

Abstract
Type 2 diabetes affects over 300 million people, causing severe complications and premature death, yet the underlying molecular mechanisms are largely unknown. Pancreatic islet dysfunction is central for type 2 diabetes pathogenesis, and therefore understanding islet genome regulation could provide valuable mechanistic insights. We have now mapped and examined the function of human islet cis-regulatory networks. We identify genomic sequences that are targeted by islet transcription factors to drive islet-specific gene activity, and show that most such sequences reside in clusters of enhancers that form physical 3D chromatin domains. We find that sequence variants associated with type 2 diabetes and fasting glycemia are enriched in these clustered islet enhancers, and identify trait-associated variants that disrupt DNA-binding and islet enhancer activity. Our studies illustrate how islet transcription factors interact functionally with the epigenome, and provide systematic evidence that dysregulation of islet enhancers is relevant to the mechanisms underlying type 2 diabetes.
Despite recent progress in mapping transcriptional regulatory elements and chromatin states1-7, our understanding of how transcription factor networks interact with chromatin to control genome function remains limited. Furthermore, little is known about how dysregulation of such networks contributes to disease. As pancreatic islet cells are pivotal in diabetes pathogenesis8-10 and genomic cis-regulatory maps in islets are still markedly underdeveloped compared to those of other cell types5,7,11-16, we have now mapped chromatin states, binding sites of key transcription factors, and transcripts in human pancreatic islets (Fig. 1a). Integrative analysis and genetic perturbations were combined to provide reference charts of cis-regulatory elements functioning in human pancreatic islets. Our studies identified enhancer domains that are central determinants of islet-specific gene activity, and linked sequence variation in these regions to the susceptibility for type 2 diabetes (T2D) and to variation in fasting glycemia (FG) levels.
Figure 1. Integrative regulatory maps of human pancreatic islet cells.

(a) transcription factor binding, active chromatin, and gene transcription maps in human islet cells.
(b) Integrative map of the NKX2.2 locus. transcription factor binding and chromatin state profiles are shown for duplicate human islet samples. RNA-Seq tracks correspond to human islets or pooled data from 14 non-pancreatic tissues to highlight islet-specific transcripts.
(c) Network topology diagram illustrating that all 5 islet transcription factors show direct auto- and cross-regulatory interactions, and frequently target adjacent genomic sites (see also Supplementary Fig. 2).
Results
Topology of an essential islet transcription factor network
To characterize transcription factor networks that control gene activity in human islet cells, we focused on five key β-cell transcription factors: FOXA2, MAFB and NKX2.2, which are expressed in insulin-producing β-cells and other major islet cell-types; NKX6.1, which is specific for β-cells; and PDX1, which is present in β-cells and more scarce somatostatin-producing islet cells17-21 (Supplementary Fig. 1a-c, and Supplementary Table 1). Mouse genetic experiments have shown that these five transcription factors are essential for differentiation or function of β-cells22-24, yet little is known about how these factors function at the level of regulatory networks, or how they interact with human islet chromatin to create cell-specific transcriptional activity. To determine the genomic binding sites of these islet transcription factors, we used chromatin immunoprecipitation and sequencing (ChIP-seq) in duplicate human islet samples and identified 3,911-32,747 high-confidence sites per transcription factor (Supplementary Figs. 1d,e, and 2a,b). We found that all five transcription factors invariably bound to multiple sites in the vicinity of their own and each other’s genes, a pattern indicating auto- and cross-regulatory interactions (Fig. 1b,c, and Supplementary Fig. 2c-f). Furthermore, all five transcription factors frequently bound to overlapping genomic sites, and consequently their genomic binding signals were highly correlated (r = 0.36-0.68 between pairs of islet transcription factors, in contrast with r = 0.03-0.11 between islet transcription factors and a control non-islet transcription factor) (Fig. 1b,c, and Supplementary Fig. 2c-h). This systematic analysis shows that, in agreement with earlier descriptions of transcription factor networks in diverse species and tissues25,26, human islet-specific transcription factors form a remarkably interconnected network (Fig. 1c). These genome-scale maps of binding sites for key islet transcription factors provide a framework to understand how transcription factors control islet-specific gene transcription.
Islet transcription factors bind to distinct chromatin states
We next sought to understand the relationship between the binding sites of islet transcription factors and the underlying chromatin states in human islets. We first mapped all accessible, or open, chromatin sites in pancreatic islets. We combined two assays that capture overlapping, though not identical, sets of accessible chromatin sites: FAIRE-Seq, which measures open (nucleosome-depleted) sites5, and ChIP-Seq for H2A.Z, a histone variant that is enriched in accessible chromatin sites27,28. We defined ~95,000 sites that were enriched in either of these two accessible chromatin types in two or more human islet samples. Next, we mapped key histone modifications (H3K4me1, H3K4me3 and H3K27ac) and CTCF binding sites in human islets, and used k medians clustering to subdivide all accessible chromatin sites into 5 classes (C1-C5) that displayed clearly recognizable enrichment patterns for histone modifications and CTCF binding1-3, namely: promoters (or “C1”, for Class 1); “poised” or inactive enhancers (“C2”); active enhancers (“C3”); CTCF-bound sites (“C4”); and additional accessible sites lacking active histone modifications or CTCF occupancy (“C5”) (Fig. 2a and Supplementary Fig. 3a-c). Consistent with the expectation that transcription factors bind to accessible DNA, 92% of the genomic sites bound by any of the five islet transcription factors mapped to sites enriched in FAIRE or H2A.Z in at least one sample, and islet transcription factors bound to all major classes of accessible chromatin (Fig. 2b, and Supplementary Fig. 4). Integrating histone marks and transcription factor binding maps thus resolved discrete classes of transcription factor-bound accessible chromatin sites in human islets.
Figure 2. transcription factor networks establish distinct types of interactions with the epigenome.

(a) k-medians clustering of 95,329 islet accessible chromatin sites (defined by FAIRE and/or H2A.Z-enrichment in two samples) shows 5 subclasses of accessible chromatin (C1-C5) that we refer to as promoter, inactive enhancer, active enhancer, CTCF-bound sites or other, based on previously defined patterns of histone modifications and CTCF binding1-3 (See also Supplementary Fig. 3a). Histone and CTCF reads were calculated in 100 bp bins across 6 Kb windows centered on merged FAIRE/H2A.Z-enriched sites.
(b) Average transcription factor binding signal distribution relative to the center of each accessible chromatin class, and percentage of sites in each class bound by at least one transcription factor. Non quantile-normalized reads were processed as described for the panel above.
(c) Density of C1, C3 or C3 accessible chromatin sites bound by two or more transcription factors in the sites surrounding the TSSs of 1,000 most islet-specific genes, 1,000 ubiquitously active genes, or 1,000 islet inactive genes.
(d,e) Examples of islet-specific transcription factors binding to the 5′ end of ubiquitously active genes TBP and PSMB1, or to the islet-specific T2D susceptibility gene SLC30A8. The regulome track depicts color-coded C1-C5 chromatin states.
(f) Co-binding by multiple transcription factors is more common in C3 (active-enhancer) chromatin. The numbers of transcription factor-bound sites consistent in two samples for each category are shown above.
(g) Co-binding by three or more transcription factors at C1 (promoter) chromatin is not associated with islet-specific activity of the adjacent gene.
