immunity as an important connection between melanocyte stress and adaptive immunity in vitiligo.
DAMPs and other endogenous proinflammatory signals – Danger from within
In contrast to the antigen specificity of adaptive immunity, the innate immune system must rapidly initiate responses without specific antigen recognition, and does so through activation of pattern recognition receptors (PRRs) by “danger signals”. Examples of PRRs include toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), and RIG-I-like receptors (RLRs). Many microbe-derived ligands for TLRs have been identified, including lipopolysaccharide (LPS) for TLR4, single-stranded viral RNA for TLR7, and unmethylated CpG bacterial DNA for TLR9. Like TLRs, NLRs and RLRs respond to a variety of ligands including bacterial flagellin and viral nucleic acids. NLRs form large multiprotein complexes known as inflammasomes, which recruit caspases through oligomerization leading to subsequent activation of caspase proteolytic activities. Recruited caspases process proinflammatory cytokine precursors into mature forms that are subsequently secreted – examples include IL-1β and IL-18 [35]. Because PRR ligands are derived from viral and bacterial pathogens, they are referred to as pathogen-associated molecular patterns (PAMPs).
More recently, molecular patterns released during sterile inflammation have been identified that activate PRRs [36]. During sterile inflammation, these patterns are not associated with pathogens but are self-derived following cellular damage, and are thus referred to as damage-associated molecular patterns (DAMPs). For example, in addition to pathogen-derived DNA, NLRP3 inflammasomes are activated in response to ROS [37] and mitochondrial stress [38]. Heat shock proteins (HSPs) are protein-folding chaperones induced in response to cellular stress and UPR activation to improve protein folding, and they activate TLR2, TLR4 and other PRRs [36]. HSP70i is induced following phenol-induced stress in melanocytes and in vitiligo patient skin. In mouse models of vitiligo, HSP70i is required for vitiligo induction [39], accelerates disease progression [40], and activates dendritic cells (DCs) in the skin [41]. Thus, ROS and HSPs generated by melanocytes in response to stress serve as DAMPs in vitiligo, activating PRRs to initiate inflammation.
DAMPs may be secreted from cells, liberated during cellular damage or death, or transported by exosomes. Exosomes are small microvessicles secreted from cells as a means of cell-cell communication [42]. Monobenzone increases exosome secretion by melanocytes, and exosomes deliver known vitiligo target antigens to DCs, contribute to their activation, and lead to the induction of autoimmune T cell responses against melanocytes and melanomas [31]. In addition to antigens, exosomes carry micro-RNAs (miRNAs), HSPs, and other proteins that act as DAMPs. Specifically, the miRNA miR-29b is induced by oxidative stress, and exosomes containing miR-29b induces activation in a macrophage cell line [43]. Tumor-derived HSP70+ exosomes stimulate both the activation and migration of human natural killer (NK) cells in vitro [44]. Therefore, exosome secretion may provide a means by which melanocytes communicate stress to the innate immune system.
Phenols that are known to induce and exacerbate vitiligo also activate the unfolded protein response (UPR) in melanocytes, resulting in induction of the transcription factor X-box-binding protein 1 (XBP1) and splicing to its activated form (XBP1s). This leads to production of IL-6 and IL-8, providing a direct link between cellular stress and immune activation [32]. IL-6 antagonizes Treg responses, and IL-6 and IL-8 both promote recruitment of immune cell populations.
Innate cell populations in vitiligo
If melanocyte stress generates DAMPs that activate PRRs and the innate immune response, one would expect evidence of increased recruitment and/or activation of innate immune cell populations in vitiligo. Macrophages, NK cells, and inflammatory dendritic cells (DCs) all infiltrate active vitiligo lesions [6,41,45], however despite the fact that IL-8 is produced by stressed melanocytes and is a chemoattractant for neutrophils, neutrophil infiltration is not characteristic of vitiligo. While the role of macrophages in vitiligo has not been further examined, evidence exists for functional roles of NK cells and inflammatory DCs.
Characterization of the transcriptome of skin from vitiligo patients revealed an innate immune signature reflecting an infiltration of NK cells in both lesional and non-lesional skin. Gene expression correlated with immunofluorescence staining patterns, as NK cells were increased almost five-fold in lesional skin of vitiligo patients when compared to healthy controls, and almost two-fold in unaffected skin. This indicates that patients have a propensity for innate immune activation in the skin, which could tip the balance towards development of autoimmune inflammation, and that NK cells may be important players in vitiligo pathogenesis [45] by directly responding to intracellular stress ligands [46].
