

Abstract
Differential transcription of the clusterin (CLU) gene yields two CLU isoforms, a nuclear form (nCLU) and a secretory form (sCLU), which play crucial roles in prostate tumorigenesis. Pro-apoptotic nCLU and anti-apoptotic sCLU have opposite effects and are differentially expressed in normal and cancer cells; however, their regulatory mechanisms at the transcriptional level are not yet known. Here, we examined the transcriptional regulation of nCLU in response to hypoxia. We identified three putative hypoxia response elements (HREs) in the human CLU promoter between positions −806 and +51 bp. Using a luciferase reporter, electrophoretic gel mobility shift, and chromatin immunoprecipitation assays, we further showed that hypoxia-inducible factor-1α (HIF-1α) bound directly to these sites and activated transcription. Exposure to the hypoxiamimetic compound CoCl2, incubation under 1% O2 conditions, or overexpression of HIF-1α enhanced nCLU expression and induced apoptosis in human prostate cancer PC3M cells. However, LNCaP prostate cancer cells were resistant to hypoxia-induced cell death. Methylation-specific PCR analysis revealed that the CLU promoter in PC3M cells was not methylated; in contrast, the CLU promoter in LNCap cells was methylated. Co-treatment of LNCaP cells with CoCl2 and a demethylating agent promoted apoptotic cell death through the induction of nCLU. We conclude that nCLU expression is regulated by direct binding of HIF-1α to HRE sites and is epigenetically controlled by methylation of its promoter region.
Keywords: HIF-1α, hypoxia response element, nuclear clusterin, promoter methylation, prostate cancer
INTRODUCTION
Clusterin (CLU), a glycoprotein also known as apolipoprotein J, is ubiquitously expressed in various tissues and is upregulated following exposure to cytotoxic stimuli (de Silva et al., 1990; Gleave and Miyake, 2005; Pucci et al., 2004; Trougakos and Gonos, 2002; Trougakos et al., 2004). The single CLU gene produces two alternatively spliced isoforms - nuclear CLU (nCLU) and secretory CLU (sCLU) - with differrent subcellular localizations and biological functions (Rosenberg and Silkensen, 1995; Shannan et al., 2006; Trougakos and Gonos, 2002; Wilson and Easterbrook-Smith, 2000). Each isoform contains a unique exon 1 fragment, but both share the remaining sequences (from exon 2 to 9). nCLU is detected as a ∼55 kDa glycosylated protein in the nucleus and induces apoptotic cell death, whereas the mature form of sCLU is a disulfide-linked heterodimeric protein (75–80 kDa) targeted for secretion. In cancer cells, sCLU exerts an anti-apoptotic function and its expression is elevated upon metastasis (Andersen et al., 2007; Bettuzzi et al., 2002; Leskov et al., 2003; Schepeler et al., 2007). However, no definitive mechanism has been proposed to account for the differential expression of these isoforms. Understanding how the expression of these isoforms is regulated may provide new strategies for the diagnosis, prevention, and treatment of cancers.
Recent reports have shown that epigenetic modifications of the CLU promoter are involved in tumorigenesis (Lund et al., 2006; Serrano et al., 2009). It has been shown that the CLU promoter is methylated in LNCaP and TRAMP C2 cells in association with downregulation of CLU expression (Rauhala et al., 2008). In contrast, the CLU promoter is reported to be unmethylated in PC3 and DU145 cells (Rauhala et al., 2008; Sala et al., 2009). Epigenetic regulation of the CLU gene appears to be important in tumor development and progression, particularly in prostate cancer.
Hypoxia, a characteristic feature of several pathological states, including cancer (Semenza, 1998), induces stabilization of hypoxia-inducible factor-1α (HIF-1α), which transcriptionally activates various genes involved in anaerobic metabolism, angiogenesis, and cell survival (Otrock et al., 2009). A recent report also showed that CLU is induced by hypoxia in human tendinopathy at both mRNA and protein levels (Millar et al., 2012), suggesting that hypoxia may directly regulate the transcription of CLU through a hypoxia-induced signaling pathway. Regulation of promoter activity in hypoxia occurs through the interaction of HIF-1α with a hypoxia response element (HRE) (Semenza, 2001). Here, we identified three putative HRE sites (A/GCGTG) in the CLU promoter using a bioinformatics analysis and demonstrated that HIF-1α bound to these sites and activated nCLU expression. Moreover, we found that nCLU expression is epigenetically regulated, suggesting that controlling methylation of the CLU promoter would be an effective approach for inducing apoptotic cell death and thus might be a good strategy for treating cancers.