(h) Genes located <25 Kb from clusters of C3 sites that are highly bound by transcription factors show enriched expression in islets and β-cells relative to 14 non-islet tissues. Boxes show interquartile range (IQR), notches are 95% confidence intervals of the median, and whiskers extend to either 1.5 times the IQR or to extreme values. Gene expression data from non-pancreatic tissues from panels g and h is broken down by individual tissues in Supplementary Figs. 5d and 7b. P-values were calculated with the Wilcoxon rank-sum test.
Cis-regulatory clusters drive islet-specific transcription
Next, we examined the functional relationship between these different classes of transcription factor-bound accessible chromatin and islet-specific gene transcription. To study islet-specific gene transcription, we analyzed RNA sequencing (RNA-seq) profiles from three human islet samples29, two fluorescence-activated cell sorting (FACS)-purified β-cell samples29, and 14 non-pancreatic tissues, and use these to define three classes of genes: (a) “islet-specific” genes, which were the 1,000 genes that showed the strongest transcript enrichment in human islet cells, (b) 1,000 “ubiquitously transcribed” genes that were similarly active in islet cells and other tissues, and (c) 1,000 inactive genes, which showed the lowest expression levels in islet cells. Contrary to our expectation that cell type-specific transcription factors should primarily bind to cell type-specific genes, islet-specific transcription factors bound to promoter (C1) accessible chromatin to a similar extent at both islet-specific and ubiquitously transcribed genes (Fig. 2c,d, and Supplementary Fig. 5a,b). Likewise, binding events of islet-specific transcription factors at C2, C4 and C5 accessible chromatin sites were not enriched at loci harboring islet-specific genes (Fig. 2c, and Supplementary Fig. 5c). In sharp contrast, islet-specific gene loci showed an increased density of islet transcription factor-bound active enhancer (C3) accessible chromatin sites (Fig. 2c,e). Islet-specific genes had a median of 3 transcription factor-bound C3 sites (Interquartile range [IQR] = 1 to 5) within 50 Kb of their transcriptional start site (TSS), whereas ubiquitously transcribed genes had 1 (IQR = 0-3), and inactive genes had a median of 0 transcription factor-bound C3 sites (IQR = 0-1) (Kruskall-Wallis P < 1 × 10−16). Consistent with studies showing that combinatorial transcription factor interactions are critical for enhancer activation30,31, 72.8% of genomic sites bound by three or more islet transcription factors were associated with C3 accessible chromatin (Fig. 2f). Although some promoter (C1) accessible chromatin sites were also bound by multiple islet transcription factors, these were not associated with islet-specific transcription, suggesting that the link between transcription factor co-occupancy and cell-specific transcription is largely confined to active enhancers (Fig. 2g, and Supplementary Fig. 5d). Taken together, these findings revealed remarkable differences between transcription factor binding at different accessible chromatin classes. They suggest that certain binding events of islet-specific transcription factors, such as those occurring at promoters, are not generally associated with cell type-specific gene transcription, which is instead tightly linked to clusters of active enhancer chromatin sites bound by multiple transcription factors.
We scanned the genome to identify all clusters of three or more enhancer sites active in islets (see methods and Supplementary Fig. 6a,b). We identified 3,677 such clusters, which mapped near genes with strong islet-enriched expression (median expression in islets of 8.3 reads per kilobase per million mapped reads (RPKM) [IQR = 2.9-19.6], non-islet tissues 4.8 RPKM [IQR = 0.7-14.2]; Wilcoxon P < 10−30), in contrast to more modest islet enrichment in genes near non-clustered (“orphan”) enhancers (median expression in islets 6.0 RPKM [IQR = 1.5-14.3], non-islet tissues 5.0 RPKM [IQR = 0.8-13.9]; Wilcoxon P < 10−5) (Supplementary Fig. 6c). Islet-enriched transcription was most pronounced near the 1,813 enhancer clusters that showed higher than median occupancy by islet transcription factors (Fig 2h, Supplementary Fig. 7a-c). Remarkably, most genes that are currently known to be important for islet cell identity, function or disease were associated with an islet enhancer cluster (92% of a manually annotated list of 65 such genes, 90% of which belonged to the high transcription factor occupancy subset, Supplementary Fig. 7d and Supplementary Table 2). These findings reinforce the view that the genomic program that underlies islet cell identity is tightly associated with clusters of enhancers bound by islet transcription factors.
To investigate the function of transcription factor-bound enhancer clusters, we first verified that individual clustered C3 sites truly act as cell type-specific enhancers. Consistent with their chromatin signature, 8 of 12 transcription factor-bound active enhancer C3 sites (but not transcription factor-bound C2 and C5) displayed β-cell-selective enhancer activity in episomal reporter assays in mouse cell lines (Fig. 3a). Five conserved, transcription factor-bound C3 sites were tested in transgenic zebrafish assays and all exhibited enhancer activity (three specifically in the pancreatic islet), whereas transcription factor-bound C2 or C5 sites showed no activity (Fig. 3b and Supplementary Fig. 8a-c). These experiments show that clustered sites have islet enhancer activity.
Figure 3. Enhancer clusters form functional 3D chromatin domains.

(a) Luciferase assays in mouse MIN6 β-cells and 3T3 fibroblasts show that 8/12 transcription factor-bound C3 sites, but not transcription factor-bound C2 and C5 sites, confer cell-specific expression to a minimal promoter (*t-test P < 0.005, compared with empty vector, n =3 transfections per condition).
(b) C3-3, a C3 element in a cluster >1 Mb from ISL1, shows selective enhancer activity in pancreatic islet (pi) of zebrafish 70 hpf embryos. The enhancer transgene (YFP) was injected into a transgenic line that exhibits fluorescence (mCherry) in insulin+ cells.
(c) MAFB knockdown in human EndoC-βH1 β cells causes downregulation of genes bound by MAFB at clustered (C3+) rather than orphan (C3−) MAFB-bound enhancers. The bar-plot illustrates Gene Set Enrichment Analysis (GSEA) False Detection Rates (FDR) for different MAFB-bound gene sets amongst genes that are downregulated after infecting two shRNAs for MAFB (MAFB KD) vs. 4 non-targeting shRNAs (NT). As a control we repeated the analysis using the same number of arrays but comparing sets of different NT control shRNAs. The horizontal dashed line signals FDR = 0.05 as a reference. See also Supplementary Fig. 9a-e.
(d) Misexpression of PDX1 with MAFA and NGN3 in HEK293 cells preferentially activates genes associated to PDX1-bound clustered (C3+) enhancers, but not genes bound by PDX1 at promoter accessible chromatin sites or those associated with orphan (C3−) PDX1-bound enhancers. The number of transcription factor-bound genes for C1-C5 is reported in methods.
(e) 4C-Seq analysis shows selective interactions between clustered transcription factor-bound enhancers and cell-specific promoters. Note, for example, interactions between the ISL1 promoter and the C3-3 enhancer tested in zebrafish transgenics and located >1 Mb away (green star). The red triangle indicates the ISL1 promoter viewpoint, pink lines highlight interactions between ISL1 and other sites. The track labeled “4C-Seq” shows aligned sequences, and “4C-Seq sites” are significant interaction sites.
(f) 4C-Seq analysis of nine loci shows that C3 sites interact more often than expected with nearby promoters, in contrast with other accessible chromatin classes. The red diamonds depict the mean overlap of different accessible chromatin sites with viewpoint-interacting sites, whereas the grey box plot shows the overlap of randomized accessible chromatin (C1-C5) sites after 1,000 iterations in the same loci.
