

Abstract
Cell adhesion to extracellular matrix is a complex process involving protrusive activity driven by the actin cytoskeleton, engagement of specific receptors, followed by signaling and cytoskeletal organization. Thereafter, contractile and endocytic/recycling activities may facilitate migration and adhesion turnover. Focal adhesions, or focal contacts, are widespread organelles at the cell-matrix interface. They arise as a result of receptor interactions with matrix ligands, together with clustering. Recent analysis shows that focal adhesions contain a very large number of protein components in their intracellular compartment. Among these are tyrosine kinases, which have received a great deal of attention, whereas the serine/threonine kinase protein kinase C has received much less. Here the status of protein kinase C in focal adhesions and cell migration is reviewed, together with discussion of its roles and potential substrates.
Keywords: Kinase, Proteoglycan, Cytoskeleton, Microfilaments, Adhesion
Background
Ever since the discovery of the protein kinase C (PKC) family as the major phorbol ester-inducible tumor promoters in the 1980s (Blumberg 1988), these serine/threonine kinases have been extensively studied. They represent a major family involved in multiple signaling pathways with well-established roles in essential cellular processes, such as proliferation, differentiation and regulation of gene transcription (Marte et al. 1994; Way et al. 2000). In addition, some members are implicated in the control of cell adhesion and migration, including responses to extracellular matrix macromolecules.
Prominent junctional organelles involved in cell-matrix adhesion are focal adhesions, also referred to as focal contacts. These are signaling organelles at the cell-matrix interface and nearly always lie at the termini of actin-containing microfilament bundles (Hotulainen and Lappalainen 2006). Some microfilament bundles are contractile (Pellegrin and Mellor 2007; Vallenius 2013) so that focal adhesions can be essential sensors of cellular tension. Signaling to the cytoskeleton is integral to focal adhesions, but they can also regulate proliferation and survival.
The major transmembrane components of focal adhesions are integrin receptors (Zaidel-Bar et al. 2007). However, the complexity of the intracellular compartment of these organelles is extraordinary, with over 160 components identified from previous literature (Zaidel-Bar and Geiger 2010). Three more recent proteomics studies using various cellular models and preparation techniques have, however, changed the landscape completely (Humphries et al. 2009; Kuo et al. 2011; Schiller et al. 2011). Combining their data yields 1683 proteins associated with integrin-mediated adhesion (Geiger and Zeidal-Bar 2012). Moreover, there are rather few proteins common to all three analyses. However, as expected, matrix proteins, cytoskeletal and adaptor proteins, receptors, endocytic components, phosphatases and kinases feature strongly. Among these, tyrosine kinases, such as focal adhesion kinase (FAK) and Src have received much attention (Brown et al. 2005; Gardel et al. 2010; Webb et al. 2004; Westhoff et al. 2004). Originally believed to be essential for their assembly, it seems more likely that FAK is involved in the turnover of these structures (Geiger et al. 2001; Ilic et al. 1995). Serine/threonine kinases have, on the other hand, received much less attention, and few representatives appear to be localized to focal adhesions. However, members of the PKC family, most notably PKCα, have been recorded on several occasions.
In this review, we focus on the PKC family and its possible roles in focal adhesion assembly and dynamics. Since focal contacts and focal adhesions are key components for adhesion and migration (Chrzanowska-Wodnicka and Burridge 1996; Kirfel et al. 2004; Ridley et al. 2003) both in vitro and in vivo, their functions are important to understand. Moreover, they are reduced or absent and may have different compositions in tumor cells, which provides further rationale for their study.
PKC Structure and Activation
The PKCs are sub-grouped into three classes, based upon structural homology and cofactor requirements for activation. The classical PKCs (cPKCs) are the α, βI, βII and γ isoforms, which are activated by Ca2+, diacylglycerol (DAG) and phosphatidylserine (PS). PKC δ, ϵ, η and θ comprise the novel PKCs (nPKCs), which lack the Ca2+ binding property, whereas the atypical PKCs (aPKCs) ζ and ι/λ respond only to PS for activation (Fig. 1). This family of 65-79 kDa kinases all have a conserved N-terminal regulatory domain, containing one or two membrane-targeting motifs and a pseudosubstrate that blocks the catalytic site of the C-terminal catalytic domain of the non-activated kinase (Newton 2001). A conserved and essential threonine phosphorylation site is present at their activation loop and threonine or serine phosphorylation sites at the turn- and hydrophobic motifs. The aPKCs are slightly different, having a negatively charged glutamic acid in place of serine or threonine at the hydrophobic site (Keranen et al. 1995).
Figure 1.

PKC family structures. Subfamily nomenclatures, corresponding regulatory- and kinase domain regions, specific cofactor requirements and binding sites, and the three priming sites are indicated. The figure is not to scale. N: N-terminus; C: C-terminus; PS: pseudosubstrate; C1, C2…: conserved region 1, 2…; V: variable region; hinge: hinge region; DAG: diacylglycerol; PtdSer: phosphatidylserine; PtdIns4,5P2: phosphatidylinositol 4,5-bisphosphate; AL: activation loop; TM: turn motif; HM: hydrophobic motif.
According to the classical view of PKC activation, the newly synthesized protein localizes at the plasma membrane (Borner et al. 1989), where it becomes phosphorylated at the activation loop. This PDK-1–dependent phosphorylation (Dutil et al. 1998) is followed by a rapid phosphorylation at the turn- and hydrophobic motifs of classical and novel PKCs (Borner et al. 1989; Keranen et al. 1995). This primed and ready-to-signal kinase now localizes in the cytosol until activated (Keranen et al. 1995). Upon activation, the kinase is recruited to specific subcellular compartments to phosphorylate downstream substrates. PDK-1–mediated phosphorylation takes place without any cofactor requirements (Dutil et al. 1998) and seems to occur as soon as PKC is expressed, as PDK-1 is a constitutively active enzyme (Mora et al. 2004), and the rate limiting factor of PKC priming phosphorylation seems to be the dissociation of PDK-1 (Gao et al. 2001). Consequently, most PKC species are found in their primed state, and dephosphorylation is probably a pathway either for their degradation or rephosphorylation. Therefore, their subcellular localization and temporal activity are considered to be the most important regulators of PKC activity.
In a canonical process for the activation of primed PKC, second messengers are produced through ligand binding to cognate receptors, such as growth factor receptors, with downstream activation of phospholipase Cγ (PLCγ). This cleaves the membrane lipid phosphatidylinositol 4,5-bisphosphate (PtdIns4,5P2) to yield the co-factors diacylglycerol and inositol trisphosphate (IP3), which can trigger Ca2+ flux into the cytosol from intracellular stores (Parekh et al. 2000). Canonical PKC activation is possible at adhesions, since phospholipase Cγ1 is listed as a focal adhesion component. This may be an indicator of convergence of growth factor/tyrosine kinase signaling with that of integrins, since integrins have no intrinsic kinase activity. Early studies implicating phospholipase in this pathway used fibronectin-coated beads as a proxy for focal adhesion formation (Plopper et al. 1995; Miyamoto et al. 1995).
In terms of focal adhesions, however, an additional mechanism of PKCα activation has been documented (Lim et al. 2003; Oh et al. 1998). The transmembrane proteoglycan receptor, syndecan-4, is frequently associated with focal adhesions, and in the presence of Ptdins4,5P2, PKCα is activated in a ternary complex (Oh et al. 1997, 1998). Consistent with this non-canonical PKCα activation, discrete binding sites for PtdIns4,5P2 have been localized in this protein (Corbalán-García et al. 2003). Furthermore, in neuroblastoma cells, PKCα was insensitive to stimulation by DAG in vivo (Raghunath et al. 2003). PtdIns4,5P2 is important for focal adhesion assembly for several reasons in addition to a potential role in PKC signaling; for example, mediating conformational changes in vinculin is required for its binding to actin and other focal adhesion components (Gilmore and Burridge 1996).