MATERIALS AND METHODS
Cell culture
The human prostate cancer cell lines, PC3M and LNCaP, were obtained from the American Type Culture Collection (USA) and were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (HyClone, Thermo, USA). Cells were maintained in a humidified 5% CO2 incubator at 37°C. For low oxygen (O2) treatment, cells were cultured in a hypoxic incubator (Sanyo Electron Corporation) adjusted to 1% O2 for 6, 9, or 24 h.
Cell viability assay
Cell viability was measured using the 3(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. MTT solution (2 mg/ml) was added to each well and incubated with cells at 37°C for 4 h. The resulting formazan crystals were dissolved in dimethyl sulfoxide, and absorbance was measured at 570 nm using a microplate reader (Tecan, Switzerland).
Immunocytochemistry
Cells were fixed in 4% formaldehyde and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS). Fixed cells were stained with primary goat anti-CLU antibody (1:250; Santa Cruz Biotechnology, USA) for 2 h, followed by incubation with Alexa Fluor488-conjugated anti-goat IgG (Invitrogen, USA) in the dark for 1 h at room temperature. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays were performed using an in situ Cell Death Detection Kit (Roche, Germany), according to the manufacturer’s protocol. Cells were mounted using ProLong Gold antifade reagent containing the nuclear stain 4′,6-diamidino-2-pheny-lindole (DAPI; Invitrogen, USA).
Immunoblotting
Cells were homogenized in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40) and Protease Inhibitor Cocktail (Sigma-Aldrich, USA). Cell lysates containing equal amounts of total proteins were electrophoresed and transferred to nitrocellulose membranes. Blots were probed with anti-HIF-1α, anti-vascular endothelial growth factor (VEGF), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibodies (Cell Signaling, USA); and anti-CLU and anti-β-actin antibodies (Santa Cruz Biotechnology). Immunoreactive antigens were detected using an Enhanced Chemiluminescence Detection Kit (Amersham Bioscience, USA). Images were analyzed using the ImageJ program.
Quantitative polymerase chain reaction
Total RNA was extracted using the RNeasy Mini kit (Qiagen, Germany) and converted into cDNA with the Reverse Transcription System (Promega, USA) according to the manufacturer’s protocol. Quantitative polymerase chain reaction (PCR) was performed on a LightCycler 480 Real-Time PCR System using Power SYBR Green PCR Master Mix (Roche, Switzerland). Relative mRNA levels were normalized to those of β-actin mRNA. The primers used in these experiments have been described in previous reports (Chen et al., 2009; Cochrane et al., 2007; Kase et al., 2010).
Plasmid construction
The CLU upstream promoter region was subcloned by PCR using genomic DNA extracted from PC3M cells as a template. The primers used for PCR were as follows: −806 KpnI forward, 5′-GAT AGG TAC CGA TTT CCT AAC TGG GAA GGC TC-3′; −480 KpnI forward, 5′-GAT AGG TAC CCG TTT CAA TAC TTC CTG TGC TCT-3′; −125 KpnI forward, 5′-GAT AGG TAC CCT TCC AGA AAG CTC CCC T-3′; −76 KpnI forward, 5′-GAT AGG TAC CAT GCA GGT TTG CAG CCA G-3′; and +51 BglII reverse, 5′-TCG CAG ATC TGT CAG CGG CAC CC-3′. The restriction sites are underlined. PCR products were digested with KpnI and BglII restriction enzymes and inserted into the pGL3 basic vector (Promega). HRE mutants were generated using site-directed mutagenesis. All constructs were confirmed by direct sequencing (Bioneer, Korea).
Luciferase assay
Cells were transfected using Lipofectamine LTX (Invitrogen), according to the manufacturer’s instructions. After 24 h, cells were washed with cold PBS and lysed in lysis buffer provided in the Luciferase Assay System (Promega). Transfection efficiency was normalized using the β-Galactosidase Enzyme Assay System (Promega), and luminescence was measured using a microplate reader (Tecan, Switzerland).