Inflammatory DCs, defined as CD11c+ CD11b+, are at increased frequencies in both the skin and peripheral blood of vitiligo patients [41]. These cells are also present in the skin of mice with vitiligo, are induced in vitro by the stress-induced protein HSP70i, and are present at even higher numbers in mice that overexpress HSP70i. These studies implicate HSP70i as a crucial signal bridging melanocyte stress and vitiligo induction through innate immune cell activation.
Genome-wide association studies identify potential stress, innate, and adaptive risk alleles in vitiligo
In support of these observations, recent genome-wide association studies (GWAS) reveal multiple genetic risk factors for vitiligo that intersect with these pathways [47]. First, both class I and class II HLA molecules (T cell antigen recognition), PTPN22 (T cell signaling), CD80 (T cell activation), IL2Rα (T cell activation/regulation), GZMB (T cell cytotoxicity), FoxP3 and BACH2 (Treg development and function [48]) implicate a role for adaptive immunity in vitiligo. Next, association of XBP1 with vitiligo supports a role for the UPR pathway in pathogenesis, although it also plays a role in antigen processing. Others found that polymorphisms in XBP1 correlate with the risk of developing Crohn’s disease, and its function is through activation of the UPR in a mouse model of inflammatory bowel disease [49]. Finally, IFIH1 (also called MDA5) and NLRP1 are both PRRs that activate innate immune responses, TICAM1 (also called TRIF) is an adaptor protein in TLR signaling, and caspase 7 is activated by inflammasomes [50], all of which implicate an important role for innate immunity in vitiligo. Future studies should focus on the interface between melanocyte stress, innate and adaptive immunity.
Conclusions and implications – a working hypothesis
In conclusion, both environmental and cell-intrinsic factors trigger melanocyte stress, which results in the production of DAMPs that activate innate immunity, followed by activation of adaptive immune cells, including autoreactive CD8+ T cells that kill melanocytes. Normal melanogenesis, which begins with the amino acid tyrosine as a substrate, generates stress that may be further exacerbated by environmental insults, including phenols and UV light. Melanocyte stress results in production of ROS, activation of the UPR, and release of exosomes. Stress-induced DAMPs, such as HSP70i, serve as ligands for PRRs and activate innate immune cells. Induction of the UPR also results in the direct release of proinflammatory cytokines from melanocytes. Inflammasomes respond to stress signals as well, and while inflammasome activity has not yet been reported within melanocytes, it has been detected in other non-myeloid derived cells. These signals deliver melanocyte-specific antigens and activate PRRs, leading to DC activation and subsequent T-cell priming for melanocyte-specific cytotoxicity (summarized in Fig 2).
Figure 2. Innate signaling pathways activated by cellular stress lead to adaptive autoimmune responses against melanocytes.

The production of melanin results in cellular stress [1]. Environmental insults, including UV light and chemical phenols such as monobenzone, exacerbate this response [2]. Melanocyte stress is characterized by intracellular ROS and activation of the UPR [3], which are both capable of activating PRRs [4] either directly, or through the production of HSP70i and antigen-containing exosomes [5]. These signals function as DAMPs to activate dendritic cells [6] and subsequent priming of CD8+ T-cells for autoimmune attack of melanocytes [7]. Stressed melanocytes secrete low levels of IL-6 and IL-8, which may recruit immune populations and/or antagonize the suppressor function of regulatory T cells (Treg). Abbreviations: DAMP – damage-associated molecular pattern, DC – dendritic cell, GWAS – genome-wide association study, HSP – heat shock protein, NLR – NOD-like receptor, PRR – pattern recognition receptor, RLR – RIG-I-like receptor, ROS – reactive oxygen species, TLR – toll-like receptor, UPR – unfolded protein response. Numbers in brackets correspond to references.
These mechanisms may have evolved as a defense against melanoma and malignancies in general, as tumor cells exhibit increased cellular stress. In support of this hypothesis, the risk of melanoma is inversely proportional to that of vitiligo [51]. The precise threshold of acceptable stress before a cytotoxic response is generated still needs to be determined, and may answer how the proper balance between self-tolerance and tumor surveillance is maintained. Cellular stress pathways may also help determine which specific tissue will be targeted in a patient with autoimmunity. Cellular stress has been implicated in other organ-specific autoimmune diseases, including type I diabetes [52] and Crohn’s disease [49], implying that mechanisms involved in translating stress to adaptive immunity in vitiligo may be shared. Future studies will be required to identify how intrinsic and extrinsic triggers of cellular stress, innate signaling pathways, and adaptive immune responses work together to initiate, propagate and maintain autoimmunity in vitiligo.