The Life Cycle of Focal Adhesions
At the leading edge of migrating cells, actin fibers polymerize and protrude through the lamellum to form nascent adhesions (Beningo et al. 2001). These very small, rounded structures contain talin, paxillin and tyrosine phosphorylated proteins (Zaidel-Bar et al. 2003, 2004). They are the first structures to monitor the extracellular matrix and, upon receiving a migratory signal, they develop into focal complexes, which additionally contain α-actinin, vinculin and FAK (Zaidel-Bar et al. 2003, 2004) and are located between the lamellum and lamellipodium (Geiger et al. 2009). Like nascent adhesions, focal complexes may govern cell migration or further mature into the larger focal adhesions or focal contacts (Beningo et al. 2001; Geiger et al. 2001) with longer turnover rates (Ivaska 2012). Focal adhesions form at the end of F-actin stress fibers and are sites where the cells experience mechanical force and molecular signaling cues from the ECM, and from where this information is transmitted into the cell interior to induce signaling cascades (Geiger et al. 2001). Large focal adhesions bind strongly to their substratum and mediate cell spreading by transducing a large contractive load onto the stress fibers, whereas smaller adhesions at the leading edge mediate traction forces required for cell migration (Pellegrin and Mellor 2007). As the trailing edge focal adhesions disassemble, the cell body is able to translocate (Kirfel et al. 2004) in the direction of cell migration. In addition to the cellular distribution of PKCs serving as important means to control focal adhesions, the temporal control of individual PKC isoform activation also represents a control mechanism.
Cell Spreading and Focal Complex Formation
When anchorage-dependent cells are seeded onto fibronectin, an interaction between integrin α5β1 and the central integrin-binding domain of fibronectin induces an outside-in activation of the integrin, leading to cell attachment through the activation of PKCϵ (Besson et al. 2001; Disatnik and Rando 1999; Disatnik et al. 2002). This was shown in mouse skeletal muscle cells lacking α5 integrin, which were unable to attach or spread onto the RGD-containing domain, as were α5-expressing cells treated with a PKC inhibitor (Disatnik and Rando 1999). PKC in this system was also potentially able to mediate inside-out activation of α4β1 integrin, initiating cell spreading through an α5β1-independent mechanism. PKCα and δ activities were not required in order for these cells to spread; these isoforms seemed to be activated later and were involved in the formation of adhesion organelles (Disatnik et al. 2002).
In the case of PKCα, this delayed activity probably reflected its importance in focal complex formation during cell spreading. Accordingly, it was reported in MEF cells that PKCα activity was required in order for these cells to form focal complexes during cell spreading (Bass et al. 2007a). This effect of PKCα was mediated through an early activation of Rac1 in cells seeded onto the α5β1-binding domain of fibronectin. Once again, cells were able to spread without PKCα engagement; however, for focal complexes to form, interaction of syndecan-4 with fibronectin to recruit PKCα-mediated Rac1 activation was required, as knock down or drug-mediated inhibition of this kinase failed to activate Rac1 and induce the maturation of focal complexes (Bass et al. 2007b).
Focal Adhesion Formation
Once cells have attached and started to spread, focal complexes start to mature into focal adhesions. In various contexts, PKCα has been discussed as a regulator.
Historically, PKCα was first recorded as a focal adhesion component by Jaken’s group in REF52 cells (Fig. 2) (Jaken et al. 1989). Subsequently, we showed this was true of primary fibroblasts (Woods and Couchman 1992). However, in 3T3 cells, focal adhesions did not stain strongly for PKCα and, in transformed cells, it was noted that PKCα was more frequently localized to the nucleus (Leach et al. 1989). More recent proteomics work with mouse fibroblasts identified PKCα, but no other family members, in the integrin adhesome, interestingly in a pool that was dependent on myosin II activity (Schiller et al. 2011). A similar study with human fibroblasts also identified the kinase, but not reproducibly (Kuo et al. 2011).
Figure 2.
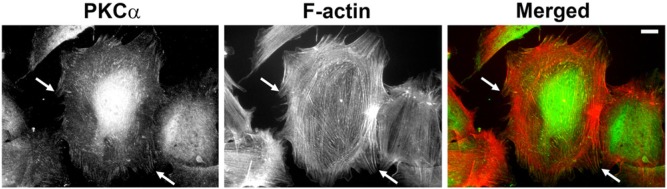
PKCα localizes to focal adhesions. Endogenous PKCα was detected in rat embryonic fibroblasts by indirect immunofluorescence microscopy. Cells were fixed in 4% paraformaldehyde containing 0.1% Tween 20 in phosphate-buffered saline followed by sequential incubations with anti-PKCα (clone M4, Millipore, Billerica, MA) and Alexa488-labelled anti mouse IgG (green; Molecular Probes, Carlsbad, CA). Filamentous actin (F-actin) was visualized using Phalloidin-Alexa568 (red; Molecular Probes). Images were captured on a Zeiss Axioplan epifluorescence microscope, and intensity and contrast were enhanced using Adobe Photoshop CS6 (Adobe Systems Inc., San Jose, CA). Arrows show co-localization of PKCα with termini of actin stress fibers. Bar = 10 μm.
Integrin β1 and PKCα have been suggested to interact directly in a single report, this being in breast carcinoma cells (Ng et al. 1999). In primary fibroblasts, however, a syndecan-4–mediated mechanism has been proposed (Bass et al. 2007a; Horowitz et al. 1999; Oh et al. 1998; Woods and Couchman 1992). These cells seeded onto the RGD-containing central domain of fibronectin will spread but form minimal focal adhesions and stress fibers (Bass et al. 2007a; Woods et al. 1986). However, the addition of the HepII domain of fibronectin, which binds to the heparan sulfate chains of syndecan-4 proteoglycan promotes recruitment to focal complexes and focal adhesions (Bass et al. 2007a; Saoncella et al. 1999; Woods et al. 1986, 2000). Clustering of the proteoglycan depends on binding of PtdIns4,5P2 (Koo et al. 2006; Oh et al. 1997). This complex is then able to recruit and activate PKCα (Oh et al. 1998), inducing a signaling cascade in which phosphorylation of RhoGDIα (on Ser34) releases its binding to the small GTPase RhoA (Dovas et al. 2006, 2010). In its GTP-bound and active state, RhoA binds ROCKII to release its auto-inhibition, leading to the phosphorylation of the myosin light chain of myosin II through myosin light-chain kinase and phosphatase activation and inhibition, respectively (Yoneda et al. 2005). Phosphorylated myosin II is able to bind actin, which activates the intrinsic ATPase activity of myosin II, leading to contraction of the cytoskeleton, and formation of stress fibers and focal adhesions (Chrzanowska-Wodnicka and Burridge 1996; Citi and Kendrick-Jones 1987; Riento and Ridley 2003).
PKCα-mediated regulation of Rho GTPase activation is, however, quite complex, and other scenarios have been reported. PKCα also seems to mediate RhoA inhibition during focal complex formation in the early stage of cell spreading. Accordingly, in the initial α5β1- and syndecan-4-mediated spreading of immortalized mouse fibroblasts onto their respective fibronectin domains, focal complex formation required a p190ARhoGap (p190A)-mediated down-regulation of RhoA, localizing this small GTPase in the cytosol (Bass et al. 2008). The ability of focal complexes to mature into focal adhesions, however, required relocation and activation of RhoA at these structures at later time-points when cells were spread. The interaction of active PKCα with syndecan-4 was required for p190A subcellular localization and for its inhibition of RhoA activity (Bass et al. 2008). Thus, spatiotemporal regulation of PKCα is essential for the formation of focal complexes and, later, their maturation into focal adhesions. In a similar way, CHO cells expressing β1 or β3 integrin do not form focal adhesions in response to cognate antibodies used as ligands but PKC and Rho activity were required additionally (Defilippi et al. 1997). Elfenbein et al. (2009) suggested a different scenario with respect to syndecan-4/PKCα and RhoGDI. In this case, evidence was presented that, in response to FGF2 binding to the heparan sulfate of syndecan-4, PKCα phosphorylated Ser96 of RhoGDI, causing release of RhoG (a Rac homolog), which, in turn, led to the polarized activation of Rac1, and cell migration (Elfenbein et al. 2009).
