

Abstract
Plant breeders have focused on improving plant architecture as an effective means to increase crop yield. Here, we identify the main-effect quantitative trait loci (QTLs) for plant shape-related traits in rice (Oryza sativa) and find candidate genes by applying whole genome re-sequencing of two parental cultivars using next-generation sequencing. To identify QTLs influencing plant shape, we analyzed six traits: plant height, tiller number, panicle diameter, panicle length, flag leaf length, and flag leaf width. We performed QTL analysis with 178 F7 recombinant in-bred lines (RILs) from a cross of japonica rice line ‘SNUSG1’ and indica rice line ‘Milyang23’. Using 131 molecular markers, including 28 insertion/deletion markers, we identified 11 main- and 16 minor-effect QTLs for the six traits with a threshold LOD value > 2.8. Our sequence analysis identified fifty-four candidate genes for the main-effect QTLs. By further comparison of coding sequences and meta-expression profiles between japonica and indica rice varieties, we finally chose 15 strong candidate genes for the 11 main-effect QTLs. Our study shows that the whole-genome sequence data substantially enhanced the efficiency of polymorphic marker development for QTL fine-mapping and the identification of possible candidate genes. This yields useful genetic resources for breeding high-yielding rice cultivars with improved plant architecture.
Keywords: agronomic trait, plant shape, quantitative trait loci, rice, whole genome re-sequencing
INTRODUCTION
The increasing world population has spawned tremendous concerns about impending global food shortages. Nevertheless, the per capita increase in global food production has been greater than the increase in population since 1960, due to enormous increases in cereal crop production (Trewavas, 2001). This dramatic increase in crop yield, known as the Green Revolution, largely resulted from the development of genetically improved high-yielding crop varieties (Hedden, 2003). Among the genetic approaches used to improve yield potential, “ideal-type breeding” was used to modify plant architecture (morphology); for example, the improved rice cultivar IR24 has more tillers, reduced plant height and erect leaves. Rice breeders have harnessed natural variation in cultivars to modify rice plant architecture. For example, incorporating the recessive gene sd1 from the Chinese variety Dee-geowoo-gen, reduced plant height (Suh and Heu, 1978), and incorporating the photoperiod-insensitive gene se1 increased rice adaptability to a wide range of latitudes (Sano, 1992; Tamura et al., 1998).
Although these genetic variants played an important role in the marked increase in crop production, the demand for rice varieties with higher yield potential and greater yield stability has continued to rise due to increasing human population and changing global climate (Khush, 2005). To further enhance the yield potential of rice over that of existing high-yielding cultivars, crop scientists proposed an ideal plant architecture that includes fewer tillers with few or no unproductive tillers, more grains per panicle, and thick and sturdy stems (Khush, 1995; Li et al., 2012). Some of the genes and QTLs regulating architecture-related traits have been reported. For example, OsSPL14, which is regulated by OsmiR156, affects architecture-related traits; a point mutation in OsSPL14 perturbs OsmiR156-mediated regulation of OsSPL14 and generates an ideal-type rice plant with fewer tillers, increased lodging resistance and enhanced grain yield (Jiao et al., 2010). Marathi et al. (2012) reported 19 novel QTLs and 15 QTL hotspots affecting plant architecture, including yield-related traits, using RILs developed from Pusa1266, a semi-dwarf, high-yielding cultivar. Further work identified several other QTLs regulating plant shape, including leaf length, leaf width, tiller number and panicle length (Farooq et al., 2010; Liu et al., 2010; Wang et al., 2010; 2011). Modifying plant architecture for architecture-based breeding will require identification of QTLs and their causal genes in different genetic backgrounds.
QTL mapping and cloning using DNA markers are useful tools to unlock the genetic basis of complex phenotypic variation. Moreover, the application of molecular markers developed from QTL analysis enhances breeding efficiency by enabling marker-assisted selection for particular agronomic traits. However, traditional approaches for QTL analysis require substantial time and labor because of the low efficiency of polymorphic marker development for high-resolution map construction and the difficulties of candidate gene identification. Whole-genome sequencing, using next-generation technology can overcome these limitations of QTL analysis. A sequencing-based geno-typing method uses single nucleotide polymorphisms (SNPs) identified by whole-genome sequencing of a mapping population (Huang et al., 2009). This approach could substantially reduce the amount of time and effort required for genotyping of a mapping population. Recent work also proposed another approach, named ‘QTL-seq’, for rapid identification of QTLs for a given phenotype. QTL-seq works by sequencing DNAs from two populations, each composed of 20-50 individuals showing extreme opposite trait values in segregating progenies (Takagi et al., 2013). Although these approaches improve the time- and cost-effectiveness of QTL mapping, approaches to increase the efficiency of candidate gene identification have not yet been proposed.
In this study, we conducted QTL mapping for six traits related to plant architecture in rice and identified 11 main-effect and 16 minor-effect QTLs. By analysis of whole-genome sequencing data and meta-expression profiles, we identified 15 strong candidate genes for the 11 main-effect QTLs. The plant-shape QTLs and their candidate genes provide useful genetic resources for breeding of high-yielding rice cultivars.
MATERIALS AND METHODS
Plant materials and growth conditions
To identify QTLs influencing plant shape in rice, we crossed a japonica cultivar ‘SNU-SG1’ with the tongil-type cultivar Milyang23 with a genetic make-up similar to indica (> 90%) (Yoo et al., 2007). Among 338 F7 RILs developed from the F2 population by single seed descent, we chose 178 F7 RILs to conduct QTL mapping; these included 40 lines showing extreme phenotypic values for each trait. F7:8 heterogeneous inbred family-near isogenic lines (H-NILs) derived from F7 RILs were used to test the allele effects of candidate gene of plant shape-related QTLs (Tuinstra et al., 1997). In 2011, the RIL population and two parents were evaluated at the Seoul National University Experimental Farm in Suwon (37° N latitude), Korea. The field experiment was conducted in a randomized complete block design with two replications in two consecutive years (2010 and 2011) and field management followed normal rice agronomic practices. Seeds were sown in the seed bed of the greenhouse on April 20 and seedlings were transplanted into the paddy field on May 19 at a spacing of 0.3 m by 0.15 m with one seedling per hill. Compound fertilizer (21N-17P-17K) was applied in the same amount (24 kg/10a) and applied three times: before transplanting and at 7 and 10 weeks after transplanting.
Evaluation of agronomic traits
For the phenotypic evaluation of the selected 178 F7 RILs, we measured these agronomic traits; plant height, tiller number, panicle diameter, panicle length, flag leaf length and flag leaf width. All of the traits were evaluated when panicles were fully emerged. Panicle diameter (0.5 cm below the panicle base) was measured by a digital caliper. Plant height was measured from the soil surface to the apex of the main stem. On the main stem, flag leaf length was measured from the junction of leaf blade and sheath to the leaf apex, and flag leaf width was measured at the widest location of the leaf. Tiller number was scored when the grains had matured fully. Of all the flowered tillers of an individual plant, the longest panicle was measured for panicle length.
Molecular markers for genotyping
Genomic DNAs were extracted from leaf tissues by a CTAB method (Murray and Thompson 1980). Of 131 DNA markers used for genotyping, 68 markers are RM-series simple sequence repeat (SSR) markers that were designed by Temnykh et al. (2001), and 35 markers are S-series sequence-tagged site (STS) markers that were designed based on the sequence differences between japonica and indica, using information from the Crop Molecular Breeding Lab, Seoul National University (Chin et al., 2007). The other 28 markers are NGS-based insertion/deletion markers designed by sequence comparison between the two parent genotypes. To develop these markers, we investigated 7 to 11 base pairs of unique insertion/deletion (InDel) regions by comparing whole-genome sequencing data of the two parents, and designed primers to get PCR product (< 150 bp) containing the InDels. The initial genetic map was constructed with SSR and STS markers, and the next-generation based InDel markers were used to further map unlinked regions in the linkage map.
Linkage map construction and data analysis
A molecular linkage map was constructed using Mapmaker 3.0 (Lander et al., 1987; Lincoln et al., 1993). Distance between markers is presented in centiMorgans (cM) using the Kosambi map function (Kosambi, 1994), and the order of markers was established using three point linkage analyses. QTL analysis was conducted with composite interval mapping implemented in Qgene 4.3.10 software (Joehanes and Nelson, 2008). Significance thresholds for QTL detection were determined with 1000 permutations, and the threshold of logarithm of odds (LOD) for each QTL ranged from 2.24 to 3.03 with 95% confidence. The QTLs explaining more than 10% of the phenotypic effect were defined as main-effect QTLs as described previously (Wang et al., 2010).