Electrophoretic gel mobility shift assay
Electrophoretic mobility shift assays (EMSAs) were performed using a DIG Gel Shift Kit (Roche), as specified by the manufacturer. The sequences of the three oligonucleotide probes containing HREs were as follows: HRE1 probe, 5′-CAT TCT TTG GGC GTG AGT CAT GCA G-3′ (−95 to −71); HRE2 probe, 5′-CGG GCT AAC CGC GTG AGA GGG GCT C-3′ (−301 to −277); and HRE3 probe, 5′-ATT AAA CCA AAC GTG GAT CTG CAA G-3′ (−714 to −690). The VEGF promoter probe was used as described previously (Chen et al., 2009). The HRE binding site, CGTG, was mutated to TCGG for each probe. For competition assays, unlabeled and mutant probes were added to the reaction mixture prior to addition of the labeled probe. The HIF-1α binding complex was electrophoresed on a 6% native polyacrylamide gel, transferred to a positively charged nylon membrane, and incubated with an alkaline phosphatase-conjugated anti-digoxigen antibody. Associated alkaline phosphatase activity was subsequently detected by incubation with the chemiluminescent substrate CSPD.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Park et al., 2007) using anti-HIF-1α antibodies. The following primer pairs were used for quantitative PCR: ChIP HRE1, 5′-CCC AGC CCG GTG CTG CAC CG-3′ (forward) and 5′-GGG CAC TGG GAG GCG CCG TA-3′ (reverse); ChIP HRE2, 5′-TTT AAA TGG GTC AAG AGA AGT GG-3′ (forward) and 5′-CAG TAG GGA AGA GCC TGA ACT C-3′ (reverse); ChIP HRE3, 5′- CTA ACT GGG AAG GCT CAG GGC-3′ (forward) and 5′-ATC TTT TTG GAA CCC AAC CAA GC-3′ (reverse).
Methylation analysis
Genomic DNA was obtained using an i-Genomic DNA Extraction Kit (Intron Biotechnology, Korea). Methylation analysis of the CLU promoter was performed by detecting restriction enzyme-sensitive sites using a PCR-based protocol, as previously described (Serrano et al., 2009). Briefly, DNA from prostate cancer cells was digested with the methylation-sen-sitive restriction enzyme, KspI, which acts on a specific sequence (CCGCGG) located in the CLU promoter. Digested genomic DNA was used as the template for PCR. Amplified products were analyzed using gel electrophoresis and ethidium bromide staining, followed by imaging with the Gel Doc EQ System (Bio-Rad Laboratories, USA).
Statistical analysis
Results are representative of three independent values presented as means ± standard deviation (SD). Significances of differences were expressed as P < 0.001 (***), P < 0.005 (**), or P < 0.01 (*) compared to controls.
RESULTS
Cobalt chloride induces apoptosis
To examine whether hypoxic conditions are cytotoxic towards PC3M prostate cancer cells, we treated cells with 200 or 400 μM cobalt chloride (CoCl2), a hypoxia-mimetic agent, for 3, 6 or 9 h, and determined cell viability relative to that of untreated cells incubated for the same times. CoCl2 induced a concentration- and time-dependent reduction in cell viability, as shown in Fig. 1A. To examine CoCl2-induced apoptosis and relate it with CLU expression in these cells, we performed TUNEL assays and immunofluorescence staining for CLU. CoCl2-treated cells showed intense nuclear TUNEL staining, which, interestingly, was associated with enhanced nuclear expression of nClu (Fig. 1B).
Fig. 1.
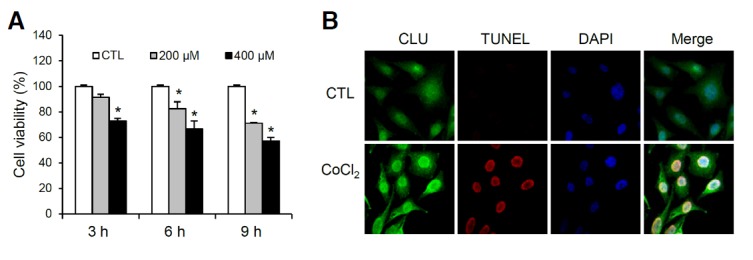
The hypoxia-mimetic compound, CoCl2, induces apoptosis in PC3M cells. (A) Viability of PC3M cells in response to CoCl2 was measured by MTT assay. PC3M cells were exposed to 0, 200, and 400 μM CoCl2 for 3, 6 and 9 h; cell viability is presented relative to controls as a percentage. (B) Immunofluorescence images of PC3M cells showing CLU (green), TUNEL (red), and DAPI (blue) staining. Cells were treated with vehicle or 400 μM CoCl2 for 6 h. *P < 0.01, compared to controls.