PKCα Substrates and Interactome
As a serine/threonine kinase, PKCα has many potential substrates. Related to the integrin adhesome, however, the list of known substrates is modest, but almost certainly incomplete. The adhesome website (www.adhesome.org) currently lists 26 interactions, though many of these relate to components that may be peripheral to focal adhesions. Others do not clearly involve the α isoform. Table 1 lists the known substrates and interacting partners with respect to focal adhesions. Prominent substrates are focal adhesion proteins; e.g., filamin A and C, vinculin, VASP and talin. Other potential substrates are tyrosine phosphatases and kinases, but overall the impact of PKC interaction and phosphorylation has yet to be fully evaluated.
Table 1.
PKCα Interacting Partners and Substrates.
| Interacting partner | Phosphorylation | Outcome | References | Notes |
|---|---|---|---|---|
| Vinculin | Tail, at phospholipid-binding site | Stabilization of the open conformation | Ziegler et al. 2002 | |
| VASP | Ser157 | ? | Chitaley et al. 2004 | |
| Talin | Multiple | ? | Litchfield and Ball 1986 | PKC from brain |
| Filamin A | Ser2152 | Facilitates caveolin 1 mediated internalization | Tigges et al. 2003; Muriel et al. 2011 | |
| Ezrin | ? | Ezrin activation | Ng et al. 2001 | |
| Fascin | Ser39 | Increased migration, focal adhesion remodeling | Anilkumar et al. 2003 | Constitutive interaction with a S39D fascin mutant |
| SHP2 | Ser576 and Ser591 | None | Strack et al. 2002 | |
| Syndecan-4 | Not phosphorylated | Kinase Activation in the presence of PtdIns4,5P2 | Oh et al. 1997; | |
| Horowitz et al. 1999 | ||||
| Integrin β1 | ? | Integrin internalization through dynamin-1 pathway | Ng et al. 1999 | |
| Caveolin 1 | ? | Internalization | Sukumaran et al. 2002 | |
| pp60src | Ser12 | ? | Gould et al. 1985 | PKC from brain |
| p72syk | ? | Enhanced tyrosine kinase activity | Borowski et al. 1998 |
In the table, ‘?’ indicates an unknown variable.
Whereas PKCα may be a focal adhesion component, the counteracting serine/threonine phosphatases have also been reported to be part of focal adhesions. In a number of different cell types, PP1δ was reported as a focal adhesion component, which could be immunoprecipitated with FAK and αV integrin (Villa-Moruzzi et al. 1998). PP1α was reported not to be a focal adhesion component, but later Eto et al. (2007) reported on the appearance of PP1α in very late-staged adhesions that contained tensin. In endothelial cells, PP2A activity was reported to oppose cell migration (Young et al. 2002). With its inhibition, paxillin serine phosphorylation increased, while its tyrosine phosphorylation decreased. This caused dissolution of FAK/Src/paxillin complexes, with increased Src activity leading to enhanced migration. One of the fibroblast adhesome studies (Kuo et al. 2011) reported the involvement of several serine/threonine phosphatases, which will be interesting to follow up. In contrast, given the important roles for FAK and Src, tyrosine phosphatases are well known modulators of integrin-mediated adhesion (Stoker 2005; Cohen and Guan 2005; Broussard et al. 2008).
In the context of focal adhesion assembly, PKCα substrates may not be limited to regulators of small G-proteins of the Rho family. Raf-1 is a direct target of PKCα, which, when activated, signals through the ERK/MAPK cascade (Kolch et al. 1993). In some cells, phosphoERK localizes to focal adhesions (Besson et al. 2001), but in these glioma cells, PKCϵ activity, not PKCα, localized phosphoERK at focal adhesions. This may be an important facet of survival signaling, a function of focal adhesions additional to theirs roles in cytoskeletal organization (Meredith and Schwartz 1997). Whereas the tyrosine kinase FAK is reported not to be a direct substrate for PKCα (Sinnett-Smith et al. 1993; Vuori and Ruoslahti 1993), in some way its activity might be regulated by PKC (Tu et al. 2001). However, the molecular basis for this is unclear. Focal adhesions contain a plethora of cytoplasmic protein components, many of which could be substrates for PKCα. The major focal adhesions adaptor protein paxillin is serine phosphorylated upon cell adhesion onto extracellular matrix substrates and PKCs may directly or indirectly mediate paxillin phosphorylation at adhesion sites (Nichilo and Yamada 1996; Bellis et al. 1997).
The interaction between PKCα and caveolin 1 is worthy of mention, as this hints at a possible role for the kinase in adhesion turnover. PKCα phosphorylation of filamin A at Ser2152 is required for caveolin 1 internalization (Muriel et al. 2011). This may be related to β1 integrin endocytosis in a dynamin- and caveolin-dependent manner, in response to syndecan-4/PKCα activation by ligand binding to the heparan sulfate chains of the syndecan (Bass et al. 2011).
Focal Adhesion Disassembly
In addition to their role in focal adhesion assembly, PKCs might therefore be implicated in the disassembly of these structures. Phosphorylation of paxillin at Ser178 is known to be important for cell migration and an Ala178 non-phosphorylatable mutant of this protein induced focal adhesions of increased size in NBTII cells, implicating a possible role for paxillin phosphorylation in focal adhesion disassembly and cell migration (Huang et al. 2003). In hepatocellular carcinoma cells, hepatocyte growth factor binding to its receptor c-Met induced cell migration through ERK-mediated paxillin phosphorylation, while also stimulating PKC activation (Hu et al. 2013). Using a PKC isotype-specific shRNA screen and drug treatments in this cell system, PKCα and δ isoforms were found to inhibit ERK-mediated phosphorylation of paxillin at Ser178, probably through mediating endocytosis and degradation of the c-Met receptor. Yet, the expression of these isoforms was still required for actin polarization and migration of this cell line. This apparent discrepancy between PKC-mediated inhibition of ERK activity and the simultaneous promotion of cell migration once again demonstrates the general theme of subtle spatiotemporal regulation of PKC activity in cell migration and focal adhesion dynamics. Accordingly, time-course experiments showed that both the membrane level and activity of PKC fluctuated three times within 24 hours, matching the phosphorylation levels of its downstream signaling proteins, as well as a temporal actin polarization (Hu et al. 2013).
Although not strictly related to cell adhesion, Violin et al. (2003) provided new insight into PKC and substrate dynamics. In a series of FRET microscopy studies, they showed an oscillatory phosphorylation/dephosphorylation of membrane-associated PKC substrates that immediately followed similar cytoplasmic Ca2+ oscillations. Moreover, a YFP-PKCβII probe also showed similar oscillations in membrane association with cycling times of minutes. These studies were carried out in HeLa cells, but whether these observations can be extrapolated to PKC in adhesions is not known. Possibly, this spatiotemporal regulation of PKC, and substrate phosphorylation, is necessary to provide focal adhesion turnover that is timed to meet the specific requirements of cell migration. It would be interesting to know whether the serine phosphorylation of paxillin or other PKC substrates in adhesions exists to the same degree at both the leading and trailing edges of cells, and whether cell migration influences the spatial regulation of PKC-dependent, focal adhesion component phosphorylation.
Connecting Focal Adhesions to the Cytoskeleton
Vinculin is one of the focal adhesion proteins known to mediate tension and, in its phosphorylated state, it is active and able to bind F-actin to connect focal adhesions with the actin cytoskeleton (Grashoff et al. 2010; Johnson and Craig 1995; Massoumi and Sjölander 2001), and possibly transduce tension monitored at focal adhesions into signaling cascades throughout the cytoskeleton. Besides its important role in focal adhesions, vinculin is also present at cell-cell contacts. Phorbol ester-induced activation of PKCα causes vinculin to dissociate from its cell-cell interaction partner, α-catenin, in intestinal epithelial cells, and induces its accumulation with PKCα at focal adhesions, leading to increased cell adhesion (Massoumi and Sjölander 2001). Although not investigated, this might also induce a migratory phenotype, which requires cell-cell contacts to become weaker and focal adhesions to grow at the leading edge of migrating cells. PKCα-mediated phosphorylation of vinculin was also seen in HeLa cells spreading on collagen (Ziegler et al. 2002). In this system, PtdIns4,5P2 was suggested to recruit vinculin to focal complexes—this interaction exposing a docking site for PKCα to phosphorylate the vinculin tail (Ziegler et al. 2002). As discussed, PtdIns4,5P2 is important for PKCα recruitment to some focal adhesion protein complexes. In this setting, however, an additional role for PtdIns4,5P2 probably exists, where it disrupts the closed intramolecular head-tail conformation of vinculin and induces a parallel oligomerization to expose its PKCα binding site (Johnson and Craig 1995; Ziegler et al. 2002).