High-throughput whole genome re-sequencing
High-throughput whole-genome sequencing was performed on the two parents, SNU-SG1 and Milyang23. The Illumina Genome Analyzer II was used to generate short reads and bases were called with the Sequence Analysis Software of Pipeline version 1.4 of the Genome Analyzer (Illumina, Inc., USA). Genomic DNAs were extracted using the QIAquick PCR Purification Kit (Qiagen) from leaf tissues. Full sequencing was processed in the National Instrumentation Center for Environmental Management (NICEM) in Seoul National University, Seoul, Korea. The sequencing yielded 26X and 31X fold coverage, as well as 9 Gbp and 11 Gbp in read length, for SNU-SG1 and Milyang23, respectively (Supplementary Tables S1 and S2). Then, the 75 bp paired-end reads of the two parents were mapped to the japonica cv. Nipponbare reference genome, which was used as the reference sequence (International Rice Genome Sequencing Program, 2005). Finally, the low-quality bases (Q score in scale < 20), and sites with conflicting genotypes among reads were excluded, and only the reads aligned to unique locations in the reference genome were used for sequence construction. The total number of SNPs was 3,202,922, which include 163,933 from SNU-SG1 and 3,038,989 from Milyang23.
Identification of candidate genes for the QTLs
Of the 27 QTLs, 11 main-effect QTLs with high LOD and R2 value were used for candidate gene analysis. We identified candidate genes harboring mutated sequences such as SNPs and InDels by the following process. First, a reference sequence (Nipponbare) corresponding to each main-effect QTL was transferred from the GRAMENE database (http://www.gramene.org/) into text in Microsoft Word. The genomic region associated with each QTL was selected by confidence interval (CI) that was determined by 1.5-LOD support interval (Dupuis and Siegmund, 1999). 1.5-LOD support intervals give a 95% probability of QTL location on the map and were calculated as the longest contiguous interval in which the LOD value was within 1.5-fold of its maximum. Coding sequence regions derived from SNU-SG1 and Milyang23 were interrogated to detect mutated sequences using the AutoHotkey macro program (http://www.autohotkey.com/). This process has two steps. First, the polymorphic sequences (< 15 bp) obtained from whole genome sequencing data of the two parents were copied and pasted into Microsoft Excel for sequence blast using Auto-Hotkey source codes 1 and 2 (Supplementary Fig. S1). Second, the sequences were blasted against the Nipponbare genome sequence to locate the genomic regions of polymorphic sequences in Microsoft Word using the AutoHotkey source code 3 (Supplementary Fig. S1). Among the genes harboring mutated sequences, only the genes for which the mutation caused amino acid changes were selected as candidate genes. Arabidopsis orthologous gene information for the candidate QTL genes was obtained from The Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org/).
Heatmap analysis or microarray data analysis
We used the information of 1150 Affymetrix array data (platform accession number is GPL2025) from the NCBI gene expression omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) to analyze the mRNA expression pattern of candidate genes in Supplementary Table S3 (Cao et al., 2012; Jung et al., 2011). From the Affymetrix anatomical meta-expression database, we generated the log2 average intensity data of 16 tissues/organs; callus, coleoptile, collar, leaf, flag leaf, root, shoot, stem, internode, shoot apical meristem (SAM), panicle, anther, stigma/ovary, palea/lemma, developing seed, and embryo. Among them, the data for stem and palea/lemma were only available for indica, the data for coleoptile, collar, internode, stigma/ovary and embryo were only for japonica, and the other 9 tissues/organs have the data for both. Then, the TIGR Multiple Experiment Viewer (MEV) software was used to prepare the Heatmap using the anatomical expression data (Supplementary Fig. S2; Table S3) (Eisen et al., 1998).
RNA extraction, reverse transcription (RT) and quantitative real-time PCR (qPCR)
Flag leaf tissues of SNU-SG1 and Milyang23 grown in paddy field were homogenized in liquid nitrogen. Total RNA extraction and RT-qPCR were performed as previously described (Yoo et al., 2009). Briefly, the RT products equivalent to 50 ng of total RNA and GoTaq qPCR Master Mix (Promega) were used for qPCR reactions using the Light Cycler 480 (Roche) and Roche Optical System software was to calculate threshold cycle (DCT) value. The rice Ubiquitin gene (Os01g22490) was used as an internal control. The primers used for qPCR are listed in Supplementary Table S4.
RESULTS
Phenotypic variation in F7 RILs from the Milyang23/SNUSG1 cross
For QTL analysis of the morphological traits that determine plant architecture, we used 178 F7 RILs from the cross of Milyang23 (indica) and SNU-SG1 (japonica) (Yoo et al., 2007). We analyzed six shape-related traits that critically affect lodging and yield: plant height, tiller number, panicle diameter, panicle length, flag leaf length, and flag leaf width. We measured phenotypic variation in the two parental lines and the RILs (Fig. 1). Most of the traits showed approximately normal distribution with bidirectional transgressive segregation. We also observed a large amount of variation between the two parental cultivars for the traits, except for plant height and tiller number. Supplementary Table S5 shows descriptive statistics of the six agronomic traits. We analyzed the correlation among the traits in the F7 RIL population (Table 1) and observed a positive correlation between flag leaf length and other traits, except for flag leaf width. We also found relatively high positive correlations between plant height and panicle length (0.64), and between panicle diameter and flag leaf width (0.53).
Fig. 1.
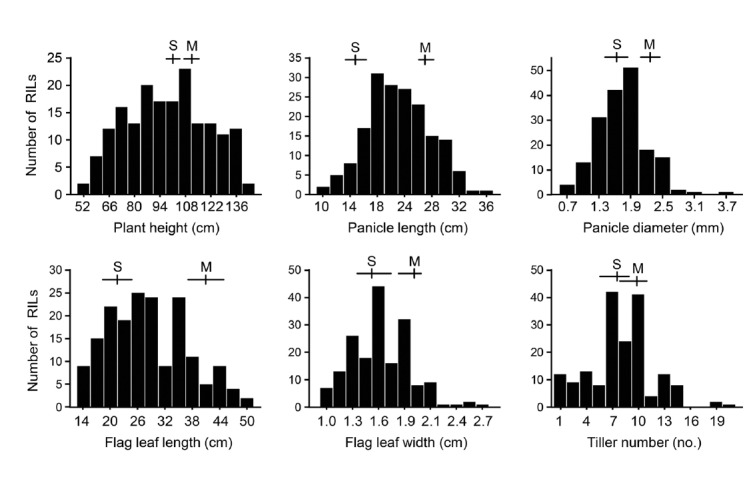
Frequency distribution of the six agronomic traits in the 178 F7 RILs derived from the cross of SNU-SG and Milyang23. The y-axis of each graph indicates the number of F7 RILs. Means and ranges of the parents are marked at the top of each histogram; S, SNU-SG1; M, Milyang23.
Table 1.
Correlations of the six agronomic traits analyzed in the F7 recombinant inbred lines
| Ph | Pl | Fll | Flw | Pd | |
|---|---|---|---|---|---|
| Pl | 0.64*** | ||||
| Fll | 0.29*** | 0.31*** | |||
| Flw | − 0.02 | 0.02 | 0.08 | ||
| Pd | 0.11 | 0.16* | 0.20** | 0.53*** | |
| Tln | 0.32 | 0.07 | 0.37*** | − 0.06 | − 0.19** |
Significance level:
P < 0.001,
< 0.01, and
< 0.05.
Ph, plant height; Pl, panicle length; Fll, flag leaf length; Flw, flag leaf width; Pd, panicle diameter; Tln, tiller number.
Linkage map construction
A coarse-scale linkage map was initially constructed over the whole rice genome using 178 F7 RILs and 68 SSR and 35 STS markers. To construct linkage map for the unlinked regions in the initial map, we designed 28 next-generation based InDel markers using the whole-genome sequencing data. We used Mapmaker 3.0 to construct the genetic linkage map, which consisted of 12 linkage groups and covered 2,451 cM (Fig. 2). The average distance between adjacent markers was 14.5 cM, which is less than 20 cM, the minimal compulsory level for QTL mapping (Lander and Botstein, 1989).
Fig. 2.