Highlights.
Melanocytes from vitiligo patients display features of elevated cellular stress
Stressed cells produce damage-associated molecular patterns (DAMPs)
DAMPs activate innate immune cell populations through pattern recognition receptors
Activated innate immune cells can promote adaptive immunity in vitiligo
Activated stress pathways appear to play a role in organ-specific autoimmunity
Acknowledgments
We are grateful for helpful discussions and critical review of the manuscript by Dr. Katherine Fitzgerald and Dr. Pranoti Mandrekar. JEH is supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number AR061437 and research grants from the Charles H. Hood Foundation and Vitiligo Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other grant organizations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ongenae K, Beelaert L, van Geel N, Naeyaert JM. Psychosocial effects of vitiligo. J Eur Acad Dermatol Venereol. 2006;20:1–8. doi: 10.1111/j.1468-3083.2005.01369.x. [DOI] [PubMed] [Google Scholar]
- 2.Alikhan A, Felsten LM, Daly M, Petronic-Rosic V. Vitiligo: a comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol. 2011;65:473–491. doi: 10.1016/j.jaad.2010.11.061. [DOI] [PubMed] [Google Scholar]
- 3.Spritz RA. Six decades of vitiligo genetics: genome-wide studies provide insights into autoimmune pathogenesis. J Invest Dermatol. 2012;132:268–273. doi: 10.1038/jid.2011.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schallreuter KU, Bahadoran P, Picardo M, Slominski A, Elassiuty YE, Kemp EH, Giachino C, Liu JB, Luiten RM, Lambe T, et al. Vitiligo pathogenesis: autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp Dermatol. 2008;17:139–140. doi: 10.1111/j.1600-0625.2007.00666_1.x. discussion 141–160. [DOI] [PubMed] [Google Scholar]
- 5.Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. The American journal of pathology. 1996;148:1219–1228. [PMC free article] [PubMed] [Google Scholar]
- 6.van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, Tigges B, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest. 2000;80:1299–1309. doi: 10.1038/labinvest.3780138. [DOI] [PubMed] [Google Scholar]
- 7.Ogg GS, Rod Dunbar P, Romero P, Chen JL, Cerundolo V. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. The Journal of experimental medicine. 1998;188:1203–1208. doi: 10.1084/jem.188.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wankowicz-Kalinska A, van den Wijngaard RM, Tigges BJ, Westerhof W, Ogg GS, Cerundolo V, Storkus WJ, Das PK. Immunopolarization of CD4+ and CD8+ T cells to Type-1-like is associated with melanocyte loss in human vitiligo. Lab Invest. 2003;83:683–695. doi: 10.1097/01.lab.0000069521.42488.1b. [DOI] [PubMed] [Google Scholar]
- *9.van den Boorn JG, Konijnenberg D, Dellemijn TA, van der Veen JP, Bos JD, Melief CJ, Vyth-Dreese FA, Luiten RM. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. 2009;129:2220–2232. doi: 10.1038/jid.2009.32. This paper showed that CD8+ T cells are necessary and sufficient for melanocyte destruction in human vitiligo, clearly implicating vitiligo as an autoimmune disease. [DOI] [PubMed] [Google Scholar]
- 10.Palermo B, Campanelli R, Garbelli S, Mantovani S, Lantelme E, Brazzelli V, Ardigo M, Borroni G, Martinetti M, Badulli C, et al. Specific cytotoxic T lymphocyte responses against Melan-A/MART1, tyrosinase and gp100 in vitiligo by the use of major histocompatibility complex/peptide tetramers: the role of cellular immunity in the etiopathogenesis of vitiligo. J Invest Dermatol. 2001;117:326–332. doi: 10.1046/j.1523-1747.2001.01408.x. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto T, Irie RF, Fujii S, Huang SK, Nizze AJ, Morton DL, Hoon DS. Anti-tyrosinase-related protein-2 immune response in vitiligo patients and melanoma patients receiving active-specific immunotherapy. J Invest Dermatol. 1998;111:1034–1039. doi: 10.1046/j.1523-1747.1998.00411.x. [DOI] [PubMed] [Google Scholar]
- 12.Kemp EH, Waterman EA, Gawkrodger DJ, Watson PF, Weetman AP. Autoantibodies to tyrosinase-related protein-1 detected in the sera of vitiligo patients using a quantitative radiobinding assay. Br J Dermatol. 1998;139:798–805. doi: 10.1046/j.1365-2133.1998.02503.x. [DOI] [PubMed] [Google Scholar]