(g) transcription factor-bound C3, but not other transcription factor-bound C-sites, establish frequent interactions with nearby promoters. The boxes show IQR and whiskers extend to 1.5 times the IQR.
We next designed experiments to directly test whether transcription factor-binding sites that reside in clusters of enhancers, but not necessarily other transcription factor binding sites, are functionally important for the activation of islet-selective genes. First, we transduced human β-cells (EndoC-β H1) with two independent interfering hairpin RNAs for MAFB or with four control hairpins (Supplementary Fig. 9a). Knockdown of MAFB caused down-regulation of genes linked to MAFB-bound enhancer clusters (Gene Set Enrichment Analysis [GSEA] Normalized Enrichment Score [NES] 1.98, FDR q < 1.5 × 10−2) (Fig. 3c), such as ROBO2 and G6PC2 (Supplementary Fig. 9b,c). However, genes bound by MAFB only at their promoters, or at other classes of accessible chromatin, were not significantly affected (Fig. 3c and Supplementary Fig. 9d,e). Next, we transfected HEK293 embryonic kidney cells with expression vectors encoding PDX1, and assessed which PDX1-bound genes were activated. We co-transfected PDX1 with MAFA and NGN3 because this transcription factor combination, which has been previously employed to activate β-cell genes32, was more efficient than PDX1 alone for gene activation in heterologous cells. We observed transcriptional enhancement of genes near PDX1-bound enhancer clusters (GSEA NES 2.2, FDR q < 1 × 10−3), whereas genes that are exclusively bound by PDX1 at promoter or other accessible chromatin classes were unaffected (GSEA NES −1.3, FDR q = 0.37) (Fig. 3d). These studies confirm that chromatin profiling can identify subsets of transcription factor binding sites that are critically important for cell type-specific gene activity.
Having shown that active enhancer clusters are pervasive at islet-specific genes, and furthermore that they are important for islet-specific gene activity, we examined the relationship between enhancer multiplicity and the formation of broad 3D chromatin structures. We selected 9 loci, 8 of which contain active enhancer clusters near an islet-enriched gene, and used human islet chromatin to perform circular chromosome conformation capture coupled with high-throughput sequencing (4C-Seq) (Fig. 3e)33. Transcription factor-bound C3 sites from these clusters displayed frequent strong interactions with the promoters of islet-enriched genes at the same loci, including examples involving C3 sites located >1 Mb from their target promoter (Fig. 3e-g, additional loci are shown in Supplementary Fig. 10a-e). By contrast, transcription factor-bound non-C3 sites at the same loci did not show greater interactions than expected by chance (P < 1 × 10−3 for transcription factor-bound C3 sites vs. randomized sites, P = 0.41 for transcription factor-bound non-C3 sites vs. randomized sites) (Fig. 3f-g). These observations suggest that islet enhancer clusters are 3D structural units in which multiple transcription factor-bound active enhancers (rather than other transcription factor-bound sites) interact with target promoters. Collectively, the results indicate that human islet-specific gene transcription is largely driven by combinatorial transcription factor binding at clusters of enhancers that form 3D chromatin structures.
The islet cis-regulatory sequence code
The identification of the genomic sites that underlie islet gene activity allowed us to explore the sequence code that is recognized by islet transcription factor networks to drive cell-specific transcription. We identified 46 highly enriched sequence motifs in clustered enhancers, including recognition sequences of key β-cell transcription factors (HNF1A, RFX, FOXA, NEUROD1, NKX6.1, PDX1, MAFA/B), as well as previously unrecognized motifs (Fig. 4, and Supplementary Table 3). We further identified combinations of these 46 motifs that were most enriched in islet enhancers relative to enhancers from 9 non-pancreatic cell types34 (Supplementary Fig. 11a-c). Motifs for RFX35 or the pioneer FOXA factors36 were present in all of the most islet-enriched combinations (Supplementary Fig. 11b), consistent with a major role of these factors in the activation of islet enhancers. To independently validate this analysis, we mapped all instances of the 10 most enriched motif combinations to the mouse genome, and found that they also located near genes that show islet-enriched expression in mice (median expression of nearby genes in mouse islets vs. non-islet tissues: 1.0 (IQR = 0.0-5.2) vs. 0.6 (IQR = 0.0-3.7 RPKM), Wilcoxon P = 9.6 × 10−31) (Supplementary Fig. 11d). In silico analysis thus expanded the range of candidate combinatorial islet transcription factor interactions, and provide a resource to discover novel transcription factors and upstream signaling pathways that control β-cell transcriptional programs.
Figure 4. Known and novel transcription factor motifs are enriched in clustered islet enhancers.
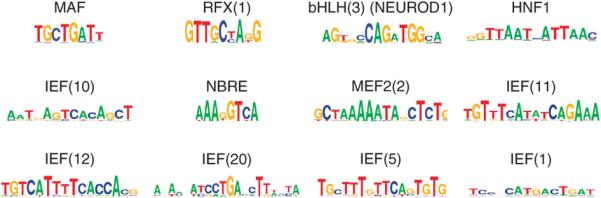
Examples of sequence motifs that are enriched in clustered enhancers at P < 1 × 10−60 (HOMER50). Several motifs match known islet transcription factor recognition sequences, whereas others are candidate binding sites for novel regulators. IEF = Islet Enriched Factor. Supplementary Table 3 shows a complete list of motifs that showed enrichment at P < 1 × 10−60 (HOMER).
Sequence variation in islet enhancers is associated with T2D
Genome-wide association (GWA) studies have identified dozens of loci associated with T2D and glycemic traits37,38, but the molecular mechanisms linking specific alleles to cellular functions remain poorly described. The catalog of islet cis-regulatory elements allowed us to explore to what extent genetic variants that contribute to T2D susceptibility and to variation in fasting glycemia (FG) in non-diabetic individuals act through islet regulatory mechanisms.
First, we examined loci with genome-wide significant association with T2D or FG in European populations, considering all variants in high linkage disequilibrium (LD; 1000 Genomes Project [1KG] CEU (Utah residents of Northern and Western European ancestry) r2 > 0.8) with a lead SNP reported in the National Human Genome Research Institute (NHGRI) GWAs catalog39. These associated variants were enriched in C3 sites, and more prominently in clustered C3 rather than orphan C3 sites (T2D: C3 P = 0.004; clustered C3 P = 0.012, orphan C3 P = 0.20; FG: C3 P < 1 × 10−5; clustered C3 P = 3 × 10−5, orphan C3 P = 0.12) (Fig. 5a). There was no significant enrichment of T2D or FG-associated variants in other accessible chromatin classes, and variants associated with other complex traits were not enriched in clustered islet C3 sites (all traits P > 0.05). Moreover, little enrichment was seen in the 200 bp sites immediately flanking C3 sites (Fig. 5a; T2D P = 0.44, FG P = 0.043) suggesting the C3 enrichment is not driven solely by the genomic context of these sites. These findings indicate that a subset of T2D and FG GWAs loci are likely to harbor functional cis-regulatory variants in islet active enhancers.
Figure 5. Islet enhancers are enriched in T2D and FG level-associated loci.