Vinculin is one of the first components to be localized at cellular adhesion sites and is a commonly used marker of both the initial focal complexes as well as focal adhesions (Geiger et al. 2001). The interaction between PKCα and vinculin was transient and only observed at early time points, suggesting a role for PKCα in initiating focal adhesion formation, at least in regards to vinculin phosphorylation (Ziegler et al. 2002). An interaction between PtdIns4,5P2 and vinculin has been shown to inhibit the association between vinculin and F-actin, while promoting vinculin interactions with focal adhesion proteins (Steimle 1999). This might explain the observed transient nature of the PKCα/vinculin interaction, in that PKCα would then be transiently recruited to phosphorylate vinculin, which then dissociates from PtdIns4,5P2, and also PKCα, to be free for its interactions with F-actin in its now phosphorylated state. Therefore, PtdIns4,5P2-mediated temporal control of PKCα localization near its vinculin substrate has important implications in connecting focal adhesions to the cytoskeleton.
Focal Adhesions and Cell Migration
When cells receive a migratory stimulus, they polarise to migrate in the stimulatory direction (Li and Gundersen 2008), a process that often involves the atypical family of PKCs (Etienne-Manneville and Hall 2001; Iden and Collard 2008). The migrating cells undergo repeated cycles of lamellipodial protrusion of their leading edge, produced through F-actin polymerization (Pellegrin and Mellor 2007), to anchor this site onto the substrate (Abercombie et al. 1971), enabling contraction of actomyosin fibres throughout the cell body (Pellegrin and Mellor 2007). The generated traction force displaces the cell body in the forward direction, due to retraction of the trailing edge and the concomitant disassembly of the trailing edge focal adhesions, releasing cells from their substratum at the cell rear (Kirfel et al. 2004; Pellegrin and Mellor 2007).
In contrast to the initial cell adhesion and spreading, during which primarily the ϵ isoform of PKC is activated (Disatnik et al. 2002), an early activation of PKCδ is required for migration of many cell types (Chaudhuri et al. 2005). This isoform phosphorylates syndecan-4 at Ser183 (Murakami et al. 2002), inhibiting syndecan-4 interaction with PtdIns4,5P2 and subsequently oligomerization (Koo et al. 2006) as well as PKCα binding and activation (Koo et al. 2006; Murakami et al. 2002). Therefore, PKCα and δ appear to have contrasting roles in cell adhesion and migration. Possibly, the PKCδ isoform is required for the initiation of migration, whereas PKCα is important for an initiated migration to proceed, where an earlier activation would promote cell spreading. Accordingly, in endothelial cells, prolonged drug-mediated activation of PKCδ negatively regulated cell migration by binding and serine phosphorylating syndecan-4 to inhibit its PKCα association and activation (Chaudhuri et al. 2005). One function of PKCδ might then be to inhibit PKCα’s effects on focal adhesions and cell spreading (Chaudhuri et al. 2005) during the initiation of migration. Additionally, PKCδ-mediated inhibition of PKCα could be required for cells to form the leading edge protrusion, which is required for cells to migrate. Interestingly, both the α and δ isoforms of PKC have been identified at nascent adhesions (Jockusch et al. 1995). Fibronectin-mediated activation of PKCα inhibits the formation of nascent adhesions and cell protrusions by phosphorylating fascin (at Ser39) to block its association with actin and thereby its functioning in F-actin polymerization (Adams 2004). A dynamic activity of PKCα is probably required in this context. Accordingly, both Ser39 phosphorylated and dephosphorylated fascin species are important for carcinoma cell protrusion and migration on laminin (Hashimoto et al. 2007). Contrasting this, though, the effect of Ser39 has been suggested to be ECM substrate-specific, as phosphorylation of this residue was required for focal adhesion formation and cell spreading specifically on fibronectin (Adams et al. 1999), with reduced cell migration (Anilkumar et al. 2003). The interaction between wild-type fascin and PKCα was, however, temporal (Anilkumar et al. 2003), which does support an involvement of PKCα in focal adhesion formation during cell spreading, but does not exclude its importance in the regulation of focal adhesion dynamics during the later stages of cell migration.
An important aspect of cell migration is the replenishment of integrin at the leading edge of cells, to control the dynamics of focal adhesions and cell polarity, through integrin-mediated regulation of downstream adhesion components (Caswell et al. 2009). The exact mechanism of integrin recycling in cell migration is, however, still a matter of investigation. A controversy seems to exist between a model where integrins from the trailing edge are internalized to be recycled at the leading edge focal adhesions or, alternately, that most of the internalization and recycling occurs adjacent to the leading edge (Bretscher 1996; Caswell and Norman 2006). As the formation of adhesions and their turnover is very dynamic at the leading edge of cells, it seems plausible that integrins at these sites would be internalized and recycled in order to regulate such dynamics.
Integrins are internalized into early endosomes to be sorted for degradation or recycling through a long or short pathway loop. Short-loop recycled integrins, such as αvβ3, are, upon stimulation, rapidly brought back to the plasma membrane through a Rab4-dependent mechanism, whereas other integrins are transported to the perinuclear recycling compartment for relocation to the membrane through a Rab11-mediated process of long-loop internalization (Caswell and Norman 2006).
PKCs are known to regulate cell-surface integrin levels in several cell types. Since the earlier studies on cell adhesion were conducted, it has more recently been suggested that PKCϵ is a key kinase in β1 integrin recycling, an essential part of the spreading and migratory process (Ivaska et al. 2002, 2005), while Besson et al. (2002) proposed an interaction between these two proteins mediated by RACK1. Evidence that PKCϵ locates to nascent or mature focal adhesions is lacking, but this is not inconsistent with the key roles for the kinase in trafficking, rather than signaling, at adhesions.
Focal Contacts and Focal Adhesions
The terms focal contact and focal adhesion are both used interchangeably to describe the same structures. However, a distinction between them was proposed many years ago (Couchman and Rees 1979): rapidly migrating cells established small, short-lived structures that would eventually give way to larger, more stable adhesions that were more compatible with anchorage. The principle that focal adhesion size is inversely related to migration speed has been revisited several times (Lauffenburger and Horwitz 1996; Huttenlocher and Horwitz 2011). In the intervening period, however, no clear biochemical distinctions between these two potential classes of adhesions have been described (Geiger et al. 2009). This underlies the continuing lack of knowledge on what governs the turnover rate of focal adhesions. In some cells, moreover, sliding of these structures with respect to the matrix-coated substrate has been reported (Goldyn et al. 2009; Smilenov et al. 1999) but, once again, it is not known how this property relates to the molecular constitution of adhesions. Whether PKC isoforms have roles to play in the stability or half-life of adhesions is unknown. Great strides have been made recently by proteomics analyses that have identified a huge number of potential components (Geiger and Zeidal-Bar 2012); some may be dispensable for focal adhesion assembly (Ilic et al. 1995), whereas others, such as, vinculin and talin (Geiger et al. 2009), appear essential.