- 13.Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol. 2010;184:1909–1917. doi: 10.4049/jimmunol.0902778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol. 2012;132:1869–1876. doi: 10.1038/jid.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambe T, Leung JC, Bouriez-Jones T, Silver K, Makinen K, Crockford TL, Ferry H, Forrester JV, Cornall RJ. CD4 T cell-dependent autoimmunity against a melanocyte neoantigen induces spontaneous vitiligo and depends upon Fas-Fas ligand interactions. Journal of immunology (Baltimore, Md.: 1950) 2006;177:3055–3062. doi: 10.4049/jimmunol.177.5.3055. [DOI] [PubMed] [Google Scholar]
- 16.Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan CC, Carroll MW, Moss B, Rosenberg SA, Restifo NP. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klarquist J, Denman CJ, Hernandez C, Wainwright DA, Strickland FM, Overbeck A, Mehrotra S, Nishimura MI, Le Poole IC. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010;23:276–286. doi: 10.1111/j.1755-148X.2010.00688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tu CX, Jin WW, Lin M, Wang ZH, Man MQ. Levels of TGF-beta(1) in serum and culture supernatants of CD4(+)CD25 (+) T cells from patients with non-segmental vitiligo. Arch Dermatol Res. 2011;303:685–689. doi: 10.1007/s00403-011-1154-8. [DOI] [PubMed] [Google Scholar]
- 19.Lili Y, Yi W, Ji Y, Yue S, Weimin S, Ming L. Global activation of CD8+ cytotoxic T lymphocytes correlates with an impairment in regulatory T cells in patients with generalized vitiligo. PLoS One. 2012;7:e37513. doi: 10.1371/journal.pone.0037513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou L, Li K, Shi YL, Hamzavi I, Gao TW, Henderson M, Huggins RH, Agbai O, Mahmoud B, Mi X, et al. Systemic analyses of immunophenotypes of peripheral T cells in non-segmental vitiligo: implication of defective natural killer T cells. Pigment Cell Melanoma Res. 2012;25:602–611. doi: 10.1111/j.1755-148X.2012.01019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puri N, Mojamdar M, Ramaiah A. In vitro growth characteristics of melanocytes obtained from adult normal and vitiligo subjects. J Invest Dermatol. 1987;88:434–438. doi: 10.1111/1523-1747.ep12469795. [DOI] [PubMed] [Google Scholar]
- 22.Jimbow K, Chen H, Park JS, Thomas PD. Increased sensitivity of melanocytes to oxidative stress and abnormal expression of tyrosinase-related protein in vitiligo. Br J Dermatol. 2001;144:55–65. doi: 10.1046/j.1365-2133.2001.03952.x. [DOI] [PubMed] [Google Scholar]
- 23.Maresca V, Roccella M, Roccella F, Camera E, Del Porto G, Passi S, Grammatico P, Picardo M. Increased sensitivity to peroxidative agents as a possible pathogenic factor of melanocyte damage in vitiligo. J Invest Dermatol. 1997;109:310–313. doi: 10.1111/1523-1747.ep12335801. [DOI] [PubMed] [Google Scholar]
- 24.Boissy RE, Liu YY, Medrano EE, Nordlund JJ. Structural aberration of the rough endoplasmic reticulum and melanosome compartmentalization in long-term cultures of melanocytes from vitiligo patients. J Invest Dermatol. 1991;97:395–404. doi: 10.1111/1523-1747.ep12480976. [DOI] [PubMed] [Google Scholar]
- 25.Schallreuter KU, Moore J, Wood JM, Beazley WD, Gaze DC, Tobin DJ, Marshall HS, Panske A, Panzig E, Hibberts NA. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J Investig Dermatol Symp Proc. 1999;4:91–96. doi: 10.1038/sj.jidsp.5640189. [DOI] [PubMed] [Google Scholar]
- 26.Shalbaf M, Gibbons NC, Wood JM, Maitland DJ, Rokos H, Elwary SM, Marles LK, Schallreuter KU. Presence of epidermal allantoin further supports oxidative stress in vitiligo. Exp Dermatol. 2008;17:761–770. doi: 10.1111/j.1600-0625.2008.00697.x. [DOI] [PubMed] [Google Scholar]
- 27.Koca R, Armutcu F, Altinyazar HC, Gurel A. Oxidant-antioxidant enzymes and lipid peroxidation in generalized vitiligo. Clin Exp Dermatol. 2004;29:406–409. doi: 10.1111/j.1365-2230.2004.01524.x. [DOI] [PubMed] [Google Scholar]
- 28.Oliver EASL, Warren LH. Occupational leukoderma. JAMA. 1939;113:927–928. [Google Scholar]