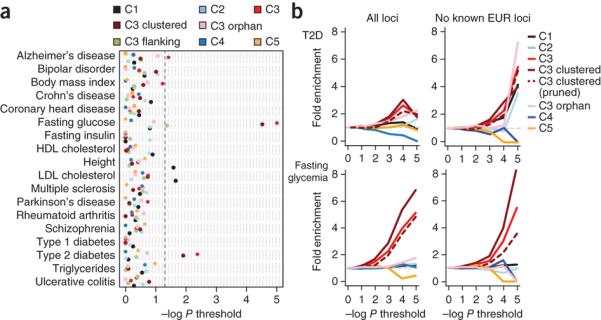
(a) Loci associated with complex traits were tested for enrichment of overlap with classes of islet sites (C1-C5, clustered C3, orphan C3) compared to matched background loci. T2D and FG-associated loci were enriched for islet active enhancer sites (C3) but not for directly flanking sites (C3 flank) or for other islet classes. The enrichment of C3 sites for both T2D and FG level was stronger when considering clustered rather than orphan (non-clustered) enhancers. The most significant enrichment for other complex diseases was C1 sites for LDL cholesterol (P = 0.022) and height (P = 0.026) and C3 sites for Alzheimer’s disease (P = 0.038). No significant enrichment of variants associated with other complex traits was seen in clustered C3 enhancers.
(b) Enrichment of islet regulome sites in T2D38 and FG37 genome wide association data. We determined the number of variants overlapping sites from each islet accessible chromatin class that surpassed a series of association significance thresholds for T2D and FG. We then calculated fold-enrichment values at each threshold by comparing to the number of matched background variants significant at that threshold. C3 sites and clustered C3 sites were more enriched for T2D and FG association at increasing p value thresholds (left panel), even after removing known European T2D/FG loci (right panel). Patterns in C3 and clustered C3 sites were maintained when pruning variants to retain a single variant in each LD block (r2 > 0.2). In all such analyses, clustered C3 sites displayed significant enrichment relative to the null distribution of permuted counts for T2D and FG association P values <0.001 (P values for these analyses are given in the text).
To further examine the contribution of islet cis-regulatory variation to T2D and FG, we considered association results for the full set of ~2.5 million variants represented in the largest available genome-wide association meta-analyses for these traits, rather than restricting analysis to signals reaching stringent genome-wide significance thresholds37,38. For each islet accessible chromatin class, we identified the overlapping variants that exceeded a series of association significance thresholds, and calculated fold-enrichment values using a null distribution of permuted variant counts (see Methods). Variants in C3 sites were collectively enriched for T2D and FG GWAs values of P < 0.001 (T2D: C3 P = 2.1 × 10−11, FG: C3 P < 1 × 10−16). This was predominantly a feature of C3 clusters rather than orphan C3 sites (T2D: clustered C3 P = 5.1 × 10−11, orphan C3 P = 0.006; FG: clustered C3 P<1 × 10−11, orphan C3 P = 0.23) (Fig. 5b, and Supplementary Fig. 12). Fold-enrichment patterns were unchanged when the genome-wide association data were aggressively pruned (r2 > 0.2) to retain only the most associated signal in each LD cluster, and remained significant for FG and T2D variants in clustered C3 sites (T2D: C3 P = 0.2, clustered C3 P = 0.031; FG: C3 P =3.6 × 10−6, clustered C3 P = 5.0 × 10−5). Enrichment was also retained when established (genome-wide significant) T2D and FG loci were removed (Fig. 5b; T2D: C3 P = 7.3 × 10−5, clustered C3 P = 1.1 × 10−5; FG: C3 P = 1.1 × 10−5, clustered C3 P = 4.1 × 10−9), suggesting that the association of variants in C3 sites (and, in the T2D analysis, of those in C1 also) extends to novel loci that have yet to be confirmed to genome wide significance.
To further examine this point, we sought examples of variants overlapping islet accessible chromatin sites that map within such “moderately” associated loci. We found that rs72695654 disrupts a sequence motif and abolishes binding of a β-cell protein complex to a C3 site that is located in an enhancer cluster within the ACSL1 gene, which encodes a long-chain fatty acyl-CoA synthase that is highly expressed in islets (Supplementary Fig. 13a-c). Long-chain fatty acyl-CoA synthesis has been implicated in β-cell stimulus-secretion coupling, survival and lipotoxicity40-42. The rs72695654 variant is in high LD (1KG CEU r2 = 0.94) with rs735949, which is strongly associated with T2D (P = 3.7 × 10−6) and, independently, with FG level in non-diabetic individuals in large genome-wide meta-analyses (P = 1.6 × 10−5)37,38 (Supplementary Fig. 13a). This finding suggests that cis regulatory maps can be used to prioritize amongst loci for which genetic evidence is strong, but not genome-wide significant, to guide hypothesis-driven experiments that define susceptibility variants.
We next attempted to define functional enhancer variants within established GWAs loci that might be causal for T2D susceptibility. We first catalogued all variants that reside within C3 sites and are in high LD with established genome-wide significant T2D and FG association signals (Supplementary Tables 4-6)37,38. We confirmed that the TCF7L2 intronic SNP43 rs7903146, previously shown to be located in an islet FAIRE-enriched site and to affect chromatin state and enhancer function5, maps to a C3 site that is bound by NKX2.2, FOXA2, and MAFB, yet does not show active chromatin marks in an extensive panel of non-islet human cell types (Supplementary Fig. 14a,b). Similar observations were made at multiple established T2D and FG regions, including SLC2A2, CDKN2A, C2CD4A/B, SLC30A8, DGKB, and PCSK1, where we identified associated variants that mapped to transcription factor-bound C3 sites, many of which disrupt islet-enriched sequence motifs (see Supplementary Fig. 15a,b, describing a public browser that facilitates visualization of T2D/FG GWAs variants and the islet regulome, and Supplementary Fig. 16). For example, at the ZFAND3 T2D locus discovered in East Asians44, SNP rs58692659 forms part of an array of variants that was highly-correlated with the reported lead SNP (rs9470794) (1KG CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) r2 = 0.96), and maps to a C3 element bound by multiple islet transcription factors within an enhancer cluster (Fig. 6a). This variant altered sequence-specific DNA binding of a key islet-enriched transcription factor, NEUROD1, and abolished enhancer activity in β-cells (Fig. 6b,c). We note that this SNP, along with many others in the region, is monomorphic in European samples (1KG CEU) and there is no T2D association at ZFAND3 in Europeans (rs9470794 DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) P = 0.8)44, consistent with rs58692659 having a causal role at this locus. These results illustrate how islet accessible chromatin maps can pinpoint functional cis-regulatory variants that are strong candidates for a causal role in driving T2D-association signals.
Figure 6. A T2D risk variant at ZFAND3 disrupts islet enhancer activity.

(a) Regional plot of 1000 Genomes variants and islet accessible chromatin elements at the ZFAND3 locus associated with T2D in East Asian individuals (r2 values based on 1KG CHB/JPT LD with rs58692659); rs9470794 (in cyan) at this locus shows strongest T2D association from Cho et al44. The SNP rs58692659 maps to a C3 site >10 Kb upstream of ZFAND3 bound by PDX1, FOXA2, NKX2.2, and NKX6.1 and (b) disrupts an islet-enriched bHLH-like motif that matches the NEUROD1 recognition site51, a known key islet transcription factor22. Electrophoretic mobility shift assay shows that the minor allele A at this variant abolishes binding of a protein complex that is supershifted by an anti-NEUROD1 antibody. Competition gradients identified by the grey triangle correspond to 5, 50, and 100-fold excess of cold probe.