Atypical PKCs and Migration
There has been much recent focus on the atypical members of the PKC family. These are PKCζ and PKCλ in the mouse and PKCζ and PKCι in human. The λ and ι isoforms are, in fact, about 98% identical, while PKCζ has approximately 70% identity to the other isoforms (Newton 1997). They cannot be activated by phorbol esters, but are activated by phospholipids such as phosphatidylinositol 3,4,5 trisphosphate and phosphatidic acid (Nakanishi et al. 1993; Limatola et al. 1994; Xiao and Liu 2013). As with other PKC enzymes, full activity also requires PDK-1 activity (Hirai and Chida 2003). Their importance for cell migration lies in part from complexes formed with members of the Rho family; e.g., Cdc42 and Rac, together with PAR6 and PAR3 (Joberty et al. 2000; Lin et al. 2000).These are directed to the leading edge of motile cells and regulate polarity. Since Cdc42 and Rac direct protrusive activity, these polarity complexes localize this activity for efficient locomotion. PAR6 interacts with the PB1 domain at the N-termini of the atypical PKCs, a domain that is unique to these isoforms (Xiao and Liu 2013). Linkage with integrins is also provided through an interaction between PAR complexes and Tiam1, a Rac guanine exchange factor. Tiam 1 also binds talin directly, and so indirectly with integrins (Wang et al. 2012). In this way, it is proposed that Rac activity is directed to integrin-mediated adhesions, consistent with adhesion turnover in cell migration.
In non-small cell lung cancer, PAR6 has been shown to associate with, and be phosphorylated by, the TGF-β receptor II (Gunaratne et al. 2013). In this way the atypical PKC/PAR complex promotes the TGF-β–driven epithelial-mesenchymal transformation widely reported to be a key aspect of cell motility acquired by carcinomas (Heldin et al. 2012). Many tumor cell types and some normal cells assemble adhesion complexes known as podosomes or invadopodia (Gimona et al. 2008). Even though they contain some cell-surface and cytoskeletal components in common with focal adhesions, they are nevertheless distinct. Moreover, invadopodia are sites of not only extracellular matrix adhesion, by virtue of integrins, but also matrix degradation, mediated largely by metalloproteinases. Atypical PKCs are not required for assembly of these organelles, where roles for PKCα could be important (Xiao et al. 2013), but PKCζ can be recruited by PKCδ, for example (Xiao et al. 2010, Xiao and Liu 2013). PKCζ is involved in the recruitment, release and activation of MMP9, an important matrix-degrading proteinase (Xiao and Liu 2013). In total, therefore, multiple PKC isoforms may be involved in the establishment and activity of podosomes and invadopodia, key organelles of extracellular matrix invasion.
In Vivo Adhesions and Disease
Structures analogous or homologous to focal adhesions have been described in vivo on many occasions (Kano et al. 1996; Turner et al. 1991). More recently, advanced microscopy has identified these structures in cells grown in three-dimensional matrices (Cukierman et al. 2001; Kubow and Horwitz 2011). However, others suggest that focal adhesions are not visible in cells grown in three-dimensional matrices (Fraley et al. 2010). Previous electron microscopy identified structures known as fibronexus in granulation tissue, associated with what most likely were myofibroblasts (Singer 1979). Dense bodies of smooth muscle are often proposed to be in vivo focal adhesion equivalents (Turner et al. 1991). However, these structures, much like the costameres of skeletal muscle, are very stable, in contrast to the transient adhesions of migratory cells. A proteomic comparison would be interesting but, so far, PKCα, although present in focal adhesions of tissue cultured cells, has not been shown as a component of costameres; in comparison, PKCϵ has been shown to be involved (Vanwinkle et al. 2002). Syndecan-4, on the other hand, which binds PKCα, is a costamere component (Vanwinkle et al. 2002), perhaps interacting with α-actinin in these structures (Okina et al. 2012), consistent with known interactions mapped in fibroblasts and by in vitro studies (Choi et al. 2008; Greene et al. 2003; Okina et al. 2012). Overall, however, it does suggest that PKCα may be associated with dynamic structures rather than very stable ones.
Knockout mice lacking PKCα have been produced and have at least two interesting phenotypes. They have skin wounding defects (Hara et al. 2005), reminiscent of both syndecan-1 (Stepp 2002) and -4 (Echtermeyer et al. 2001) knockout mice, which also show defects in postnatal repair. In addition, PKCα null mice, or those subject to pharmacological inhibition of the enzyme, show dramatic protection from cardiac failure (Liu and Molkentin 2011). Whether this relates to cell-matrix interactions is less clear, but it is interesting that recent data suggest that syndecan-4 is essential for compensatory hypertrophy in the pressure-overloaded heart (Finsen et al. 2011). This was linked to signaling through the phosphatase calcineurin and nuclear factor of activated T-cell (NFAT), which is reduced in the absence of the proteoglycan. This, and related data suggest that syndecan-4 can be a tension sensor independent of other focal adhesion components (Li and Chaikoff 2002). Further work is required to dissect this pathway, but it will be interesting to ascertain whether syndecan-4 is unique among the family in regard to this property.
Conclusions
Protein kinase C has central roles in signaling in response to many extracellular ligands, and can influence many aspects of cell behavior. Its association with focal adhesions, particularly with regard to PKCα, has been recorded several times but, although its interactions are known, the specific responses and impact on adhesion dynamics still await elucidation. Within adhesions there are a plethora of potential substrates, and it will take much further work to understand its molecular functions.
Footnotes
Declaration of Conflicting Interest: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: Support from the Danish National Research Foundation, Lundbeck Fonden, the Novo Nordisk Fonden, and the department of Biomedical Sciences at the University of Copenhagen are gratefully acknowledged.
References
- Abercombie M, Heaysman J, Pegrum S. (1971). The Locomotion of Fibroblasts in Culture. Exp Cell Res 67:359-367 [DOI] [PubMed] [Google Scholar]
- Adams JC. (2004). Roles of fascin in cell adhesion and motility. Curr Opin Cell Biol 16:590-596 [DOI] [PubMed] [Google Scholar]
- Adams JC, Clelland JD, Collett GD, Matsumura F, Yamashiro S, Zhang L. (1999). Cell-matrix adhesions differentially regulate fascin phosphorylation. Mol Biol Cell 10:4177-4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar N, Parsons M, Monk R, Ng T, Adams JC. (2003). Interaction of fascin and protein kinase Cα: a novel intersection in cell adhesion and motility. EMBO J 22:5390-5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass M, Morgan M, Humphries M. (2007a). Integrins and syndecan-4 make distinct, but critical, contributions to adhesion contact formation. Soft Matter 3:372-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass MD, Roach KA, Morgan MR, Mostafavi-Pour Z, Schoen T, Muramatsu T, Mayer U, Ballestrem C, Spatz JP, Humphries MJ. (2007b). Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J Cell Biol 177:527-538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass MD, Morgan MR, Roach KA, Settleman J, Goryachev AB, Humphries MJ. (2008). p190RhoGAP is the convergence point of adhesion signals from alpha 5 beta 1 integrin and syndecan-4. J Cell Biol 181:1013-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, Humphries MJ. (2011). A syndecan-4 hair trigger initiates wound healing through caveolin- and RhoG-regulated integrin endocytosis. Dev Cell. 21:681-93. 10.1016/j.devcel.2011.08.007. Epub 2011 Oct 6. Erratum in: Dev Cell 2012 Nov 13;23(5):1081-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellis SL, Perrotta JA, Curtis MS, Turner CE. (1997). Adhesion of fibroblasts to fibronectin stimulates both serine and tyrosine phosphorylation of paxillin. Biochem J 325:375-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beningo K, Dembo M, Kaverina I, Small JV, Wang YL. (2001). Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol 153:881-888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Davy A, Robbins SM, Yong VW. (2001). Differential activation of ERKs to focal adhesions by PKC epsilon is required for PMA-induced adhesion and migration of human glioma cells. Oncogene 20:7398-7407 [DOI] [PubMed] [Google Scholar]