- 29.Mosher DB, Parrish JA, Fitzpatrick TB. Monobenzylether of hydroquinone. A retrospective study of treatment of 18 vitiligo patients and a review of the literature. Br J Dermatol. 1977;97:669–679. doi: 10.1111/j.1365-2133.1977.tb14275.x. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh S, Mukhopadhyay S. Chemical leucoderma: a clinico-aetiological study of 864 cases in the perspective of a developing country. Br J Dermatol. 2009;160:40–47. doi: 10.1111/j.1365-2133.2008.08815.x. [DOI] [PubMed] [Google Scholar]
- **32.van den Boorn JG, Picavet DI, van Swieten PF, van Veen HA, Konijnenberg D, van Veelen PA, van Capel T, Jong EC, Reits EA, Drijfhout JW, et al. Skin-depigmenting agent monobenzone induces potent T-cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J Invest Dermatol. 2011;131:1240–1251. doi: 10.1038/jid.2011.16. This study was the first to reveal the mechanism of monobenzone in vitiligo induction and identified exosomes as potential carriers of DAMPs following the induction of stress in melanocytes. Together with 33 and 48, it provides a plausible link between melanocyte stress and inflammation. [DOI] [PubMed] [Google Scholar]
- **33.Toosi S, Orlow SJ, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J Invest Dermatol. 2012;132:2601–2609. doi: 10.1038/jid.2012.181. This study demonstrated that activation of the UPR results in the production of proinflammatory cytokines IL-6 and IL-8 directly from melanocytes, and together with 32 and 48, provides a plausible link between melanocyte stress and inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyskens FL, Jr, Farmer P, Fruehauf JP. Redox regulation in human melanocytes and melanoma. Pigment Cell Res. 2001;14:148–154. doi: 10.1034/j.1600-0749.2001.140303.x. [DOI] [PubMed] [Google Scholar]
- 34.Shi F, Kong BW, Song JJ, Lee JY, Dienglewicz RL, Erf GF. Understanding mechanisms of vitiligo development in Smyth line of chickens by transcriptomic microarray analysis of evolving autoimmune lesions. BMC Immunol. 2012;13:18. doi: 10.1186/1471-2172-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–332. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 38.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 39.Mosenson JA, Zloza A, Klarquist J, Barfuss AJ, Guevara-Patino JA, Poole IC. HSP70i is a critical component of the immune response leading to vitiligo. Pigment Cell Melanoma Res. 2012;25:88–98. doi: 10.1111/j.1755-148X.2011.00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denman CJ, McCracken J, Hariharan V, Klarquist J, Oyarbide-Valencia K, Guevara-Patino JA, Le Poole IC. HSP70i accelerates depigmentation in a mouse model of autoimmune vitiligo. J Invest Dermatol. 2008;128:2041–2048. doi: 10.1038/jid.2008.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **42.Mosenson JA, Zloza A, Nieland JD, Garrett-Mayer E, Eby JM, Huelsmann EJ, Kumar P, Denman CJ, Lacek AT, Kohlhapp FJ, et al. Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Sci Transl Med. 2013;5:174ra128. doi: 10.1126/scitranslmed.3005127. This paper identified HSP70i as a DAMP in vitiligo that has the capacity to activate DCs from humans and mice and to accelerate depigmentation in a mouse model of vitiligo. Together with 32 and 33, it provides a plausible link between melanocyte stress and inflammation, and with 48, helps to implicate innate immune cell populations in vitiligo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bang C, Thum T. Exosomes: new players in cell-cell communication. Int J Biochem Cell Biol. 2012;44:2060–2064. doi: 10.1016/j.biocel.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 43.Thulasingam S, Massilamany C, Gangaplara A, Dai H, Yarbaeva S, Subramaniam S, Riethoven JJ, Eudy J, Lou M, Reddy J. miR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264. 7 cells. Mol Cell Biochem. 2011;352:181–188. doi: 10.1007/s11010-011-0752-2. [DOI] [PubMed] [Google Scholar]