(c) Luciferase assay shows reduced enhancer activity of allele A compared to G of rs58692659 in MIN6 β-cells. The data are presented as mean ± s.d. Three independent experiments were performed in triplicate. P values were calculated by a two-sided t-test.
Discussion
Our work provides reference cis-regulatory maps for ongoing efforts to dissect the transcriptional program of pancreatic β-cells45 and the molecular mechanisms of human T2D10. We show that islet-specific transcription factors establish widespread binding to accessible chromatin sites where they are apparently not engaged in cis regulation of cell-specific gene transcription. We further demonstrate that binding events that do drive the transcription of genes underlying islet cell identity reside in clusters of transcriptional enhancers. These clusters mirror previously reported clusters of accessible chromatin in human islets5, and although they are groups of discrete enhancers, they share many features and may thus represent the same phenomenon as recently reported large enhancers (super-enhancers)46,47. Our 4C-Seq analysis allows us to speculate that enhancer multiplicity has an architectural role by creating active chromatin structures in genomic domains that are transcriptionally silent in most non-islet cell types. These findings have therefore uncovered central cis-regulatory determinants of islet-cell gene transcription.
Our systematic analysis implicates sequence variation at islet enhancer clusters in the susceptibility for T2D and in variation of FG levels. Recently, SNPs associated with common polygenic diseases were shown to be enriched in noncoding genomic elements defined by DNaseI hypersensitivity, which marks accessible chromatin regions such as active promoters, insulators, repressors, poised and active enhancers48, or by H3K4me3, which is enriched in promoters and more weakly in enhancers49. We have now defined for the first time functionally distinct transcription factor-bound genomic sites in human islets, and thereby disclosed that T2D susceptibility is specifically associated with allelic variation in pancreatic islet distant enhancers. Our results therefore link islet cis-regulatory networks to the mechanisms underlying T2D susceptibility and glycemic variation. The availability of integrated cis-regulatory maps in pancreatic islets will facilitate hypothesis-driven experiments to establish the exact manner in which common and lower frequency genetic variants impact islet cells in human diabetes.
The islet regulome, including transcription factor occupancy, chromatin states, motifs, enhancer clusters, and GWAs significance P values for T2D and FG can be accessed and visualized online (see Supplementary Fig. 15 for a description of this browser and URL).
Online Methods
Human islets
Islets were isolated from donors without a history of glucose intolerance52, shipped in culture medium, and then cultured for three days prior to analysis. β-cells were FACS-purified as described53. Islet samples were selected from a set of 120 islet samples based on ascertainment of minimal exocrine contamination, using dithizone staining and qRT-PCR of lineage-specific markers (Supplementary Table 7). High purity was also ascertained by strong H3K4me3 and H3K27ac-enrichment at key β-cell specific genes, including INS which shows no enrichment of histone marks in publically available human islet regulatory maps (Supplementary Fig. 2i)11,12,14.
Ethics
Human pancreata were harvested from brain-dead organ donors after obtaining informed consent from family members. Islet isolation centers had permission to use islets for scientific research if they were insufficient for clinical islet transplantation, in accordance with national laws and institutional ethical requirements. Ethical approval for the project was given by the Clinical Research Ethics Committee of Hospital Clinic de Barcelona.
FAIRE and ChIP
Human islets processed for FAIRE and ChIP as described5,54,55 using 1-1.5 μg antibodies against H3K4me1 (ab-8895), H3K4me3 (Upstate 05-745), H3K27ac (Abcam ab4729), CTCF (Millipore, 07-729), H2A.Z (Abcam ab4174, Millipore 09-862), Pdx1 (BCBC, AB2027), FOXA2 (Santa Cruz, sc-6554), Nkx2.2 (Sigma, HPA003468), MafB (Sigma, HPA005653), Nkx6.1 (Sigma, HPA036774). Expected cell-specific and subcellular patterns of formalin-fixed epitopes were ascertained by western blotting on human islet extracts and dual immunofluorescence of human pancreas (Supplementary Fig. 1b,c).
RNA-Seq
PolyA+ RNA-Seq from three highly purified human islet samples, and two from FACS-purified human β-cells have been described29. Non-pancreatic RNA-Seq reads were from the Illumina human BodyMap2 project (see URL). ~2.5 billion 50 bp paired-end reads from 14 human tissues or cell types (adipose, adrenal glands, breast, colon, heart, kidney, liver, lung, lymphatic node, muscle, ovary, prostate, thyroid gland and white blood cells) were processed identically as the islet data.
We defined (a) 1000 islet-specific non-redundant RefSeq transcripts with highest median expression ratio in islets vs. non-islet samples, and expressed in islets at >10 RPKM, (b) 1000 ubiquitously expressed transcripts with median islets vs. non-islet expression ratios closest to 1, and >10 RPKM islet expression, and (c) 1,000 inactive transcripts with the lowest median RPKM expression in islets.
ChIP-seq analysis
Sequencing was performed on Illumina HiSeq or GAII platforms. Quality-filtered 36-51 bp single-end reads were aligned to the human genome (NCBI36/hC18) using Bowtie v0.11.356, allowing only one mismatch and unique mapping. Clonal reads were removed. To calculate genome coverage densities reads were extended in silico to a final length equal to the fragment size estimation based on MACS57, and averaged by the number of aligned reads in each ChIP-seq experiment. Transcription factor and H2A.Z-enriched sites were detected with MACS57 using default parameters. Transcription factor peaks were called at P < 1 × 10−10 and H2A.Z peaks at P < 1 × 10−5 over a background model derived by sequencing input DNA (Supplementary Table 8). We only retained enrichment sites found in replicate experiments. To calculate false discovery rates (FDR) of replicated peaks we balanced the number of input and sample reads with MACS, and ascertained that >94% of peaks called at these thresholds showed FDR <0.01. For FAIRE we tuned the local background noise to λlocal=max λ5000, λ125000 and used a cut-off of P < 1 × 10−3, retaining only enrichment sites found in replicate experiments. We considered peaks to be overlapping if they shared a minimum of one base.
Accessible chromatin classes
Accessible chromatin was defined by merging all FAIRE and H2A.Z-enriched sites that were found in at least two islet samples. These two established marks of accessible chromatin5,28 proved to be complementary, because FAIRE enrichment was weak in promoters, yet readily identified central regions of enhancers and CTCF-binding sites, whereas H2A.Z-enrichment was strongest in promoters, and also marked enhancers and CTCF-bound sites (Fig 2a, Supplementary Fig. 3a). We generated 6 Kb windows centered on the accessible chromatin site, and computed the read coverage for each feature (H2A.Z, FAIRE, H3K4me3, H3K4me1, H3K27ac, CTCF) over 100 bp bins. Cluster358 was used for K-medians clustering using HI32, a single human islet sample for which there was availability for all marks. For C1, two clusters that showed nearly identical histone modification enrichment patterns were artificially merged into a single class. Enrichment patterns at clusters (or accessible chromatin “classes”) were concordant in replicate samples (Supplementary Fig. 3b).
Genome scan of enhancer clusters
To define enhancer clusters, we first created 1,000 iterations of randomized C3 sites in the mappable genome of individual chromosomes. We then calculated for each chromosome the 25th percentile of inter-site distances of randomized C3 sites (Supplementary Fig. 6a). Next, we defined clusters of islet C3 sites as any group of ≥3 C3 sites in which all adjacent C3 sites were separated by less than the abovementioned 25th percentile distance for randomized sites in the same chromosome. The distribution of islet C3 clusters differed from that of clusters generated with randomized C3 sites (Supplementary Fig. 6b).