- Besson A, Wilson TL, Yong VW. (2002). The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. J Biol Chem 277:22073-22084 [DOI] [PubMed] [Google Scholar]
- Blumberg PM. (1988). Protein Kinase C as the receptor for the phorbol ester tumor promoters: sixth Rhoads memorial award lecture. Cancer Res 48:1-8 [PubMed] [Google Scholar]
- Borner C, Filipuzzi I, Wartmann M, Eppenberger U, Fabbro D. (1989). Biosynthesis and posttranslational modifications of protein kinase C in human breast cancer cells. J Biol Chem 264, 13902-13909 [PubMed] [Google Scholar]
- Borowski P, Heiland M, Kornetzky L, Medem S, Laufs R. (1998). Purification of catalytic domain of rat spleen p72syk kinase and its phosphorylation and activation by protein kinase C. Biochem J 331( Pt 2):649-657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher MS. (1996). Moving membrane up to the front of migrating cells. Cell 85:465-467 [DOI] [PubMed] [Google Scholar]
- Broussard JA, Webb DJ, Kaverina I. (2008). Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol 20:85-90 [DOI] [PubMed] [Google Scholar]
- Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE. (2005). Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell 16:4316-4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell PT, Norman JC. (2006). Integrin trafficking and the control of cell migration. Traffic 7:14-21 [DOI] [PubMed] [Google Scholar]
- Caswell PT, Vadrevu S, Norman JC. (2009). Integrins: masters and slaves of endocytic transport. Nature Rev Mol Cell Biol 10:843-853 [DOI] [PubMed] [Google Scholar]
- Chaudhuri P, Colles SM, Fox PL, Graham LM. (2005). Protein kinase Cdelta-dependent phosphorylation of syndecan-4 regulates cell migration. Circ Res 97:674-681 [DOI] [PubMed] [Google Scholar]
- Chitaley K, Chen L, Galler A, Walter U, Daum G, Clowes AW. (2004). Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. FEBS Lett 556:211-21514706852 [Google Scholar]
- Choi Y, Kim S, Lee J, Ko SG, Lee W, Han IO, Dovas A, Oh E-S. (2008). The oligomeric status of syndecan-4 regulates syndecan-4 interaction with alpha-actinin. Eur J Cell Biol 87:807-815 [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. (1996). Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol 133:1403-1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citi S, Kendrick-Jones J. (1987). Regulation of non-muscle myosin structure and function. Bioessays 7:155-159 [DOI] [PubMed] [Google Scholar]
- Cohen LA, Guan JL. (2005). Mechanisms of focal adhesion kinase regulation. Curr Cancer Drug Targets 5:629-643 [DOI] [PubMed] [Google Scholar]
- Corbalán-García S, García-García J, Rodríguez-Alfaro J, Gómez-Fernández JC. (2003). A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Calpha. J Biol Chem 278:4972-4980 [DOI] [PubMed] [Google Scholar]
- Couchman JR, Rees D. (1979). Actomyosin organisation for adhesion, spreading, growth and movement in chick fibroblasts. Cell Biol Int Rep 3:431-439 [DOI] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM. (2001).Taking cell-matrix adhesions to the third dimension. Science 294:1708-1712 [DOI] [PubMed] [Google Scholar]
- Defilippi P, Venturino M, Gulino D, Duperray A, Boquet P, Fiorentini C, Volpe G, Palmieri M, Silengo L, Tarone G. (1997). Dissection of Pathways Implicated in Integrin-mediated Actin Cytoskeleton Assembly. J Biol Chem 272:21726-21734 [DOI] [PubMed] [Google Scholar]
- Disatnik M-H, Rando T. (1999). Integrin-mediated muscle cell spreading: The role of protein kinase C in outside-in and inside-out signaling and evidence of integrin cross-talk. J Biol Chem 274:32486-32492 [DOI] [PubMed] [Google Scholar]
- Disatnik M-H, Boutet SC, Lee CH, Mochly-Rosen D, Rando TA. (2002). Sequential activation of individual PKC isozymes in integrin-mediated muscle cell spreading: a role for MARCKS in an integrin signaling pathway. J Cell Sci 115:2151-2163 [DOI] [PubMed] [Google Scholar]
- Dovas A, Yoneda A, Couchman JR. (2006). PKCalpha-dependent activation of RhoA by syndecan-4 during focal adhesion formation. J Cell Sci 119:2837-2846 [DOI] [PubMed] [Google Scholar]
- Dovas A, Choi Y, Yoneda A, Multhaupt HAB, Kwon S-H, Kang D, Oh E-S, Couchman JR. (2010). Serine 34 phosphorylation of rho guanine dissociation inhibitor (RhoGDIalpha) links signaling from conventional protein kinase C to RhoGTPase in cell adhesion. J Biol Chem 285:23296-23308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutil EM, Toker A, Newton AC. (1998). Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr Biol 8:1366-1375 [DOI] [PubMed] [Google Scholar]
- Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, Goetinck P. (2001). Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest 107:R9-R14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfenbein A, Rhodes JM, Meller J, Schwartz M, Matsuda M, Simons M. (2009). Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCalpha in a Rac1 activation pathway. J Cell Biol 186:75-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. (2001). Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 106:489-498 [DOI] [PubMed] [Google Scholar]
- Eto M, Kirkbride J, Elliott E, Lo SH, Brautigan DL. (2007). Association of the tensin N-terminal protein-tyrosine phosphatase domain with the alpha isoform of protein phosphatase-1 in focal adhesions. J Biol Chem 282:17806-17815 [DOI] [PubMed] [Google Scholar]
- Finsen AV, Lunde IG, Sjaastad I, Østli EK, Lyngra M, Jarstadmarken HO, Hasic A, Nygård S, Wilcox-Adelman SA, Goetinck PF, Lyberg T, Skrbic B, Florholmen G, Tønnessen T, Louch WE, Djurovic S, Carlson CR, Christensen G. (2011). Syndecan-4 is essential for development of concentric myocardial hypertrophy via stretch-induced activation of the calcineurin-NFAT pathway. PloS One 6:e28302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley SI, Feng Y, Krishnamurthy R, Kim DH, Celedon A, Longmore GD, Wirtz D. (2010). A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol 12:598-604. 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Toker A, Newton AC. (2001). The carboxyl terminus of protein kinase C provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J Biol Chem 276:19588-19596 [DOI] [PubMed] [Google Scholar]
- Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. (2010). Mechanical integration of actin and adhesion dynamics in cell migration. Ann Rev Cell Dev Biol 26:315-333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. (2001). Transmembrane extracellular matrix– cytoskeleton crosstalk. Nature Rev Mol Cell Biol 2:793-805 [DOI] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. (2009). Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol 10:21-33 [DOI] [PubMed] [Google Scholar]
- Geiger T, Zaidel-Bar R. (2012). Opening the floodgates: proteomics and the integrin adhesome. Curr Opin Cell Biol 4:562-8. 10.1016/j.ceb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Gilmore A, Burridge K. (1996). Regulation of vinculin bindning to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature 381:531-535 [DOI] [PubMed] [Google Scholar]
- Gimona M, Buccione R, Courtneidge SA, Linder S. (2008). Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol 20:235-241 [DOI] [PubMed] [Google Scholar]
- Goldyn AM, Rioja BA, Spatz JP, Ballestrem C, Kemkemer R. (2009). Force-induced cell polarisation is linked to RhoA-driven microtubule-independent focal-adhesion sliding. J Cell Sci 122:3644-3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould KL, Woodgett JR, Cooper JA, Buss JE, Shalloway D, Hunter T. (1985). Protein kinase C phosphorylates pp60src at a novel site. Cell 42:849-857 [DOI] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. (2010). Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466:263-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene DK, Tumova S, Couchman JR, Woods A. (2003). Syndecan-4 associates with alpha-actinin. J Biol Chem 278:7617-7623 [DOI] [PubMed] [Google Scholar]