CoCl2 promotes the expression of HIF-1α target genes and nCLU
To further examine whether hypoxic conditions affect CLU expression in PC3M cells, we treated cells with CoCl2 and measured CLU mRNA and protein levels. CoCl2 treatment is known to stabilize HIF-1α protein and promote expression of its target genes. Using real-time PCR analyses, we confirmed an increase in the mRNA levels of HIF-1α target genes, including VEGF and GAPDH. CoCl2 treatment induced a concentration-and time-dependent increase (2-3-fold) in nCLU mRNA levels (Fig. 2A). We also examined changes in the protein levels of HIF-1α and its known target genes, as well as nCLU, by Western blot analysis. As expected, protein levels of HIF-1α, VEGF, and GADH were increased by CoCl2 treatment. nCLU, detected as a 55 kDa band, was also increased by CoCl2 treatment (Fig. 2B). These results suggest that treatment of PC3M cells with CoCl2 reduces cell viability through increased expression of HIF-1α-responsive genes, including nCLU, which is a known pro-apoptotic protein. To demonstrate that HIF-1α is directly responsible for CLU gene expression, we also transiently transfected PC3M cells with a green fluorescent protein (GFP)-tagged HIF-1α expression plasmid. Overexpression of GFP-tagged HIF-1α, confirmed by fluorescence microscopy, induced an increase in the levels of nCLU mRNA and protein (Figs. 2C and 2D), supporting the interpretation that HIF-1α directly promotes the expression of nCLU.
Fig. 2.
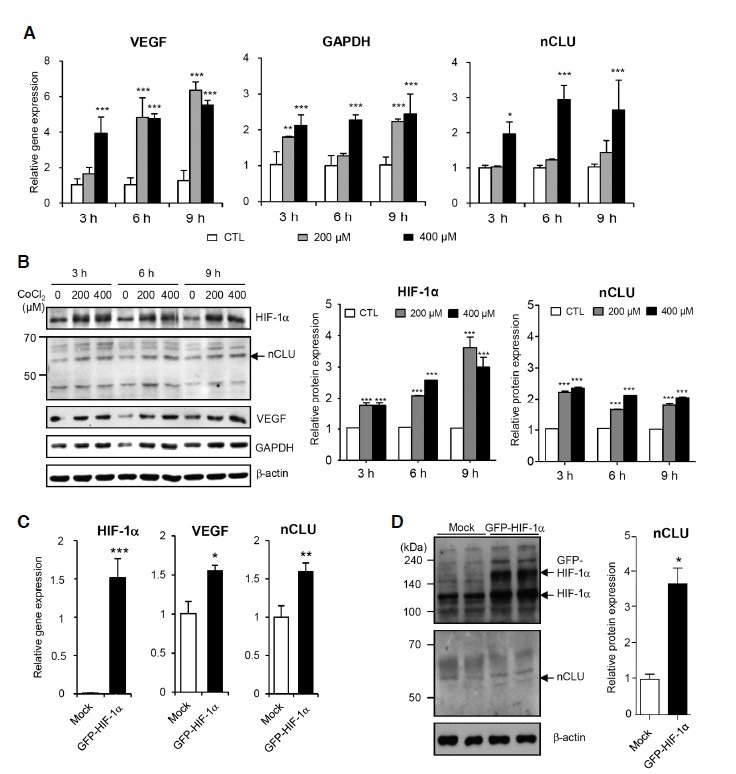
Treatment with CoCl2 and overexpression of HIF-1α induce nCLU expression in PC3M cells. (A, B) PC3M cells were treated with the indicated concentration of CoCl2 for the times shown. HIF-1α mRNA (A) and protein (B) were detected by real-time PCR and Western blotting, and quantified. (C, D) PC3M cells were transiently transfected with a GFP-tagged human HIF-1α expression vector. The mRNA (C) and protein (D) were detected by real-time PCR and Western blotting, and quantified. Relative levels were normalized to that of β-actin. ***P < 0.001, **P < 0.005, *P < 0.01, compared to controls.