We next created a score to measure the average transcription factor occupancy of islet enhancer (C3) clusters. For each cluster, we summed up the number of binding sites for each transcription factor at C3 sites, and divided this by the number of C3 sites of the cluster. Clusters with higher average transcription factor occupancy were associated with genes with enriched transcription in islets and β cells (Supplementary Fig. 7a). We defined high occupancy clusters as those showing scores in the top two quartiles (Fig. 2h, Supplementary Fig. 7a,b).
For comparisons of RNA expression in islets and non-islet samples, we defined genes containing a transcription factor-bound enhancer cluster within 25 Kb of their annotated TSS (Fig. 2h, and Supplementary Fig. 7b).
For sequence motif analysis we examined all 19,624 C3 sites that form clusters, including C3 sites not bound by the transcription factors we had profiled because we have only profiled a subset of all islet factors. This set of clustered enhancers was also associated with islet-enriched transcription (Supplementary Fig. 6c), and only 46% overlapped enhancer sites from 9 non-pancreatic cells34.
We used Genomic Regions Enrichment of Annotations Tool (GREAT)59 with default parameters to study enriched functional categories amongst genes linked to transcription factor-bound enhancer clusters (Supplementary Fig. 7c). We also used GREAT to determine which clusters were associated with a list of 65 genes with established importance for islet specialized function and identity based on a systematic literature search (Supplementary Fig. 7d and Supplementary Table 2).
Transcription factor occupancy, chromatin state, and islet-specific transcription
To compare transcription factor occupancy at different chromatin states in islet-specific, ubiquitous, vs. inactive genes, we determined sites bound by >1 transcription factor at each chromatin class, and calculated the density of such sites at +/−100 Kb from the TSS of the 3 groups of genes. This level of transcription factor occupancy was chosen to highlight that even high transcription factor occupancy at promoters is not selective for islet-specific genes, although conclusions remained unaltered using different numbers of transcription factors..
To directly examine genes bound by islet transcription factors at different chromatin states, we compared quantile-normalized RNA expression in human islets, β-cells and 14 non-pancreatic tissues. Non-pancreatic tissues were pooled in Fig. 2g,h, and broken down by individual tissues in Supplementary Figs. 5b,d and 7b. We compared RNA expression of genes bound by transcription factors at C1 but lacking transcription factor-bound C3 chromatin within 25 Kb of the TSS vs. all other genes, or else genes containing transcription factor-bound C3 clusters within 25 Kb vs. all other genes.
Enhancer function
Human sequences were cloned upstream hsp70 zebrafish core promoter60 or gata2 promoter61 linked to a Venus reporter and used to inject >200 eggs from wild-type (AB*) and Tg(ins:mCherry)jh2 Zebrafish in three independent experiments. Reporter expression patterns were documented using NIS-Elements software (Nikon). Expression was quantified by counting the number of embryos showing Venus+ cells in different tissues (Supplementary Table 9). Islet-specific expression patterns from transient transgenics were confirmed in stable transgenic lines (Supplementary Figure 8). Transcription factor-bound C3 sites were also cloned in Gateway-adapted PGL4.23 and cotransfected in triplicate wells with pRL in MIN6 and 3T3 cells, and luciferase activity was measured after 48 hr. Results were expressed as luciferase/renilla ratios in vectors carrying putative enhancers, relative to the ratio in empty PGL4.23 vector. (Fig. 3a). See Supplementary Table 10 for oligonucleotides.
Transcription factor functional studies
Human EndoC-βH1 β-cells62 were independently transduced with lentiviral vectors expressing two independent shRNAs that target MAFB mRNA or four negative control nontargeting shRNA sequences. Each shRNA was transduced in duplicate. See Supplementary Table 10 for oligonucleotides. MAFB shRNAs led to 64% and 55% inhibition of MAFB mRNA. We note that MAFB was chosen as a transcription factor target based on our ability to design two efficient inhibitory shRNAs.
HEK293T cells were transfected with pcDNA3-PDX1, pcDNA-NGN3 and pcDNA3.1-MAFA vectors or a control empty vector using Lipofectamine2000 (Invitrogen). After 72 hr RNA was extracted (Trizol,Invitrogen) and hybridized to GeneChip Human Genome U133 Plus2.0 Arrays. To assess enrichment of predefined gene sets in transcription factor perturbations, RMA-normalized data was analyzed with Gene Set Enrichment Analysis (GSEA)63 using default parameters over 1000 permutations to calculate FDR’s. The same analysis was carried in parallel for an identical number of arrays that compared 2 NT control vs. 3 NT control shRNAs. The gene sets for GSEA were created with genes whose TSS is closest to C1-C5 sites bound by either MAFB or PDX1. We thus created the following 5 non-mutually exclusive sets of genes for each transcription factor: MAFB-bound C1 (n = 363), C2 (n = 511), C3 (n = 1,891), C4 (n = 155), and C5 (n = 16); PDX1-bound C1 (n = 830), C2 (n = 878), C3 (n = 2,874), C4 (n = 247), and C5 ( n = 65). Similar results were observed using mutually exclusive gene sets (i.e., only sites bound at C3 showed NES>1, and FDR q <0.05). We also compared the behavior of two mutually exclusive sets of genes, one associated with orphan C3 sites only, and another that was only associated with clusters of C3. Gene set sizes were as follows: MAFB-bound orphan C3 (n = 577), clustered C3 (n = 657), PDX1-bound orphan C3 (n = 1,233), clustered C3 (n = 1,331).
4C-Seq
Chromosome conformation capture assays were performed as described64, and adapted for 4C-Seq as described33,65. Human islets (~5 × 106 cells) were treated with lysis buffer (10 mM Tris-HCl pH 8, 10 mM NaCl, 0.3% IGEPAL-CA-630 [Sigma-Aldrich]), 1X protease inhibitor (Complete, Roche)). Nuclei were digested with DpnII (New England Biolabs) and ligated with T4 DNA ligase (Promega). Csp6I endonuclease (Fermentas) was used in a second round of digestion, and DNA was religated. Locus-specific primers containing Illumina adaptors (Supplementary Table 10) were designed with primer3 v. 0.4.066 using the following gene promoters as viewpoints: ISL1, GNAS, C2CD4A, C2CD4B, TM4SF4, TM4SF1, PDX1, MAFB, G6PC2. Eight loci, with the exception of TM4SF1, contained enhancer clusters linked to islet-enriched genes. PCRs were performed with Expand Long Template PCR System (Roche) for each viewpoint and pulled together for sequencing. 4C-Seq was analyzed adapting a previously described procedure33. Briefly, 4C-sequencing reads were sorted, aligned, translated to restriction fragments. A moving average of 30 fragments per window was used for smoothening reads. Next, we calculated for each fragment the Poisson probability of containing a given number of “smoothened reads”. To this end, all aligned fragments were randomized 1,000 times in a 2 Mb window centered on the viewpoint and smoothened in the same way. We then identified significant interactions in the 4C-Seq experiment as those with a Poisson probability < 1 × 10−10. We tested two C3-promoter interactions by 3C capture, both of which were confirmed. To assess overlaps with chromatin sites, for all 9 loci we computed the overlap of different classes of accessible chromatin with 4C-Seq interaction sites located in a 2 Mb window centered on the viewpoint, but excluding 40 Kb on each side of the viewpoint. To contrast expected vs. observed overlap (Fig3 d,e) we randomized different chromatin sites or transcription factor-bound sites 1,000 times in mappable genomic sequences of the same 2 Mb window, and calculated the overlap of 4C-Seq enrichment sites with these positions.