- Gunaratne A, Thai BL, Di Guglielmo GM. (2013). Atypical protein kinase C phosphorylates Par6 and facilitates transforming growth factor β-induced epithelial-to-mesenchymal transition. Mol Cell Biol 33:874-886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Saito Y, Hirai T, Nakamura K, Nakao K, Katsuki M, Chida K. (2005). Deficiency of protein kinase Calpha in mice results in impairment of epidermal hyperplasia and enhancement of tumor formation in two-stage skin carcinogenesis. Cancer Res 65:7356-7362 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Parsons M, Adams J. (2007). Dual actin-bundling and protein kinase C-binding activities of fascin regulate carcinoma cell migration downstream of Rac and contribute to metastasis. Mol Biol Cell 18:4591-4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C-H, Vanlandewijck M, Moustakas A. (2012). Regulation of EMT by TGFβ in cancer. FEBS Lett 586:1959-1970 [DOI] [PubMed] [Google Scholar]
- Hirai T, Chida K. (2003). Protein kinase Czeta (PKCzeta): activation mechanisms and cellular functions. J Biochem (Tokyo) 133:1-7 [DOI] [PubMed] [Google Scholar]
- Horowitz A, Murakami M, Gao Y, Simons M. (1999). Phosphatidylinositol-4,5-bisphosphate mediates the interaction of syndecan-4 with protein kinase C. Biochemistry 38:15871-15877 [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Lappalainen P. (2006). Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol 173:383-394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C-T, Cheng C-C, Pan S-M, Wu J-R, Wu W-S. (2013). PKC mediates fluctuant ERK-paxillin signaling for hepatocyte growth factor-induced migration of hepatoma cell HepG2. Cell Signal 25:1457-1467 [DOI] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. (2003). JNK phosphorylates paxillin and regulates cell migration. Nature 424:219-223 [DOI] [PubMed] [Google Scholar]
- Humphries JD, Byron A, Bass MD, Craig SE, Pinney JW, Knight D, Humphries MJ. (2009). Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci Signal 2(87):ra51. 10.1126/scisignal.2000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR. (2011). Integrins in cell migration. Cold Spring Harb Perspect Biol 3(9):a005074. 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iden S, Collard JG. (2008). Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol 9:846-859 [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. (1995). Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377:539-544 [DOI] [PubMed] [Google Scholar]
- Ivaska J. (2012). Unanchoring integrins in focal adhesions. Nat Cell Biol 14:981-983 [DOI] [PubMed] [Google Scholar]
- Ivaska J, Whelan RDH, Watson R, Parker PJ. (2002). PKCϵ controls the traffic of β1 integrins in motile cells. EMBO J 21:3608-3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. (2005). PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J 24:3834-3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaken S, Leach K, Klauck T. (1989). Association of type 3 protein kinase C with focal contacts in rat embryo fibroblasts. J Cell Biol 109:697-704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. (2000). The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol 2:531-539 [DOI] [PubMed] [Google Scholar]
- Jockusch BM, Bubeck P, Giehl K, Kroemker M, Moschner J, Rothkegel M, Rüdiger M, Schlüter K, Stanke G, Winkler J. (1995). The molecular architecture of focal adhesions. Ann Rev Cell Dev Biol 11:379-416 [DOI] [PubMed] [Google Scholar]
- Johnson RP, Craig SW. (1995). F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature 373:261-264 [DOI] [PubMed] [Google Scholar]
- Kano Y, Katoh K, Masuda M, Fujiwara K. (1996). Macromolecular composition of stress fiber-plasma membrane attachment sites in endothelial cells in situ. Circ Res 79:1000-1006 [DOI] [PubMed] [Google Scholar]
- Keranen LM, Dutil EM, Newton AC. (1995). Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol 5:1394-1403 [DOI] [PubMed] [Google Scholar]
- Kirfel G, Rigort A, Borm B, Herzog V. (2004). Cell migration: mechanisms of rear detachment and the formation of migration tracks. Eur J Cell Biol 83:717-724 [DOI] [PubMed] [Google Scholar]
- Kolch W, Heidecker G, Kochs G, Hummel R, Vahidl H, Mischak H, Finkenzeller G, Marme D, Rapp U. (1993). Protein kinase Cα activates RAF-1 by direct phosphorylation. Nature 364:249-252 [DOI] [PubMed] [Google Scholar]
- Koo B-K, Jung YS, Shin J, Han I, Mortier E, Zimmermann P, Whiteford JR, Couchman JR, Oh E-S, Lee W. (2006). Structural basis of syndecan-4 phosphorylation as a molecular switch to regulate signaling. J Mol Biol. 355:651-663 [DOI] [PubMed] [Google Scholar]
- Kubow KE, Horwitz AR. (2011). Reducing background fluorescence reveals adhesions in 3D matrices. Nat Cell Biol 13:3–5; 10.1038/ncb0111-3. Erratum in: Nat Cell Biol 2012, 14(12):1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JC, Han X, Hsiao CT, Yates JR, 3rd, Waterman CM. (2011). Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol 13:383-93. 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. (1996). Cell migration: a physically integrated molecular process. Cell 84:359-369 [DOI] [PubMed] [Google Scholar]
- Leach KL, Powers EA, Ruff VA, Jaken S, Kaufmann S. (1989). Type 3 protein kinase C localization to the nuclear envelope of phorbel ester-treated NIH 3T3 cell. J Cell Biol 109:685-695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chaikof EL. (2002). Mechanical stress regulates syndecan-4 expression and redistribution in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 22:61-68 [DOI] [PubMed] [Google Scholar]
- Li R, Gundersen GG. (2008). Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol 9:860-873 [DOI] [PubMed] [Google Scholar]
- Lim S-T, Longley RL, Couchman JR, Woods A. (2003). Direct binding of syndecan-4 cytoplasmic domain to the catalytic domain of protein kinase C alpha (PKC alpha) increases focal adhesion localization of PKC alpha. J Biol Chem 278:13795-13802 [DOI] [PubMed] [Google Scholar]
- Limatola C, Schaap D, Moolenar WH, van Blitterswijk WJ. (1994). Phosphatidic acid activation of protein kinase C-zeta overexpressed in COS cells: comparison with other protein kinase C isotypes and other acidic lipids. Biochem J 304:1001-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. (2000). A mammalian PAR-3-PAR-6 complex implicated in CDC42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol 2:540-547 [DOI] [PubMed] [Google Scholar]
- Litchfield DW, Ball EH. (1986). Phosphorylation of the cytoskeletal protein talin by protein kinase C. Biochem Biophys res Commun 134:1276-1283 [DOI] [PubMed] [Google Scholar]
- Liu Q, Molkentin JD. (2011). Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol 51:474-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marte BM, Meyer T, Stabel S, Standke GJ, Jaken S, Fabbro D, Hynes NE. (1994). Protein kinase C and mammary cell differentiation: involvement of protein kinase C α in the induction of β-casein expression. Cell Growth Diff 5:239-247 [PubMed] [Google Scholar]
- Massoumi R, Sjölander A. (2001). Leukotriene D(4) affects localisation of vinculin in intestinal epithelial cells via distinct tyrosine kinase and protein kinase C controlled events. J Cell Sci 114:1925-1934 [DOI] [PubMed] [Google Scholar]
- Meredith JE, Schwartz MA. (1997). Integrins, adhesion and apoptosis. Trends Cell Biol 7:146-150 [DOI] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DMF, Alessi DR. (2004). PDK1, the master regulator of AGC kinase signal transduction. Sem Cell Dev Biol 15:161-170 [DOI] [PubMed] [Google Scholar]
- Murakami M, Horowitz A, Tang S, Ware JA, Simons M. (2002). Protein kinase C (PKC) delta regulates PKCalpha activity in a Syndecan-4-dependent manner. J Biol Chem 277:20367-20371 [DOI] [PubMed] [Google Scholar]
- Muriel O, Echarri A, Hellriegel C, Pavón DM, Beccari L, Del Pozo MA. (2011). Phosphorylated filamin A regulates actin-linked caveolae dynamics. J Cell Sci 124(Pt 16):2763-76. 10.1242/jcs.080804. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada KM. (1995). Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science 267:883-885 [DOI] [PubMed] [Google Scholar]
- Newton AC. (1997). Regulation of protein kinase C. Curr Opin Cell Biol 9:161-167 [DOI] [PubMed] [Google Scholar]
- Newton AC. (2001). Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101:2353-2364 [DOI] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ. (1999). PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 18:3909-3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Parsons M, Hughes WE, Monypenny J, Zicha D, Gautreau A, Arpin M, Gschmeissner S, Verveer PJ, Bastiaens PI, Parker PJ. (2001). Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J 20:2723-2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichilo MO, Yamada KM. (1996). Integrin alpha(v)beta(5)-dependent Serine Phosphorylation of Paxillin in Cultured Human Macrophages Adherent to Vitronectin. J Biol Chem 271:11016-11022 [DOI] [PubMed] [Google Scholar]