Differential regulation of nCLU expression in PC3M and LNCaP cells following exposure to low O2
To monitor transcriptional regulation of nCLU in response to genuine hypoxia, we exposed cells to 1% O2 for up to 24 h and analyzed the expression levels of HIF-1α and nCLU by real-time PCR and Western blot analysis (Fig. 3). To assess the role of differential epigenic modification in regulating CLU expression, we compared the two prostate cancer cell lines, PC3M and LNCaP, which differ with respect to CLU promoter methylation. In both cell lines, 1% O2 maximally induced HIF-1α after 6 h; however, nCLU expression was induced in PC3M, but not in LNCaP cells, at both mRNA (Fig. 3A) and protein levels (Fig. 3B). Moreover, hypoxia induced a more significant decrease in cell viability in PC3M than in LNCaP cells (Fig. 3C). These results suggest that hypoxia induces nCLU expression and subsequent cell death and that this regulation may be epigenetically controlled, as previously reported (Rauhala et al., 2008; Sala et al., 2009).
Fig. 3.
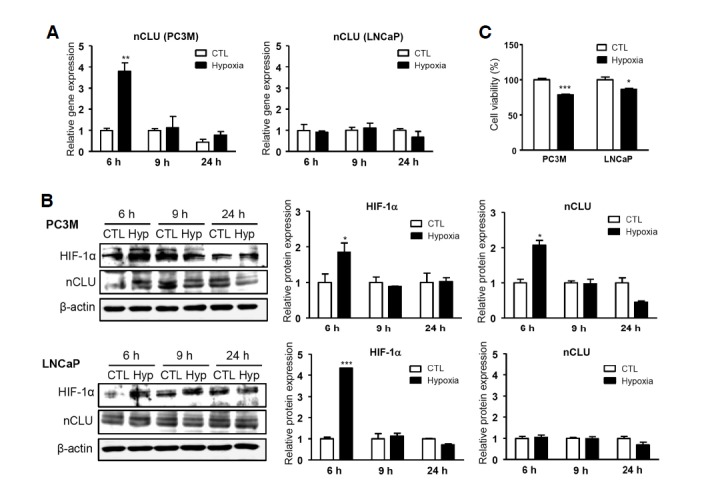
Effects of hypoxia on the differential regulation of nCLU expression and cell viability in PC3M and LNCaP cells. PC3M and LNCaP cells were exposed to 1% O2 for 6, 9, and 24 h. Expression of HIF-1α and nCLU were analyzed by real-time PCR (A) and Western blotting (B). (C) Cell viability was measured by MTT assay after exposure of cells to 1% O2 for 24 h. ***P < 0.001, **P < 0.005, *P < 0.01, compared to controls.
Putative HRE sites identified in the human CLU promoter function in the transcriptional regulation of CLU
An analysis of the genomic sequences of human CLU identified three putative HRE sites in the promoter region (Fig. 4A). Based on the locations of HRE sites, the sequences from −806 to +51 relative to the start site of HIF-1α were cloned into the promoter region of a luciferase reporter plasmid. The functionality of each HRE site was assessed by deleting multiple or individual HRE sites (Fig. 4B). Luciferase activity was determined in the presence or absence of 400 μM CoCl2 as well as under conditions of HIF-1α overexpression. The transcriptional activity of constructs containing HRE1, HRE1 and HRE2, or all three HRE sites was significantly increased under hypoxia-mimetic (CoCl2 treatment) conditions (Fig. 4C) and with transient HIF-1α overexpression (Fig. 4D). To further confirm the respective activity of the three HRE sites, we generated mutant reporter constructs by site-directed mutagenesis (Fig. 4E). All three HRE mutants exhibited decreased luciferase activity (Fig. 4F), indicating that all HRE sites in the promoter contribute to the transcriptional activation of CLU.
Fig. 4.