Electrophoretic mobility shift assay (EMSA)
EMSA with mouse MIN6 β-cell nuclear extracts were performed as described67. We used 1 μl of NEUROD1 (Santa Cruz, sc-1084) antibody for supershifts. Findings were confirmed with binding experiments carried out on a separate day. See Supplementary Table 10 for oligonucleotides.
Motif analysis
De novo motif discovery was performed with HOMER50, using a 500 bp window centered in FAIRE or H2A.Z peaks of clustered C3 sites. We searched for motifs of length = 6-20 bp and retained 46 non-redundant matrices with P < 1 × 10−60. Motifs were annotated using HOMER50, TOMTOM68 and manual comparisons (Supplementary Table 3). All possible combinations of 3 motifs from the 46 enriched motifs were computed in clustered islet C3 vs. analogous genomic sites (H3K4me1- and H3K27ac-enriched, not H3K4me3 enriched, referred to as strong enhancers by Ernst et al 34) in 9 non-pancreatic cell lines (GM12878, HepG2, HSMM, HUVEC, ESC, HMEC, K562, NHEK and NHLF). We limited the motif search window to +/− 250 bp from the center of the genomic site, and computed the log2 of ratios between motif combination frequencies in islet vs. non-islet sites as a metric of islet enrichment. We focused on combinations enriched at a Chi-square P < 10−3. To ensure that results were not affected by differences in the data types used to define enhancers, we also analyzed enhancers from 4 non-pancreatic cell lines (GM12878, HepG2, HUVEC and K562) for which the same chromatin marks were available, including FAIRE and H2A.Z 69. We defined strong enhancer sites that overlap FAIRE or H2A.Z sites, and calculated the density of the 100 most islet-enriched motif combinations in enhancers from islet and non-islet cells in a 2 Kb window centered on merged FAIRE and/or H2A.Z enrichment sites (Supplementary Fig. 11c).
To compute the ability of motif combinations to predict islet gene activity in mice, we used HOMER to scan the mouse genome (mm9) for all instances of the 10 most islet-enriched motif combinations. We then identified all possible 3-motif combinations spanning <500 bp, extended sequences on both sides of motif combinations to create 500 bp segments, and filtered for non-redundant combinations. We linked these to nearby genes using GREAT with default parameters59. Then, RNA-Seq RPKM values for mouse islets29 and 9 non-islet tissues (bone marrow, cerebellum, heart, kidney, liver, lung, embryonic fibroblast, embryonic stem cell, spleen, obtained from ENCODE/LICR70) were aligned and processed as described for human RNA-Seq, and expression values where quantile normalized across all tissues71 (Supplementary Fig. 11d).
We also performed de novo motif discovery in all accessible chromatin classes (C1-C5) as described above for GWAs variant analyses.
Regulome Browser
To facilitate exploitation of integrated datasets we created a browser (Supplementary Fig. 15a,b) that enables data downloads and visualization at desired levels of resolution for islet transcription factor binding, chromatin states, motifs, and MAGIC37 or DIAGRAM P values38 (see URL).
GWAs analysis
We identified SNPs with genome-wide association (P < 5 × 10−8) to any trait in European individuals present in the GWAs catalog. Index SNPs were pruned (CEU r2 > 0.2) so that each independent ‘locus’ was only represented by one index SNP. Each index SNP was then used to identify variants in 1000 Genomes Project (1KG) pilot 1 data in CEU r2 > 0.8.. Thus, an associated ‘locus’ consists of an index SNP and the set of 1KG SNPs in high CEU LD. We then created a background set containing all qualifying loci binned based on number of total variants (index SNP + high LD SNPs) in the locus.
For each C3 site, 200 bp regions directly flanking the left and right ends were obtained. For each trait listed in Fig. 5a, we calculated the number of loci in the background set associated with that trait containing a variant overlapping a site for each class (C1-C5) as well as clustered C3, orphan C3, and C3 flanking sites. We then permuted the set of loci by drawing from matching bins in the background set and recalculated the number of loci with a variant overlapping a site for that class. The significance of each overlap is the number of permuted sets with greater overlap than the observed set divided by the number of permutations. We ran 10,000 permutations except for FG for which we ran 100,000 permutations.
We then identified a comprehensive set of T2D and FG index SNPs identified in any population. For T2D, we used all genome-wide significant lead SNPs reported in Morris et al. 38, and further identified lead SNPs for loci not genome-wide significant in that study but reported as such by previous studies. For FG, we used all genome wide significant lead SNPs reported in Scott et al.37 for both FG level and FG level adjusted for BMI loci (the former was retained when different lead SNPs at the same locus were reported).
For all lead SNPs we identified variants in high LD (r2 > 0.8) from 1KG phase I data in CEU samples (for European loci), CHB/JPT samples (for Asian loci) or YRI samples (for African loci).
HapMap enrichment analysis
We obtained p-values for all HapMap variants from DIAGRAM38 and MAGIC37 studies for T2D and FG, respectively. Variants were pruned using LD from HapMap CEU samples as follows: first, all variants in r2 > 0.2 with a lead SNP for a trait in Europeans were removed; second, remaining variants were sorted by p-value and variants were included if not in r2 > 0.2 with a more significant variant. Both sets of variants (all HapMap and LD-pruned HapMap) were binned based on CEU minor allele frequency and distance to the closest GENCODEv12 TSS. For analyses excluding known associated loci, variants in the 500 Kb region surrounding each European lead SNP were removed.
A series of trait association p-value thresholds were set and, at each threshold, the number of variants overlapping an islet chromatin class (C1-C5, clustered C3, orphan C3) was counted. For each variant, a matching variant was then selected at random from the same bin, and the number of ‘matched’ variants with a p-value below the same threshold was counted. Fold-enrichment was calculated for each threshold by dividing the observed counts by the ‘matched’ counts averaged over 1,000 permutations.
To evaluate the significance of these enrichments, we focused on variants overlapping each chromatin class that attained P < 0.001 in T2D or FG meta-analyses. We obtained counts of matched variants significant at the same trait p-value threshold across all permutations. As the matched variant counts for these replicates were normally distributed, we calculated a Z-score for the observed islet class counts using the mean and standard deviation from the distribution of permuted counts. Reported p-values were then obtained from Z-scores using a one-sided test.
Supplementary Material
Acknowledgements
We thank J. Rios (IDIBAPS) for expert statistical advice, M. Parsons (Johns Hopkins University) for Tg(ins:mCherry)jh2 transgenics and R. Stein (Vanderbilt University), J. Habener (Harvard University) and G. Gradwohl (Institute of Genetics and Molecular and Cellular Biology) for MAFA, IDX1, and NGN3 DNA constructs. We thank the DIAGRAM and MAGIC consortia, the Singapore Prospective Study Program, the Singapore Consortium of Cohort Studies, the Singapore Indian Eye Study, the Singapore Malay Eye Study and Y.Y. Teo, E.S. Tai, T.Y. Wong, W.Y. Lim and X. Wang (National University of Singapore; funded by the National Medical Research Council of Singapore, Singapore Translational Researcher Award schemes, the Biomedical Research Council of Singapore and the National Research Foundation (NRF) Fellowship scheme). This work was carried out in part at the Centre Esther Koplowitz. This work was funded by grants from a European Foundation for the Study of Diabetes Lilly fellowship (L. Pasquali), the Ministerio de Economía y Competitividad (SAF2011-27086 to J.F., BFU2010-14839 and CSD2007-00008 to J.L.G.S.), the Innovative Medicines Initiative (DIRECT to M.I.M. and J.F.), the Andalusian Government (CVI-3488 to J.L.G.S.), the Biology of Liver and Pancreatic Development and Disease Marie Curie Initial Training Network (F.M. and J.F.), the Wellcome Trust (090532, 98381 and 090367 to M.I.M., 095101 to A.L.G., 101033 to J.F.), Juvenile Diabetes Research Foundation (31-2012-783 to T.B., F.P. and L. Piemonti) and Framework Programme 7 (HEALTH-F4-2007-201413 to M.I.M.).