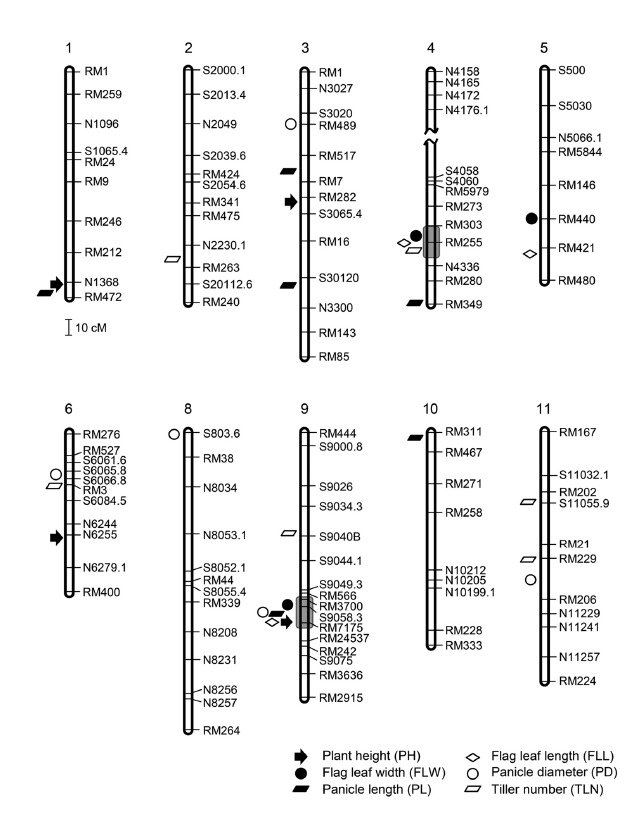
Chromosomal locations of QTLs detected by Q-genes in the F7 RIL population of SNU-SG1/Milyang23. Chromosomes are numbered at the top and markers are listed on the right of each chromosome. Wave marks in chromosome 4 represent the unlinked region. Various geometric figures indicate the location of peak LODs of the six agronomic traits. Chromosome 7 and 12 are not shown because we detected no QTLs on these two chromosomes.
QTL analysis for the six agronomic traits
To identify QTLs, we used a LOD threshold value and phenotypic effect (R2) (see “Materials and Methods”). As a result, we identified a total of 27 QTLs for the six traits, with 7.4 to 33.5% phenotypic effect (R2) and 2.9 to 15.8 LOD values, across all the chromosomes except chromosomes 7 and 12 (Table 2). Thus, chromosomes 7 and 12 were excluded from Fig. 2, because they did not carry any significant QTL above the threshold. A QTL with highest LOD (15.8) and phenotypic effect (33.5%) was detected for plant height on the long arm of chromosome 1 (Table 2; Fig. 2). Overall, we detected 3 to 6 QTLs for each agronomic trait. Of the 27 QTLs, 11 QTLs explained more than 10% of the phenotypic effect and were defined as the main-effect QTLs, which include Fll4 for flag leaf length, Flw5 and Flw9 for flag leaf width, Pl1 and Pl9 for panicle length, Ph1, Ph3 and Ph9 for plant height, and Tln2, Tln11-1 and Tln11-2 for tiller number (Table 2). Especially, three QTLs (Pl9, Ph1 and Tln2) explained more than 15% of the phenotypic effect, suggesting that these genetic materials provide a valuable resource for the identification of major genes affecting plant architecture.
Table 2.
QTL identification for the six agronomic traits by composite interval mapping using 178 F7 recombinant inbred lines
| Trait | QTL | Chr.a | Markerb | Ac | LOD | LOD peak (cM) | R2 d (%) | 95% CIe (cM) | Perf |
|---|---|---|---|---|---|---|---|---|---|
| Flag leaf length | Fll4 | 4 | RM255 | 2.08 | 4.35 | 202.7 | 10.7 | 190.–218.3 | 2.49 |
| Fll5 | 5 | RM421-RM480 | 2.43 | 3.04 | 181.8 | 7.6 | 166.2–199.7 | 2.26 | |
| Fll9 | 9 | RM7175 | −2.64 | 3.68 | 141.8 | 9.1 | 119.6–153 | 2.42 | |
| Flag leaf width | Flw4 | 4 | RM303-RM255 | 0.12 | 3.47 | 195.3 | 8.6 | 174.7–218.1 | 2.62 |
| Flw5 | 5 | RM146-RM440 | −0.13 | 4.21 | 145.5 | 10.3 | 128.5–171.1 | 2.28 | |
| Flw9 | 9 | S9058.3 | 0.08 | 4.12 | 131.9 | 10.1 | 1195-132 | 2.61 | |
| Panicle diameter | Pd3 | 3 | RM489 | 0.14 | 3.75 | 54.9 | 9.2 | 45.4–63.3 | 3.03 |
| Pd6 | 6 | S6065.8-S6066.8 | −0.19 | 2.96 | 42.5 | 7.4 | 25.9–70.4 | 2.60 | |
| Pd8 | 8 | S803.6-RM38 | −0.14 | 2.87 | 2.8 | 7.1 | 0.1–12.1 | 2.38 | |
| Pd9 | 9 | RM3700-RM7175 | 0.13 | 3.65 | 143 | 8.8 | 133.3–153.1 | 2.72 | |
| Pd11 | 11 | RM229-RM206 | 0.12 | 2.91 | 94.9 | 7.2 | 78.5–12.1 | 2.63 | |
| Panicle length | Pl1 | 1 | N1368-RM472 | 1.43 | 4.31 | 224.6 | 10.5 | 217.9–229.9 | 2.58 |
| Pl3-1 | 3 | RM517-RM7 | −1.16 | 3.53 | 113.0 | 8.7 | 91.8–140.9 | 2.58 | |
| Pl3-2 | 3 | S30120-N3300 | −1.17 | 2.88 | 218.4 | 7.2 | 210.3–263.3 | 2.58 | |
| Pl4 | 4 | RM349 | 0.39 | 3.34 | 269.3 | 8.3 | 244.4–269.3 | 2.56 | |
| Pl9 | 9 | S9058.3-RM7175 | −2.39 | 9.60 | 136.1 | 22.0 | 119.8–142.7 | 2.37 | |
| Pl10 | 10 | RM311-RM467 | 0.79 | 3.15 | 9.7 | 7.8 | 0.1–33.6 | 2.24 | |
| Plant height | Ph1 | 1 | N1368-RM472 | 14.79 | 15.8 | 222.7 | 33.5 | 217.5–226.8 | 2.38 |
| Ph3 | 3 | RM282- S3065.4 | −7.21 | 4.45 | 130.6 | 10.9 | 128.5–143.4 | 2.45 | |
| Ph6 | 6 | N6255-S6279.1 | −6.73 | 4.03 | 106 | 9.9 | 85.1–125.5 | 2.36 | |
| Ph9 | 9 | RM7175 | −8.30 | 5.68 | 143.4 | 13.7 | 130.1–151.9 | 2.39 | |
| Tiller number | Tln2 | 2 | N2230.1-RM263 | 1.45 | 6.33 | 184.7 | 15.3 | 163.3–199.6 | 2.49 |
| Tln4 | 4 | RM255-N4336 | −1.30 | 3.36 | 212.6 | 8.4 | 184–232.1 | 2.85 | |
| Tln6 | 6 | RM3 | 0.74 | 3.79 | 51.1 | 9.4 | 43.2–51.3 | 2.64 | |
| Tln9 | 9 | S9034.4-S9040B | 0.37 | 3.01 | 76.1 | 7.6 | 56.2–78.1 | 2.57 | |
| Tln11-1 | 11 | RM202-S11055.9 | −1.18 | 4.33 | 48.7 | 10.7 | 26.8–74.3 | 2.45 | |
| Tln11-2 | 11 | RM229 | −1.22 | 5.61 | 94.3 | 13.7 | 91.2–104.2 | 2.45 |
Chromosome number and marker intervals
Positive and negative values indicate additive effects contributed by SNU-SG1 and Milyang23 alleles, respectively.
Phenotype variation explained by the detected QTL
95% confidence interval for the detected QTL; cM represents centiMorgans.
LOD thresholds raised by permutation tests, p = 0.05
QTL hotspots, genomic locations associated with multiple traits, may harbor critical regulators of various agronomic traits (Neto et al., 2012). Two QTL hotspots were identified on chromosomes 4 and 9 (grey blocks in Fig. 2). One, in a window of around 8 cM on the long arm of chromosome 9, harbors three main-effect QTLs (Flw9, Ph9 and Pl9 for flag leaf width, panicle height, panicle length, respectively) and two minor-effect QTLs (Pd9 and Fll9 for panicle diameter and flag leaf length) (Fig. 2). The other, in a window of around 5 cM on chromosome 4, carries three QTLs, including one main-effect QTL (Fll4 for flag leaf length) and two minor-effect QTLs (Flw4 for flag leaf width and Tln4 tiller number) (Fig. 2).
To compare the physical locations of the QTLs identified in this study and previously reported QTLs, we searched the literature for cases in which mapping populations derived from the crosses of indica and japonica cultivars were used for QTL identification. By using flanking DNA markers, we compared the physical locations between the QTLs in the current study and the previously published QTLs. Among 27 QTLs identified in this study, 13 QTLs were in accord with the chromosomal regions carrying the published QTLs (Table 3). The remaining 14 QTLs did not overlap with the published QTLs, indicating that these are potentially novel QTLs for the traits. Notably, the QTLs for panicle diameter are all newly discovered. Among the new 14 QTLs, 6 QTLs are the main-effect QTLs with above 10% of phenotypic effect, including Fll4, Flw5, Pl9, Ph3, Tln11-1 and Tln11-2 (Table 2).