- 44.Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, Multhoff G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005;65:5238–5247. doi: 10.1158/0008-5472.CAN-04-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *46.Yu R, Broady R, Huang Y, Wang Y, Yu J, Gao M, Levings M, Wei S, Zhang S, Xu A, et al. Transcriptome analysis reveals markers of aberrantly activated innate immunity in vitiligo lesional and non-lesional skin. PLoS One. 2012;7:e51040. doi: 10.1371/journal.pone.0051040. Through gene expression profiling of skin from vitiligo patients, this paper identified a potential role for NK cell infiltration in vitiligo and, together with 48, helps to implicate innate immune cell populations in vitiligo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Long EO, Rajagopalan S. Stress signals activate natural killer cells. J Exp Med. 2002;196:1399–1402. doi: 10.1084/jem.20021747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *48.Spritz RA. Modern vitiligo genetics sheds new light on an ancient disease. J Dermatol. 2013;40:310–318. doi: 10.1111/1346-8138.12147. This review summarizes the recent advances in identifying human genetic risk alleles for vitiligo, which together support melanocyte stress, innate immunity, and adaptive immunity in vitiligo pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roychoudhuri R, Hirahara K, Mousavi K, Clever D, Klebanoff CA, Bonelli M, Sciume G, Zare H, Vahedi G, Dema B, et al. BACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. 2013;498:506–510. doi: 10.1038/nature12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *50.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. This study identified a hypomorphic allele of XBP1 as a risk allele for patients with Crohn’s disease, demonstrated that XBP1 played a critical role in normalizing stress in the gut, and revealed that mice developed spontaneous colitis in its absence. It is the first to functionally link the UPR stress pathway to organ-specific autoimmunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erener S, Petrilli V, Kassner I, Minotti R, Castillo R, Santoro R, Hassa PO, Tschopp J, Hottiger MO. Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-kappaB target genes. Mol Cell. 2012;46:200–211. doi: 10.1016/j.molcel.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 51.Spritz RA. The genetics of generalized vitiligo: autoimmune pathways and an inverse relationship with malignant melanoma. Genome Med. 2010;2:78. doi: 10.1186/gm199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia. 2013;56:234–241. doi: 10.1007/s00125-012-2762-3. [DOI] [PubMed] [Google Scholar]
- 53.Cotter MA, Thomas J, Cassidy P, Robinette K, Jenkins N, Florell SR, Leachman S, Samlowski WE, Grossman D. N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet-induced melanoma in mice. Clin Cancer Res. 2007;13:5952–5958. doi: 10.1158/1078-0432.CCR-07-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu N, Zhang S, Zuo F, Kang K, Guan M, Xiang L. Cultured human melanocytes express functional toll-like receptors 2–4, 7 and 9. J Dermatol Sci. 2009;56:113–120. doi: 10.1016/j.jdermsci.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288:859–872. doi: 10.1074/jbc.M112.412783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901–1916. doi: 10.1016/j.bbamcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 57.Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- 58.Pawaria S, Binder RJ. CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat Commun. 2011;2:521. doi: 10.1038/ncomms1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–1546. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 60.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol. 2009;183:3170–3176. doi: 10.4049/jimmunol.0803721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooper MA, Fehniger TA, Ponnappan A, Mehta V, Wewers MD, Caligiuri MA. Interleukin-1beta costimulates interferon-gamma production by human natural killer cells. Eur J Immunol. 2001;31:792–801. doi: 10.1002/1521-4141(200103)31:3<792::aid-immu792>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 64.Chaix J, Tessmer MS, Hoebe K, Fuseri N, Ryffel B, Dalod M, Alexopoulou L, Beutler B, Brossay L, Vivier E, et al. Cutting edge: Priming of NK cells by IL-18. J Immunol. 2008;181:1627–1631. doi: 10.4049/jimmunol.181.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 66.Kumar H, Koyama S, Ishii KJ, Kawai T, Akira S. Cutting edge: cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J Immunol. 2008;180:683–687. doi: 10.4049/jimmunol.180.2.683. [DOI] [PubMed] [Google Scholar]