Footnotes
URLs: Illumina human BodyMap2 project, http://www.ebi.ac.uk/arrayexpress/browse.html?keywords=E-MTAB-513; Islet regulome browser, http://www.isletregulome.org.
Accession code: Raw data is available at ArrayExpress under accession number E-MTAB-1919.
Competing financial interests: Philippe Ravassard is shareholder and consultant for Endocells. None of the authors report any other conflict of interest relevant to the content of this report.
References
- 1.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–83. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonn S, et al. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet. 2012;44:148–56. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- 5.Gaulton KJ, et al. A map of open chromatin in human pancreatic islets. Nat Genet. 2010;42:255–9. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen Y, Yue F, McCleary DF, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunham I, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahn SE. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–58. doi: 10.1210/jcem.86.9.7713. [DOI] [PubMed] [Google Scholar]
- 9.Lyssenko V, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–32. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 10.Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–71. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhandare R, et al. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res. 2010;20:428–33. doi: 10.1101/gr.102038.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stitzel ML, et al. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010;12:443–55. doi: 10.1016/j.cmet.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khoo C, et al. Research resource: the pdx1 cistrome of pancreatic islets. Mol Endocrinol. 2012;26:521–33. doi: 10.1210/me.2011-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein BE, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–8. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, et al. Genome-wide Chromatin State Transitions Associated with Developmental and Environmental Cues. Cell. 2013;152:642–54. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tennant BR, et al. Identification and analysis of murine pancreatic islet enhancers. Diabetologia. 2013;56:542–52. doi: 10.1007/s00125-012-2797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Artner I, et al. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104:3853–8. doi: 10.1073/pnas.0700013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–8. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sander M, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000;127:5533–40. doi: 10.1242/dev.127.24.5533. [DOI] [PubMed] [Google Scholar]
- 20.Sund NJ, et al. Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev. 2001;15:1706–15. doi: 10.1101/gad.901601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sussel L, et al. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. 1998;125:2213–21. doi: 10.1242/dev.125.12.2213. [DOI] [PubMed] [Google Scholar]
- 22.Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia. 2004;47:597–613. doi: 10.1007/s00125-004-1368-9. [DOI] [PubMed] [Google Scholar]
- 23.Wilson ME, Scheel D, German MS. Gene expression cascades in pancreatic development. Mech Dev. 2003;120:65–80. doi: 10.1016/s0925-4773(02)00333-7. [DOI] [PubMed] [Google Scholar]
- 24.Oliver-Krasinski JM, Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22:1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–87. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kyrmizi I, et al. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev. 2006;20:2293–305. doi: 10.1101/gad.390906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin C, et al. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat Genet. 2009;41:941–5. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–93. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moran I, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435–48. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefflova K, et al. Cooperativity and rapid evolution of cobound transcription factors in closely related mammals. Cell. 2013;154:530–40. doi: 10.1016/j.cell.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zinzen RP, Girardot C, Gagneur J, Braun M, Furlong EE. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noordermeer D, et al. The dynamic architecture of Hox gene clusters. Science. 2011;334:222–5. doi: 10.1126/science.1207194. [DOI] [PubMed] [Google Scholar]
- 34.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith SB, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature. 2010;463:775–80. doi: 10.1038/nature08748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaret KS, et al. Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spring Harb Symp Quant Biol. 2008;73:119–26. doi: 10.1101/sqb.2008.73.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott RA, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hindorff LA, et al. A Catalog of Published Genome-Wide Association Studies
- 40.El-Assaad W, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology. 2003;144:4154–63. doi: 10.1210/en.2003-0410. [DOI] [PubMed] [Google Scholar]
- 41.Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A. 1998;95:2498–502. doi: 10.1073/pnas.95.5.2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu L, et al. Regulation of lipolytic activity by long-chain acyl-coenzyme A in islets and adipocytes. Am J Physiol Endocrinol Metab. 2005;289:E1085–92. doi: 10.1152/ajpendo.00210.2005. [DOI] [PubMed] [Google Scholar]
- 43.Helgason A, et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet. 2007;39:218–25. doi: 10.1038/ng1960. [DOI] [PubMed] [Google Scholar]
- 44.Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2011;44:67–72. doi: 10.1038/ng.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490–4. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trynka G, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. 2013;45:124–30. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seo S, Lim JW, Yellajoshyula D, Chang LW, Kroll KL. Neurogenin and NeuroD direct transcriptional targets and their regulatory enhancers. EMBO J. 2007;26:5093–108. doi: 10.1038/sj.emboj.7601923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bucher P, et al. Assessment of a novel two-component enzyme preparation for human islet isolation and transplantation. Transplantation. 2005;79:91–7. doi: 10.1097/01.tp.0000147344.73915.c8. [DOI] [PubMed] [Google Scholar]
- 53.McCulloch LJ, et al. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol Genet Metab. 2011;104:648–53. doi: 10.1016/j.ymgme.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 54.Servitja JM, et al. Hnf1alpha (MODY3) controls tissue-specific transcriptional programs and exerts opposed effects on cell growth in pancreatic islets and liver. Mol Cell Biol. 2009;29:2945–59. doi: 10.1128/MCB.01389-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Arensbergen J, et al. Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 2010;20:722–32. doi: 10.1101/gr.101709.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 59.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blechinger SR, et al. The heat-inducible zebrafish hsp70 gene is expressed during normal lens development under non-stress conditions. Mech Dev. 2002;112:213–5. doi: 10.1016/s0925-4773(01)00652-9. [DOI] [PubMed] [Google Scholar]
- 61.Meng A, Tang H, Ong BA, Farrell MJ, Lin S. Promoter analysis in living zebrafish embryos identifies a cis-acting motif required for neuronal expression of GATA-2. Proc Natl Acad Sci U S A. 1997;94:6267–72. doi: 10.1073/pnas.94.12.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravassard P, et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–97. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tena JJ, et al. An evolutionarily conserved three-dimensional structure in the vertebrate Irx clusters facilitates enhancer sharing and coregulation. Nat Commun. 2011;2:310. doi: 10.1038/ncomms1301. [DOI] [PubMed] [Google Scholar]
- 65.Splinter E, de Wit E, van de Werken HJ, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: from fixation to computation. Methods. 2012;58:221–30. doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 66.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 67.Boj SF, Parrizas M, Maestro MA, Ferrer J. A transcription factor regulatory circuit in differentiated pancreatic cells. Proc Natl Acad Sci U S A. 2001;98:14481–6. doi: 10.1073/pnas.241349398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenbloom KR, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.ENCODE Project Consortium. Myers RM, et al. A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13:204–16. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.