Table 3.
Comparison of the QTLs identified in current and previous studies using the mapping populations derived from crosses of indica and japonica cultivars
| Traits | QTL of this study
|
QTL of previous studies
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| QTL | Physical position (Mb) | QTL | Marker region | Physical position (Mb) | PTa | PSb | Mapping parents japonica (J)/indica (I) | References | |
| Plant height | Ph1c | 36.8–39.6 | QTLph1 | C86-C742 | 39.5–42.7 | BIL | 98 | Nippnbare (J)/Kasalath (I) | Ishimaru et al. (2004) |
| Qph1.2 | E60551-RM1387 | 35.0–40.5 | F2 | 301 | Nipponbare (J)/IR1545-339 (I) | Lin et al. (2011) | |||
| Ph6 | 25.5–27.9 | Qph6.2 | RM3879-RM340 | 25.9–28.5 | F2 | 301 | Nipponbare (J)/IR1545-339 (I) | Lin et al. (2011) | |
| Qph6f | RM162-RM30 | 24.0–27.3 | RIL | 226 | Zhonghui9308 (IxJ)/Xieqingzao B (I) | Liang et al. (2011) | |||
| Ph9 | 16.8–18.1 | Qph9 | RM3912-RM278 | 10.8–27.1 | F2 | 301 | Nipponbare (J)/IR1545-339 (I) | Lin et al. (2011) | |
| Flag leaf length | Fll5 | 23.9–27.3 | qFLL-5 | BIN | 27.8–28.6 | RIL | 150 | Nippnbare (J)/9311(I) | Wang et al. (2010) |
| QFll5 | RM480-RM334 | 27.5–28.6 | RIL | 180 | IRAT109 (J)/Zhenshan 97(I) | Yue et al. (2006) | |||
| Fll9 | 15.9–16.8 | qFll9 | RM24424-RM24434 | 16.2–16.5 | F2 | 176 | SN265 (J)/LTH(J) | Huang et al. (2010) | |
| Flag leaf width | Flw4 | 28.5–30.7 | qFlw4 | RM17483-RM17486 | 31.0–31.2 | RIL F7 | 190 | D50 (J)/HB277(I) | Chen et al. (2012) |
| Qflw4 | RM255-RM349 | 28.5–31.6 | RIL F10 | 180 | IRAT109 (J)/Zhenshan97(I) | Yue et al. (2006) | |||
| Flw9 | 15.9 | qFLA9 | RM6839B-RM257 | 14.5–17.9 | BRIL | 244 | 9311 (I)/Zhenshan97B(I) | Wang et al. (2012) | |
| Tiller number | Tln2 | 23.0–25.8 | Tln2-2 | RM526–RM425 | 26.6–32.2 | SSSL | 78 | IAPAR9 (J)/Hua-Jing-Xian74 (I) | Liu et al. (2012) |
| tp2b | RM561-RM6318 | 19.8–24.4 | BCRIL | 244 | Zhenshan97B (I)/9311(I) | Liu et al. (2008) | |||
| Tln4 | 30.7–33.6 | qTLN4 | RM303-RM349 | 27.5–32.4 | RIL-F11 | 254 | Xiushui 79 (J)/C Bao(J) | Jiang et al. (2012) | |
| Tln 4-3 | RG143-RG620 | 34.2–34.6 | DHd | 123 | Azucena (J)/IR64(I) | Yan et al. (1998) | |||
| Tln6 | 19.4 | pn6 | S6070 | 20.1 | F2 | 146 | Junambyeo (J)/IR71033(I) | Rahman et al. (2008) | |
| Tln9 | 10.5–12.6 | Tln9 | RM105–RM278 | 12.5–19.3 | SSSL | 91 | Hua-Jing-Xian74 (I)/Amol3(I) | Liu et al. (2012) | |
| Panicle length | Pl1 | 36.8–39.6 | qPL-1 | RM472–RM104 | 37.8–40.1 | RIL | 187 | IRAT109 (J)/Zheshan97B (I) | Liu et al. (2008) |
| Pl4 | 32.4 | QPl4b | RM349-RM280 | 32.4–34.9 | BC2F5 | 59 | Tarome Molaei (J)/IR64 (I), | Ahamadi et al. (2008) | |
PT, population type
PS, population size
Numbers at the end of QTL represent chromosome number
DH, double haploid population
Sequencing analysis for candidate gene identification
We carried out whole-genome sequencing of the two parentacultivars, SNU-SG1 and Milyang23, to obtain a cultivar-specific nucleotide polymorphism data set (see “Materials and Methods”). The polymorphic nucleotide sequences, including In-Dels, were obtained by comparing the whole genome sequences of SNU-SG1 and Milyang23 with the reference Nipponbare sequence using Pipeline version 1.4 (see “Materials and Methods”). Of the total 363,845 SNPs identified by comparing with the reference sequence, 330,317 were identified from Milyang23, and 33,528 from SNU-SG1. Sequence variation between indica- and japonica-type subspecies contributed to the large number of SNPs in Milyang23. For identification of candidate genes associated with the QTLs, we analyzed all the SNPs (including InDels) derived from SNU-SG1, but only In-Dels capable of impairing protein function were considered for Milyang23 to filter out non-causal subspecies sequence variation between indica and japonica genomes.
To perform candidate gene analysis for 11 main-effect QTLs, we analyzed the genomic region that was selected based on confidence interval for each QTL peak (Table 2). Confidence intervals were determined by 1.5-LOD support interval, which provides a QTL coverage probability of approximately 95% (Dupuis and Siegmund, 1999). The confidence intervals of the QTLs detected in the integrated map using composite interval mapping are given in Table 2. We determined the specific locations of polymorphic sequences in the individual genes by BLAST to the Nipponbare reference sequence. By these coding sequence analyses, we identified 54 genes with 656 nucleotide sequence polymorphisms in their open reading frames within the 11 main-effect QTL regions (data not shown). Among them, only the 308 SNPs that caused amino acid variation were considered as candidate genes. Thus, by analysis of the whole-genome sequencing data, we isolated 54 candidate genes carrying at least one amino acid polymorphism for the six traits (Supplementary Table S3).
Strong candidate genes for the main-effect QTLs
Among the 54 candidate genes for 11 main-effect QTLs, we further isolated strong candidate genes by analyzing their amino acid sequences and meta-expression profiles. First, by protein coding sequence analysis, we selected 11 genes as strong candidates, because they harbor potential causal SNPs leading to a premature stop codon or frameshift in their amino acid sequences (Table 4). These 11 strong candidate genes included candidates for all agronomic traits examined, except for panicle diameter, with one to seven genes for each trait. These included single genes for panicle length, plant height and flag leaf width, two genes for flag leaf length, and seven genes for tiller number (Table 4). One candidate gene (Os01g68510), encoding peptide transporter 2 (PTR2), was identified for two QTLs related to panicle length and plant height on the distal part of chromosome 1 (Table 4). The largest effect QTL Ph1 for plant height was also detected with 33.5% R2 value in this location on chromosome 1 (Tables 2 and 3). Meanwhile, QTL effect can be also caused by differences in gene expression between alleles due to mutations in regulatory regions including promoters and introns (Meuwissen, 2010). Thus, we next analyzed meta-expression profiles of the 54 candidate genes using Affymetrix array data (Supplementary Fig. S2), which provide the indica or japonica-specific gene expression (Cao et al., 2012; Jung et al., 2011). As we used the indica (Milyang23) and japonica (SNU-SG1) cultivars to create the QTL mapping population, the genes expressed differentially in the indica and japonica rice cultivars would be considered as strong candidates for the corresponding traits. By Heatmap analysis, we found that seven genes are differentially expressed in at least one tissue type with more than two-fold differences between indica and japonica (Table 5). Interestingly, all the probes corresponding to the seven genes were expressed preferentially in the flag leaves of japonica rice. To test whether these genes are differentially expressed in SNU-SG1 and Milyang23, we performed RT-qPCR analysis using flag leaves. Of the seven genes, five genes were preferentially expressed in SNU-SG1 with more than two-fold difference compared to Milyang23 (Supplementary Fig. S3), in accordance with meta-expression profile. However, we failed to amplify the other two genes (Os02g40190 and Os02g40240), possibly due to very low transcript abundance. This result strongly supports the idea that at least five genes identified by meta-expression profile analysis are strong candidate genes for the corresponding QTLs. Notably, three genes (Os02g40190, Os02g40200 and Os02g40240) for Tln2 were identified as strong candidates by both coding sequence and meta-expression analyses, and encode known receptor-like protein kinase precursors (Tables 4 and 5). Thus, taken together our data provide a total of 15 strong candidate genes by two different analyses.
Table 4.