- Oh E-S, Woods A, Couchman JR. (1997). Multimerization of the Cytoplasmic Domain of Syndecan-4 Is Required for Its Ability to Activate Protein Kinase C. J Biol Chem 272:11805-11811 [DOI] [PubMed] [Google Scholar]
- Oh E-S, Woods A, Lim S-T, Theibert AW, Couchman JR. (1998). Syndecan-4 proteoglycan cytoplasmic domain and phosphatidylinositol 4,5-bisphosphate coordinately regulate protein kinase C activity. J Biol Chem 273:10624-10629 [DOI] [PubMed] [Google Scholar]
- Okina E, Grossi A, Gopal S, Multhaupt HAB, Couchman JR. (2012). Alpha-actinin interactions with syndecan-4 are integral to fibroblast-matrix adhesion and regulate cytoskeletal architecture. Int J Biochem Cell Biol 44:2161-2174 [DOI] [PubMed] [Google Scholar]
- Parekh DB, Ziegler W, Parker PJ. (2000). Multiple pathways control protein kinase C phosphorylation. EMBO J 19:496-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin S, Mellor H. (2007). Actin stress fibres. J Cell Sci 120:3491-3499 [DOI] [PubMed] [Google Scholar]
- Plopper GE, McNamee HP, Dike LE, Bojanowski K, Ingber DE. (1995). Convergence of integrin and growth factor receptor signalling pathway within the focal adhesion complex. Mol Biol Cell 6:1349-1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunath A, Ling M, Larsson C. (2003). The catalytic domain limits the translocation of protein kinase Cα in response to increases in Ca2+ and diacylglycerol. Biochem J 370:901-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. (2003). Cell migration: integrating signals from front to back. Science 302:1704-1709 [DOI] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. (2003). Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446-456 [DOI] [PubMed] [Google Scholar]
- Saoncella S, Echtermeyer F, Denhez F, Nowlen JK, Mosher DF, Robinson SD, Hynes RO, Goetinck PF. (1999). Syndecan-4 signals cooperatively with integrins in a Rho-dependent manner in the assembly of focal adhesions and actin stress fibers. Proc Nat Acad Sci U S A 96:2805-E2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller HB, Friedel CC, Boulegue C, Fässler R. (2011). Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep 12:259-266. 10.1038/embor.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer II. (1979). The fibronexus: a transmembrane association of fibronectin-containing fibers and bundles of 5 nm microfilaments in hamster and human fibroblasts. Cell 16:675-685 [DOI] [PubMed] [Google Scholar]
- Sinnett-Smith J, Zachary I, Valverde AM, Rozengurt E. (1993). Bombesin stimulation of p125 focal adhesion kinase tyrosine phosphorylation. J Biol Chem 268:14261-14268 [PubMed] [Google Scholar]
- Smilenov LB, Mikhailov A, Pelham RJ, Marcantonio EE, Gundersen GG. (1999). Focal adhesion motility revealed in stationary fibroblasts. Science 286:1172-1174 [DOI] [PubMed] [Google Scholar]
- Steimle PA. (1999). Polyphosphoinositides inhibit the interaction of vinculin with actin filaments. J Biol Chem 274:18414-18420 [DOI] [PubMed] [Google Scholar]
- Stepp MA. (2002). Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J Cell Sci 115:4517-4531 [DOI] [PubMed] [Google Scholar]
- Stoker AW. (2005). Protein tyrosine phosphatases and signalling. J Endocrinol 185:19-33 [DOI] [PubMed] [Google Scholar]
- Strack V, Krützfeldt J, Kellerer M, Ullrich A, Lammers R, Häring HU. (2002). The Protein-tyrosine-phosphatase SHP2 is phosphorylated on serine residues 576 and 591 by protein kinase C isoforms alpha, beta 1, beta 2, and eta. Biochemistry 41:603-608 [DOI] [PubMed] [Google Scholar]
- Sukumaran SK, Quon MJ, Prasadarao NV. (2002). Escherichia coli K1 internalization via caveolae requires caveolin-1 and protein kinase Calpha interaction in human brain microvascular endothelial cells. J Biol Chem 277:50716-50724 [DOI] [PubMed] [Google Scholar]
- Tigges U, Koch B, Wissing J, Jockusch BM, Ziegler WH. (2003). The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase C alpha. J Biol Chem 278:23561-23569 [DOI] [PubMed] [Google Scholar]
- Tu LC, Chou CK, Chen HC, Yeh SF. (2001). Protein kinase C-mediated tyrosine phosphorylation of paxillin and focal adhesion kinase requires cytoskeletal integrity and is uncoupled to mitogen-activated protein kinase activation in human hepatoma cells. J Biomed Sci 8:184-190 [DOI] [PubMed] [Google Scholar]
- Turner C, Kramarcy N, Sealock R, Burridge K. (1991). Localization of paxillin, a focal adhesion protein, to smooth muscle dense plaques, and the myotendinous and neuromuscular junctions of skeletal muscle. Exp Cell Res 192:651-655 [DOI] [PubMed] [Google Scholar]
- Vallenius T. (2013). Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol 3:130001. 10.1098/rsob. 130001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanwinkle WB, Snuggs MB, De Hostos EL, Buja LM, Woods A, Couchman JR. (2002). Localization of the transmembrane proteoglycan syndecan-4 and its regulatory kinases in costameres of rat cardiomyocytes: a deconvolution microscopic study. Anat Rec 268:38-46 [DOI] [PubMed] [Google Scholar]
- Villa-Moruzzi E, Tognarini M, Cecchini G, Marchisio PC. (1998). Protein phosphatase 1 delta is associated with focal adhesions. Cell Adhes Commun 5:297-305 [DOI] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC. (2003). A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol 161:899-909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. (1993). Activation of Protein Kinase C Precedes α5β1 Integrin-mediated Cell Spreading on Fibronectin. J Biol Chem 268:21459-21462 [PubMed] [Google Scholar]
- Wang S, Watanabe T, Matsuzawa K, Katsumi A, Kakeno M, Matsui T, Ye F, Sato K, Murase K, Sugiyama I, Kimura K, Mizoguchi A, Ginsberg MH, Collard JG, Kaibuchi K. (2012). Tiam1 interaction with the PAR complex promotes talin-mediated Rac1 activation during polarized cell migration. J Cell Biol 199:331-345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL. (2000). Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21:181-187 [DOI] [PubMed] [Google Scholar]
- Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. (2004). FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol 6:154-161 [DOI] [PubMed] [Google Scholar]
- Westhoff M, Serrels B, Fincham V, Frame M, Carragher N. (2004). Src-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol 24:8113-8133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Couchman JR. (1992). Protein kinase C involvement in focal adhesion formation. J Cell Sci 101:277-290 [DOI] [PubMed] [Google Scholar]
- Woods A, Couchman JR, Johansson S, Höök M. (1986). Adhesion and cytoskeletal organisation of fibroblasts in response to fibronectin fragments. EMBO J 5:665-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Longley RL, Tumova S, Couchman JR. (2000). Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch Biochem Biophys 374:66-72 [DOI] [PubMed] [Google Scholar]
- Xiao H, Bai XH, Kapus A, Lu WY, Mak AS, Liu M. (2010). The protein kinase C cascade regulates recruitment of matrix metalloproteinase 9 to podosomes and its release and activation. Mol Cell Biol 30:5545-5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Liu M. (2013). Atypical protein kinase C in cell motility. Cell Mol Life Sci 70:3057-3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A, Multhaupt HAB, Couchman JR. (2005). The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol 170:443-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MR, Kolesiak K, Meisinger J. (2002). Protein phosphatase-2A regulates endothelial cell motility and both the phosphorylation and the stability of focal adhesion complexes. Int J Cancer 100:276-282 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Geiger B. (2010). The switchable integrin adhesome. J Cell Sci 123:1385-1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Ballestrem C, Kam Z, Geiger B. (2003). Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J Cell Sci 116:4605-4613 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Cohen M, Addadi L, Geiger B. (2004). Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc Trans 32:416-420 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. (2007). Functional atlas of the integrin adhesome. Nat Cell Biol 9:858-867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler WH, Tigges U, Zieseniss A, Jockusch BM. (2002). A lipid-regulated docking site on vinculin for protein kinase C. J Biol Chem 277:7396-7404 [DOI] [PubMed] [Google Scholar]