List of 11 strong candidate genes for the main-effect QTLs obtained by sequence analysis of whole genome re-sequencing data for the two parent cultivars, Milyang23 (indica) and SNU-SG1 (japonica)
| QTL | Typea | Gene ID | Mutationb | Description | Orthologc |
|---|---|---|---|---|---|
| Pl1, Ph1 | Milyang23 | Os01g68510 | 133/601 | Peptide transporter PTR2 | AT1G69870 (NRT1.7) |
| Tln2 | SNU-SG1 | Os02g40100 | 398/791 | DUF869 domain containing protein | AT2G23360 |
| Tln2 | SNU-SG1 | Os02g40190 | 33/240 | Receptor kinase | - |
| Tln2 | SNU-SG1 | Os02g40200 | 485/1033 | Receptor-like protein kinase precursor | - |
| Tln2 | SNU-SG1 | Os02g40240 | 675/1002 | Receptor-like protein kinase precursor | - |
| Tln2 | SNU-SG1 | Os02g40280 | 273/370 | Piwi domain containing protein | AT1G69440 (AGO7) |
| Tln2 | SNU-SG1 | Os02g41610 | 642/745 | Expressed protein, Coiled coil | - |
| Fll4 | SNU-SG1 | Os04g51450 | 35/331 | Glycosyl hydrolases family 16 | AT2G18800 (XTH21) |
| Fll4 | SNU-SG1 | Os04g53120 | 6 A.S. | XA1, NB-ARC domain containing protein | - |
| Flw5 | SNU-SG1 | Os05g31230 | 5 A.S. | N-acetyl transferase ESCO1 | AT4G31400 |
| Tln11-1 | Milyang23 | Os11g18194 | 560/761 | Cycloartenol synthase | AT2G07050 (CAS1) |
Parent type harboring mutant alleles, which were determined by sequence comparison with reference genome japonica cv. Nipponbare.
The position of amino acid in which premature stop codon occurred/total protein length, A.S. amino acid substitutions.
Accession number of Arabidopsis orthologs were obtained from The Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org/).
Table 5.
Seven strong candidate genes for the main-effect QTLs obtained by expression profile analysis of Affymetrix array data
| Gene IDa | QTL Nameb | Description | Log2 fold change (japonica/indica)c
|
|||||
|---|---|---|---|---|---|---|---|---|
| Flag leaf | Root | Shoot | SAMd | Panicle | Anther | |||
| Os09g26530 | Flw9, Ph9, Pl9 | Ser/Cys_Pept_Trypsin-like | 2.9 | 2.3 | 1.6 | 2.6 | 2.4 | 2.1 |
| Os09g26554 | Flw9, Ph9, Pl9 | Ser/Cys_Pept_Trypsin-like | 3.4 | 2.5 | 2.0 | 3.0 | 3.2 | 3.5 |
| Os03g26080 | Ph3 | Nucleoside triphosphatase | 2.3 | 1.1 | 1.0 | 1.3 | 1.1 | 1.0 |
| Os11g20239 | Tln11-1 | Hydroxyproline-rich glycoprotein DZ-HRGP precursor | 2.3 | 2.0 | 2.3 | 2.8 | 1.9 | 2.0 |
| Os02g40190 | Tln2 | Receptor kinase | 2.4 | 1.1 | 1.9 | 0.8 | 0.7 | 1.1 |
| Os02g40200 | Tln2 | Receptor-like protein kinase precursor | 2.4 | 1.4 | 2.3 | 2.1 | 0.9 | 0.8 |
| Os02g40240 | Tln2 | Receptor-like protein kinase precursor | 4.4 | 1.9 | 2.9 | 1.0 | 1.5 | 1.5 |
Locus identifier (ID) annotated by Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/) team
QTL name queried from Gramene QTL database (www.gramene.org/qtl/)
Log2 fold change of japonica/indica in Affymetrix anatomical meta-expression profile
SAM represents shoot apical meristem.
We further investigated the orthologs of these candidate genes in Arabidopsis thaliana, using Gramene (http://www.gramene.org/) and TAIR (http://www.arabidopsis.org/) databases. We found six Arabidopsis orthologs in which four genes have been functionally characterized (Table 4). In Pl1/Ph1 QTL, At1g69870 encodes a low affinity nitrate transporter (NRT1.7), which functions in nitrate remobilization in leaf (Fan et al., 2009). In Tln2 QTL, At1g69440 encodes ARGONAUTE 7, a member of the ARGONAUTE family, which functions in the regulation of developmental timing (Adenot et al., 2006). In Fll4 QTL, At2g 18800 encodes Xyloglucan endotransglycosylase/hydrolases (XTHs) which functions in carbohydrate metabolism (Paul and Janet, 1999). In Tln1-1 QTL, At2g07050 encodes cycloartenol synthase 11 (Corey et al., 1993) which functions in the biosynthesis of brassinosteroids (Asami and Yoshida, 1999). This identification of promising candidate genes provides useful genetic resources for the further isolation of the causal gene associated with each QTL.
Validation of QTL regions using H-NILs
To further validate the QTL regions associated with 15 strong candidate genes, we developed H-NILs that segregate for the phenotypes of the corresponding traits (Tuinstra et al., 1997). The genotypic and phenotypic analyses of the progeny H-NILs allow identification of the candidate genes underlying QTLs (Routaboul et al., 2012). In 178 F7 RILs, we selected heterogeneous lines for at least one of the flanking marker loci in the QTL regions (Fig. 3). By phenotypic analysis using 40 F7:8 individual plants derived from each heterogeneous line, we found several H-NILs for six main-effect QTLs that harbor strong candidate genes (Supplementary Table S6). Most of the H-NILs, including F7 RIL #72-derived ones which showed highest variation in tiller number, displayed transgressive segregation (Fig. 3, Supplementary S4). These support that the target genes are localized within the corresponding QTL regions, enhancing the fidelity of strong candidate genes. Further linkage analysis of the markers and traits in the H-NIL mapping populations may lead to isolation of the genes underlying the QTLs.
Fig. 3.

Development of heterogeneous inbred family-near isogenic lines (H-NILs) for TLN2. The rows represent the genotypes of seven F7 RILs that contain heterozygous genomic regions for the TLN2 QTL. Shaded regions indicate chromosomal segments that are homo-zygous SNU-SG1 (black), homo-zygous Milyang23 (white), and heterozygous Milyang23/H143 (grey). 40 plants of F7:8 H-NILs derived from each F7 RIL were investigated for progeny tests (on the right). Maximum (Max) and minimum (Min) tiller numbers for F7:8 H-NILs are shown with standard deviation (SD).
DISCUSSION
Next-generation sequencing data enhance the efficiency of QTL analysis
Agronomically important crop traits are generally controlled by multiple genes (QTLs) that produce continuous variation in phenotype. Conventional QTL analysis remains time-consuming and labor-intensive, mainly because it requires development of polymorphic markers for linkage analysis. To overcome this limitation, several research groups have recently introduced next-generations sequencing technology as a new strategy for genetic mapping. For example, high-throughput genotyping of recombinant populations can be performed by whole-genome sequencing of individual RILs (Huang et al., 2009). This sequence-based genotyping has proven faster for genetic map construction and more accurate for determination of recombination breakpoints than marker-based genotyping methods (Wang et al., 2011). Another group performed whole genome re-sequencing of 50 rice cultivars and investigated genome-wide variation in high-quality candidate SNPs obtained from re-sequencing data for each cultivar (Xu et al., 2012). In this study, we modified conventional mapping methods by whole-genome sequencing of the two parental cultivars to improve QTL mapping efficiency. To develop polymorphic DNA markers for linkage analysis, we generated InDel markers using whole-genome sequencing data; these markers were designed based on nucleotide sequence polymorphisms between the two parents. The InDel markers showed much higher efficiency (74.5%) in polymorphic marker development compared to conventional SSR (18.1%) and STS (21.1%) markers, which were designed from sequence polymorphisms between japonica and indica genomes (Fig. 4). These results indicate that application of a whole-genome sequencing approach to the two parental cultivars substantially enhanced the efficiency of QTL mapping with relatively lower cost for whole-genome sequencing compared to high-throughput approaches.
Fig. 4.

Comparison of efficiencies of polymorphic marker development between next-generation sequencing based InDel (NID) markers and conventional SSR and STS markers. The numbers in parentheses represent the numbers of polymorphic or non-polymorphic markers out of total DNA markers that were tested for marker development. Polymorphic markers represent the markers obviously distinguishable in PCR product size by 3% agarose gel electrophoresis. NID markers were used to construct a linkage map for unlinked regions in the initial map.
High-resolution mapping to identify candidate genes for the detected QTLs is another time-consuming and laborious step in conventional QTL analysis. Thus, instead of performing fine mapping to narrow down the QTL region to less than 100 kb, we directly analyzed the genomic regions for the QTLs that were selected based on a 1.5-LOD confidence interval (Table 2), and identified candidate genes for each QTL of interest. Our sequence analysis found an average of five candidate genes harboring 23 causal SNPs in the confidence interval-based QTL regions by comparing genomic sequences of SNU-SG1 and Milyang23 with the reference Nipponbare. In conventional QTL analysis, fine mapping has to be conducted to narrow down the QTL region to 50 to 100 kb through development of large numbers of polymorphic DNA markers in each QTL region as well as development of mapping populations such as heterogeneous inbred family-near isogenic lines (Tisne et al., 2008) and backcross inbred lines (Li et al., 2011). Whole-genome sequencing-based QTL analysis enabled us to isolate candidate genes for all the QTLs identified from initial mapping population at the same time, while conventional QTL approaches need to develop a high resolution-mapping population for each QTL to find strong candidate gene(s). Furthermore, the combination of whole-genome sequencing-based QTL and meta-expression profile analyses enable us to isolate candidate genes for the previously reported QTLs by sequencing of the two parents that were used to construct mapping populations. Therefore, we propose that this advanced QTL analysis approach, sequencing two parent cultivars, is an efficient method for candidate gene identification, as it skips the fine-mapping procedure.
Comparison of the identified QTLs with previously reported QTLs for agronomic traits
Some QTLs were identified here and in previous studies of plant architecture that examined traits such as tiller number and panicle diameter. For example, the four QTLs, Tln2, Tln4, Tln6 and Tln9, which influence tiller number, shared a similar genomic location with QTLs identified in previous studies (Jiang et al., 2012; Liu et al., 2012; Qu et al., 2012; Rahman et al., 2008; Yan et al., 1998) (Table 3). This consistency in the QTL regions associated with the tiller number among independent studies indicates that the locations of alleles for the trait are conserved across different genetic and environmental backgrounds. By contrast, the other two QTLs, Tln11-1 and Tln11-2, have not been previously identified. All the QTLs we identified for panicle diameter (Pd3, Pd6, Pd8, Pd9 and Pd11) are novel, with no correspondence to previously reported QTLs for this trait (Table 2), indicating that these QTLs may be a potentially new set of alleles specific to this genetic background.
Analysing candidate genes and inferring their functions from Arabidopsis homologs
By the whole-genome sequencing data analysis, we found 54 candidate genes in all the detected QTL regions related to six agronomic traits (Supplementary Table S3). By further sequence and meta-expression profile analyses, We designated 15 out of 54 genes as strong candidates for 11 QTLs. Of the 15 strong candidate genes, 11 genes identified by amino acid sequence analysis carry polymorphic SNPs that lead to a frameshift or premature stop codon in the coding sequence (Table 4). For example, in the Pl1 and Ph1 QTL region (Fig. 5A), five genes containing polymorphic SNPs in their coding sequences were initially found by sequence analysis (Supplementary Table S3). Of these SNPs, a 1-bp deletion in the coding region of PTR2 occurred in the Milyang23 allele, leading to a premature stop codon (Fig. 5C), whereas simple SNPs, which do not cause premature stop codon, were found for the other four genes. These suggest that PTR2 is the strongest candidate gene for the Pl1 and Ph1 QTL (Supplementary Table S3). In the same way, we identified 10 strong candidate genes for the other five agronomic traits (Figs. 4B and 4D; Supplementary Table S3).
Fig. 5.

Genomic locations of two main-effect QTLs and their candidate genes. (A, B) Os01g68510 (A) and Os11g18194 (B) genes located at two main-effect QTLs, Ph1 and Tln11-1, respectively. (C, D) Gene structures and natural variations o f Os01g68510 (C) and Os11g18194 (D). Sequence variations between SNU-SG1 and Milyang23 are indicated. Black dots represent the absence of the corresponding bases.
In addition to mutations in coding sequences, nucleotide mutations in the promoter, 5′-UTR, 3′-UTR and intron of many eukaryotic genes affect morphological traits by regulating gene expression at transcriptional or post-transcriptional levels (Lu et al., 2008; Rose et al., 2008; Yu et al., 2007). For example, TAC1, which affects tiller angle, is highly expressed in the indica rice line ‘IR24’ which displays a relatively spread-out plant architecture, but the expression of TAC1 is low in the introgressed line, IL55, derived from the japonica rice ‘Asominori’ which displays extremely erect tillers (Yu et al., 2007). This differential expression results from a nucleotide change in an intron, which affects 3′-UTR splicing. Recently, Jung et al. (2013) analyzed publicly available Affymetrix array data and identified 490 genes preferentially expressed in japonica rice and 104 genes in indica rice. Some of the genes that are preferentially expressed in each subspecies involve development of morphological traits such as plant height, panicle, culm and root (Jung et al., 2013). Thus, using the Affymetrix array data, we analyzed the expression patterns of 54 candidate genes in the indica and japonica rice samples of various tissue/organs and found seven genes that are differentially expressed between japonica and indica; this japonica-preferred expression was confirmed in SNU-SG1 by RT-qPCR analysis (Supplementary Fig. S3). Notably, three of the differentially expressed genes (Os02g40190, Os02g40200 and Os02g40240) were identified by both the sequence and expression analyses (Tables 5 and Supplementary Fig. S3). Together with 11 candidate genes that were identified by sequence analysis, an additional four genes from the meta-expression analysis between japonica and indica cultivars would be strong candidates for the causal genes for the corresponding QTLs.
The functions of the uncharacterized genes in rice can generally be predicted from the functions of orthologous genes in Arabidopsis, as proteins in these species generally have conserved molecular and biochemical functions. For example, the functions of CO and FT, flowering promoters in photoperiod pathway, are well conserved between Arabidopsis and rice and most likely in other species as well (Hayama and Coupland, 2004). Among the 15 strong candidate genes proposed in this study, the functions of four Arabidopsis orthologs have been reported (Corey et al., 1993; Fan et al., 2009; Maris et al., 2009) (Table 4). The Arabidopsis nitrate transporter NRT1.7 (Fan et al., 2009), an ortholog of PTR2, plays an important role in source-to-sink remobilization of nitrate. In addition, a rice gene, Short Panicle 1 (SP1) encoding a putative peptide transporter (PTR) has been reported to regulate inflorescence branch elongation; the knock-down sp1 mutant is defective in rice panicle elongation, producing short-panicle phenotypes (Li et al., 2009). These also indicate that PTR2 is a promising candidate gene related to the two QTLs detected for panicle length and plant height in this study. Another candidate gene Os11g18194, located in the region of Tln11-1 (Fig. 5B), is a rice ortholog of Arabidopsis CAS1, encoding a cycloartenol synthase, which is involved in the biosynthesis of brassinosteroids (Corey et al., 1993). Mutations in the genes involved in the BR biosynthesis pathway modify tiller number (Fujii et al., 1991), leaf size (Morinaka et al., 2006) and yield per plant (Choe et al., 2001). Moreover, increased levels of brassinosteroids cause a significant effect, increasing tiller number and seed weight (Wu et al., 2008). These suggest that OsCAS1 is a promising candidate gene for Tln11-1 related to tiller number. Lastly, another candidate gene Os04g51450, located in the region of Fll4, has an orthologous gene At2g18800 (XTH21), encoding a member of the xyloglucan endotransglucosylase/hydrolases (XTH), which is involved in cell loosing process at the preliminary stage of cell elongation in shoot and root meristems (Liu et al., 2007). Genes related to cell elongation, such as ANGUSTIFOLIA in Arabidopsis, play a role in leaf longitudinal development (Tomohiko et al., 1996). This supports the possible involvement of Os04g51450 in Fll4 QTL regulating flag leaf length.
In this study, we developed an improved approach for DNA marker development and candidate gene identification by whole-genome sequencing of the two parent cultivars of our RILs. This approach substantially increased the efficiency of linkage analysis by enabling marker development by simple comparison of sequence data from the two parents. In addition, the application of whole-genome sequencing to candidate gene analysis reduced the cost and time for candidate gene identification by skipping the high-resolution mapping step. By QTL analysis, we detected a total of 27 QTLs for six agronomic traits. The 15 novel QTLs, two QTL hotspots and 15 strong candidate genes identified in this study provide valuable resources for molecular breeding of new high-yielding plant varieties in rice.
Supplementary Materials - Lim et al.
Acknowledgments
We thanks Prof. Hee-Jong Koh for the PCR primer information of S-series makers for QTL mapping. This work was supported by a grant from the Next-Generation BioGreen 21 Program (Plant Molecular Breeding Center No. PJ008128), Rural Development Administration, Republic of Korea.
Note:
Supplementary materials are available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Adenot X., Elmayan T., Lauressergues D., Boutet S., Bouche N., Gasciolli V., Vaucheret H. DRB4-dependent TAS3 trans-acting siRNAs control leaf morphology through AGO7. Curr. Biol. 2006;16:927–932. doi: 10.1016/j.cub.2006.03.035. [DOI] [PubMed] [Google Scholar]
- Asami T., Yoshida S. Brassinosteroid biosynthesis inhibitors. Trends Plant Sci. 1999;4:348–353. doi: 10.1016/s1360-1385(99)01456-9. [DOI] [PubMed] [Google Scholar]
- Cao P., Jung K.H., Choi D.S., Hwang D.H., Zhu J., Ronald P.C. The rice oligonucleotide array database an atlas of rice gene expression. Rice. 2012;5:17. doi: 10.1186/1939-8433-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Luo J., Shao G., Wei X., Tang S., Sheng Z., Song J., Hu P. Fine mapping of a major QTL for flag leaf width in rice, qFLW4, which might be caused by alternative splicing of NAL1. Plant Cell Rep. 2012;31:863–872. doi: 10.1007/s00299-011-1207-7. [DOI] [PubMed] [Google Scholar]
- Chin J.H., Kim J.H., Jiang W., Chu S.H., Woo M.O., Han L., Brar D., Koh H.J. Identification of subspecies-specific STS markers and their association with segregation distortion in rice (Oryza sativa L.) J. Crop Sci. Biotech. 2007;10:175–184. [Google Scholar]
- Choe S., Fujioka S., Noguchi T., Takatsuto S., Yoshida S., Feldmann K.A. Overexpression of DWARF4 in the brassinosteroid biosynthetic pathway results in increased vegetative growth and seed yield in Arabidopsis. Plant J. 2001;26:573–582. doi: 10.1046/j.1365-313x.2001.01055.x. [DOI] [PubMed] [Google Scholar]
- Corey E.J., Seiichi P.T., Bartel B. Isolation of an Arabidopsis thaliana gene encoding cycloartenol synthase by functional expression in a yeast mutant lacking lanosterol synthase by the use of a chromatographic screen. Proc. Natl. Acad. Sci USA. 1993;90:11628–11632. doi: 10.1073/pnas.90.24.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis J., Siegmund D. Statistical methods for mapping quantitative trait loci from a dense set of markers. Genetics. 1999;151:373–386. doi: 10.1093/genetics/151.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S.C., Lin C.S., Hsu P.K., Lin S.H., Tsay Y.F. The Arabidopsis nitrate transporter NRT1.7, expressed in phloem, is responsible for source-to-sink remobilization of nitrate. Plant Cell. 2009;21:2750–2761. doi: 10.1105/tpc.109.067603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq M., Tagle A.G., Santos R.E., Ebron L.A., Fujita D., Kobayashi N. Quantitative trait loci mapping for leaf length and leaf width in rice cv. IR64 derived lines. J. Integr. Plant Biol. 2010;52:578–584. doi: 10.1111/j.1744-7909.2010.00955.x. [DOI] [PubMed] [Google Scholar]
- Fujii S., Hirai K., Saka H. Growth-regulating action of brassinolide in rice plants. In: Cutler H.G., Yokota T., Adam G., editors. Brassinosteroids. Chemistry, Bioactivity, and Application. Vol. 74. American Chemical Society; Washington DC: 1991. pp. 306–311. [Google Scholar]
- Hayama R., Coupland G. The molecular basis of diversity in the photoperiodic flowering responses of Arabidopsis and rice. Plant Physiol. 2004;135:677–684. doi: 10.1104/pp.104.042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama R., Yokoi S., Tamaki S., Yano M., Shimamoto K. Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature. 2003;422:719–722. doi: 10.1038/nature01549. [DOI] [PubMed] [Google Scholar]
- Hedden P. The genes of the Green Revolution. Trends Genet. 2003;19:5–9. doi: 10.1016/s0168-9525(02)00009-4. [DOI] [PubMed] [Google Scholar]
- Huang X., Feng Q., Qian Q., Zhao Q., Wang L., Wang A., Guan J., Fan D., Weng Q., Huang T., et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Jiang S.K., Feng L.L., Xu Z.J., Chen W.F. Analysis of QTLs for mesocotyl length in rice (Oryza sativa L.) Acta Argon. Sin. 2010;36:1108–1113. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- Ishimaru K., Ono K., Kashiwagi T. Identification of a new gene controlling plant height in rice using the candidate-gene strategy. Planta. 2004;218:388–395. doi: 10.1007/s00425-003-1119-z. [DOI] [PubMed] [Google Scholar]
- Jiang J.H., Liu Q.M., Zhang H., Liu J., Lu C., Ni W.L., Hong D.L. Tiller morphogenesis and mortality were controlled by different loci in rice (Oryza sativa L.) Afr. J. Agric. Res. 2012;7:3964–3977. [Google Scholar]
- Jiao Y., Wang Y., Xue D., Wang J., Yan M., Liu G., Dong G., Zeng D., Lu Z., Zhu X., et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010;42:541–544. doi: 10.1038/ng.591. [DOI] [PubMed] [Google Scholar]
- Joehanes R., Nelson J.C. QGene 4.0, an extensible Java QTL-analysis platform. Bioinformatics. 2008;24:2788–2789. doi: 10.1093/bioinformatics/btn523. [DOI] [PubMed] [Google Scholar]
- Jung K.H., Jeon J.S., An G. Web tools for rice transcriptome analyses. J. Plant Biol. 2011;54:65–80. [Google Scholar]
- Jung K.H., Gho H.J., Giong H.K., Chandran A.K.N., Nguyen Q.N., Choi H.B, Zhang T., Wang W., Kim J.H., Choi H.K., et al. Genome-wide identification and analysis of Japonica and Indica cultivar-preferred transcripts in rice using 983 Affymetrix array data. Rice. 2013;6:19. doi: 10.1186/1939-8433-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khush G.S. Breaking the yield frontier of rice. Geo J. 1995;35:329–332. [Google Scholar]
- Khush G.S. What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol. Biol. 2005;59:1–6. doi: 10.1007/s11103-005-2159-5. [DOI] [PubMed] [Google Scholar]
- Kosambi D. The estimation of map distance from recombination values. Ann. Eugen. 1994;12:172–175. [Google Scholar]
- Lander E.S., Botstein D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S., Green P., Abrahamson J., Barlow A., Daly M.J., Lincoln S.E., Newberg L.A. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987;1:174–181. doi: 10.1016/0888-7543(87)90010-3. [DOI] [PubMed] [Google Scholar]
- Li S., Qian Q., Fu Z., Zeng D., Meng X., Kyozuka J., Maekawa M., Zhu X., Zhang J., Li J., et al. Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J. 2009;58:592–605. doi: 10.1111/j.1365-313X.2009.03799.x. [DOI] [PubMed] [Google Scholar]
- Li Y., Fan C., Xing Y., Jiang Y., Luo L., Sun L., Shao D., Xu C., Li X., Xiao J., et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011;43:1266–1269. doi: 10.1038/ng.977. [DOI] [PubMed] [Google Scholar]
- Li S., Xie K., Li W., Zou T., Ren Y., Wang S., Deng Q., Zheng A., Zhu J., Liu H., et al. Resequencing and genetic variation identification of a rice line with ideal plant architecture. Rice. 2012;5:18. doi: 10.1186/1939-8433-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.S., Gao Z.Q., Shen X.H., Zhan X.D., Zhang Y.X., Wu W.M., Cao L.Y., Cheng S.H. Mapping and comparative analysis of QTL for rice plant height based on different sample sizes within a single line in RIL population. Rice Sci. 2011;18:265–272. [Google Scholar]
- Lin Y.R., Wu S.C., Chen S.E., Tseng T.H., Chen C.S., Kuo S.C., Wu H.P., Hsing Y.C. Mapping of quantitative trait loci for plant height and heading date in two inter-subspecific crosses of rice and comparison across Oryza genus. Bot. Stud. 2011;52:1–14. [Google Scholar]
- Lincoln S., Daly M., Lander E. 3rd (ed) Whitehead Institute for Biomedical Research; 1993. Constructing genetic linkage maps with MAPMAKER/EXP version 3.0. A tutorial and reference manual. Technical Report. [Google Scholar]
- Liu G.L., Mei H.W., Yu X.Q., Zou G.H., Liu H.Y., Hu S.P., Li M.S., Wu J.H., Chen L., Luo J. QTL analysis of panicle neck diameter, a trait highly correlated with panicle size, under well-watered and drought conditions in rice (Oryza sativa L.) Plant Sci. 2008;174:71–77. [Google Scholar]
- Liu Y.B., Lu S.M., Zhang J.F., Liu S., Lu Y.T. A xyloglucan endotransglucosylase/hydrolase involves in growth of primary root and alters the deposition of cellulose in Arabidopsis. Planta. 2009;226:1547–1560. doi: 10.1007/s00425-007-0591-2. [DOI] [PubMed] [Google Scholar]
- Liu G.F., Li M., Wen J., Du Y., Zhang Y.M. Functional mapping of quantitative trait loci associated with rice tillering. Mol. Genet. Genomics. 2010;284:263–271. doi: 10.1007/s00438-010-0566-z. [DOI] [PubMed] [Google Scholar]
- Liu G., Zhu H., Zhang G., Li L., Ye G. Dynamic analysis of QTLs on tiller number in rice (Oryza sativa L.) with single segment substitution lines. Theor. Appl. Genet. 2012;125:143–153. doi: 10.1007/s00122-012-1822-x. [DOI] [PubMed] [Google Scholar]
- Lu J., Sivamani E., Azhakanandam K., Samadder P., Li X., Qu R. Gene expression enhancement mediated by the 5′ UTR intron of the rice rubi3 gene varied remarkably among tissues in transgenic rice plants. Mol. Genet. Genomics. 2008;279:563–572. doi: 10.1007/s00438-008-0333-6. [DOI] [PubMed] [Google Scholar]
- Marathi B., Guleria S., Mohapatra T., Parsad R., Mariappan N., Kurungara V.K., Atwal S.S., Prabhu K.V., Singh N.K., Singh A.K. QTL analysis of novel genomic regions associated with yield and yield related traits in new plant type based recombinant inbred lines of rice (Oryza sativa L.) BMC Plant Biol. 2012;12:137. doi: 10.1186/1471-2229-12-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris A., Suslov D., Fry S.C., Verbelen J.P., Vissenberg K. Enzymic characterization of two recombinant xyloglucan endotransglucosylase/hydrolase (XTH) proteins of Arabidopsis and their effect on root growth and cell wall extension. J. Exp. Bot. 2009;60:3959–3972. doi: 10.1093/jxb/erp229. [DOI] [PubMed] [Google Scholar]
- Meuwissen T. Use of whole genome sequence data for QTL mapping and genomic selection. Proceedings of the 9th World Congress on Genetics Applied to Livestock Production; 2010. [Abstract ID 0018, ISBN 978-3-00-031608-1] [Google Scholar]
- Morinaka Y., Sakamoto T., Inukai Y., Agetsuma M., Kitano H., Ashikari M., Matsuoka M. Morphological alteration caused by brassinosteroid insensitivity increases the biomass and grain production of rice. Plant Physiol. 2006;141:924–931. doi: 10.1104/pp.106.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M.G., Thompson W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8:4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto E.C., Keller M.P., Broman A.F., Attie A.D., Jansen R.C., Broman K.W., Yandell B.S. Quantile-based permutation thresholds for quantitative trait loci hotspots. Genetics. 2012;191:1355–1365. doi: 10.1534/genetics.112.139451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C., Janet B. In vitro activities of four xyloglucan endotransglycosylases from Arabidopsis. Plant J. 1999;18:371–382. doi: 10.1046/j.1365-313x.1999.00459.x. [DOI] [PubMed] [Google Scholar]
- Qu Z., Li L., Luo J., Wang P., Yu S., Mou T., Zheng X., Hu Z. QTL mapping of combining ability and heterosis of agronomic traits in rice backcross recombinant inbred lines and hybrid crosses. PLoS One. 2012;7:e28463. doi: 10.1371/journal.pone.0028463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman L., Khanam M.S., Kho H.J. QTL analysis for yield related traits using populations derived from an indicajaponica hybrid in rice (Oryza sativa L.) Czech J. Genet. Plant Breed. 2008;3:93–104. [Google Scholar]
- Rose A.B., Elfersi T., Parra G., Korf I. Promoter-proximal introns in Arabidopsis thaliana are enriched in dispersed signals that elevate gene expression. Plant Cell. 2008;20:543–551. doi: 10.1105/tpc.107.057190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routaboul J.M., Dubos C., Beck G., Marquis C., Bidzinski P., Loudet O., Lepiniec L. Metabolite profiling and quantitative genetics of natural variation for flavonoids in Arabidopsis. J. Exp. Bot. 2012;63:3749–3764. doi: 10.1093/jxb/ers067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y. Genetic comparisons of chromosome 6 between wild and cultivated rice. Jpn. J. Breed. 1992;42:561–572. [Google Scholar]
- Suh H., Hue M. The segregation mode of plant height in the cross of rice varieties. XI. Linkage analysis of the semi-dwarfness of the rice variety ‘Tongil’. Korean J. Breed. 1978;10:1–6. [Google Scholar]
- Takagi H., Abe A., Yoshida K., Kosugi S., Natsume S., Mitsuoka C., Uemura A., Utsushi H., Tamiru M., Takuno S., et al. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013;74:174–183. doi: 10.1111/tpj.12105. [DOI] [PubMed] [Google Scholar]
- Tamura K., Nomura K., Oshima I., Namai H., Yano M., Sasaki T., Kikuchi F. Identification of restriction fragment length polymorphism markers tightly linked to a major photoperiod sensitivity gene, Se-1, and to a blast resistance gene, Pi-zt, in rice. SABRAO J. Breed. Genet. 1998;30:61–67. [Google Scholar]
- Temnykh S., DeClerck G., Lukashova A., Lipovich L., Cartinhour S., McCouch S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001;11:1441–1452. doi: 10.1101/gr.184001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisne S., Reymond M., Vile D., Fabre J., Dauzat M., Koornneef M., Granier C. Combined genetic and modeling approaches reveal that epidermal cell area and number in leaves are controlled by leaf and plant developmental processes in Arabidopsis. Plant Physiol. 2008;148:1117–1127. doi: 10.1104/pp.108.124271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomohiko T., Hirokazu T., Hirofumi U. Two independent and polarized processes of cell elongation regulate leaf blade expansion in Arabidopsis thaliana (L.) Heynh. Development. 1996;122:1589–1600. doi: 10.1242/dev.122.5.1589. [DOI] [PubMed] [Google Scholar]
- Trewavas A.J. The population/biodiversity paradox. Agricultural efficiency to save wilderness. Plant Physiol. 2001;125:174–179. doi: 10.1104/pp.125.1.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuinstra M., Ejeta G., Goldsbrough P. Heterogeneous inbred family (HIF) analysis: a method for developing near-isogenic lines that differ at quantitative trait loci. Theor. Appl. Genet. 1997;95:1005–1011. [Google Scholar]
- Wang L., Wang A., Huang X., Zhao Q., Dong G., Qian Q., Sang T., Han B. Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor. Appl. Genet. 2010;122:327–340. doi: 10.1007/s00122-010-1449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Zhou G., Yu H., Yu S. Fine mapping a major QTL for flag leaf size and yield-related traits in rice. Theor. Appl. Genet. 2011;123:1319–1330. doi: 10.1007/s00122-011-1669-6. [DOI] [PubMed] [Google Scholar]
- Wang P., Zhou G., Cui K., Li Z., Yu S. Clustered QTL for source leaf size and yield traits in rice (Oryza sativa L.) Mol. Breed. 2012;29:99–113. [Google Scholar]
- Wu C.Y., Trieu A., Radhakrishnan P., Kwok S.F., Harris S., Zhang K., Wang J., Wan J., Zhai H., Takatsuto S., et al. Brassinosteroids regulate grain filling in rice. Plant Cell. 2008;20:2130–2145. doi: 10.1105/tpc.107.055087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Liu X., Ge S., Jensen J.D., Hu F., Li X., Dong Y., Gutenkunst R.N., Fang L., Huang L., et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012;30:105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- Yan J.Q., Zhu J., He C.X., Benmousa M., Wu P. Quantitative trait loci analysis for the developmental behavior of tiller number in rice (Oryza sativa L.) Theor. Appl. Genet. 1998;97:267–274. [Google Scholar]
- Yoo S.C., Cho S.H., Zhang H., Paik H.C., Lee C.H., Li J., Yoo J.H., Lee B.W., Koh H.J., Seo H.S., et al. Quantitative trait loci associated with functional stay-green SNU-SG1 in rice. Mol Cells. 2007;24:83–94. [PubMed] [Google Scholar]
- Yoo S.C., Cho S.H., Sugimoto H., Li J., Kusumi K., Koh H.J., Iba K., Paek N.C. Rice virescent3 and stripe1 encoding the large and small subunits of ribonucleotide reductase are required for chloroplast biogenesis during early leaf development. Plant Physiol. 2009;150:388–401. doi: 10.1104/pp.109.136648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.S., Lin Z.W., Li H.X., Li X.J., Li J.Y., Wang Y.H., Zhang X., Zhu Z.F., Zhai W.X., Wang X.K., et al. TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J. 2007;52:891–898. doi: 10.1111/j.1365-313X.2007.03284.x. [DOI] [PubMed] [Google Scholar]
- Yue B., Xue W.Y., Luo L.J., Xing Y.Z. QTL analysis for flag leaf characteristics and their relationships with yield and yield traits in rice. Acta Genet. Sin. 2006;33:824–832. doi: 10.1016/S0379-4172(06)60116-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.