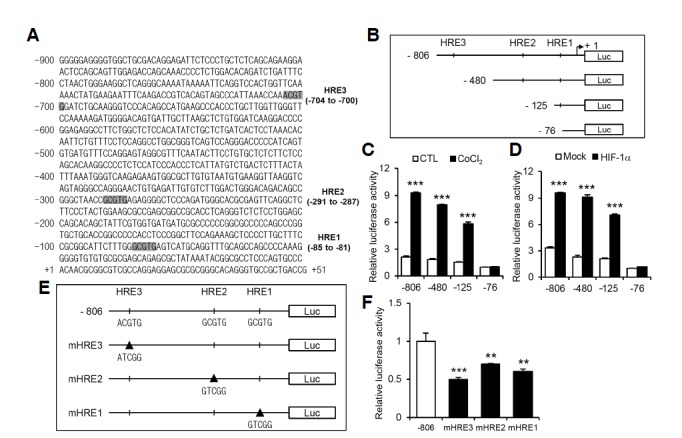
Putative HRE sites are identified in the CLU promoter and mediate luciferase reporter activity. (A) Three potential HRE sites (HRE1, -2, and -3), starting at positions -81, -287 and -700 bp, respectively, in the human CLU promoter were identified by sequence analysis. (B) Reporter gene constructs are denoted −806 Luc (−806 to +51), which contains all three HRE sites; −480 Luc (−480 to +51), which contains HRE1 and HRE2; −125 Luc (−125 to +51), which contains HRE1 only; and −76 Luc (−76 to +51), which contains no HREs. (C) HEK 293T cells were transfected with each reporter construct and stimulated with 400 μM CoCl2 for 6 h. (D) Cells were co-transfected with each reporter construct and a HIF-1α expression vector. (E, F) Mutant luciferase constructs were designed for each of the three HRE sites (mHRE1, -2, and -3) by site-direct mutagenesis using the −806 Luc reporter plasmid as a backbone. The relative luciferase activity was measured and normalized to the enzymatic activity of β-galactosidase. ***P < 0.001, **P < 0.005, *P < 0.01, compared to controls.
The three HRE sites in the CLU promoter bind HIF-1α
To demonstrate direct binding of HIF-1α to the CLU promoter, we performed EMSAs using oligonucleotide probes containing each HRE consensus sequence (Fig. 5A). Binding of HIF-1α to individual HRE sites was detected in reactions containing each labeled probe and nuclear proteins. In competition experiments, gel shifts were suppressed by excess unlabeled wild-type probe, but were not out-competed by mutant probes. Similar results were obtained in positive control EMSAs performed using a nucleotide probe derived from the VEGF promoter, which is known to bind HIF-1α. To further confirm the specificity of HIF-1α binding, antibody supershift assays were performed using a HIF-1α antibody. We observed broad and dense bands following the addition of HIF-1α antibody rather than a typical supershifted band. In addition, the HIF-1α antibody failed to alter band intensity at the HRE2 site compared with that at other HRE sites, indicating that this antibody may be not suitable for this assay. Despite these technical limitations, the observations that excess wild-type HRE-containing DNA probes, but not HRE-specific mutant probes, inhibited complex formation between wild-type probes and a nuclear factor in EMSAs, and an anti-HIF-1α antibody generated a supershifted band (albeit atypical) in these assays suggest that the nuclear factor that binds to HRE sites in the CLU promoter is HIF-1α.
Fig. 5.
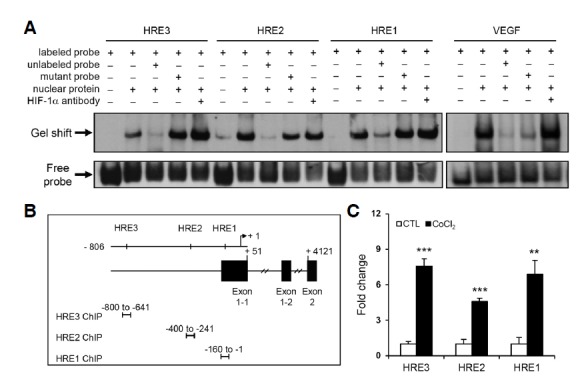
The three putative HRE sites in the CLU promoter bind to HIF-1α. (A) EMSAs were performed using DIG-labeled oligonucleotide probes containing consensus HRE sites. The labeled probe was incubated with ∼25–50 μg of nuclear extract; unlabeled probes were used as competitors. An oligonucleotide probe derived from the VEGF promoter was used as a positive control. (B) A schematic drawing of HRE sites and locations of PCR primer pairs for each HRE site-specific ChIP analysis. (C) PC3M cells were treated with 400 μM CoCl2 for 6 h, and nuclear fractions were immunoprecipitated with IgG or anti-HIF-1α antibody. Extracted chromatin fragments were analyzed by real-time PCR using the indicated primer sets to amplify sequences corresponding to each HRE site. ***P < 0.001, **P < 0.005, *P < 0.01, compared to controls.
ChIP assays were further performed to demonstrate direct binding of HIF-1α to endogenous HRE sites in the CLU promoter in intact cells. The locations of PCR primers specific to each HRE site are indicated in Fig. 5B. Chromatin immunoprecipitates recruited by the HIF-1α antibody were amplified with each primer sets and their products were quantified (Fig. 5C). Consistent with the direct regulation of CLU transcriptional activity by HIF-1α binding, our ChIP data showed that HIF-1α bound to endogenous HRE sites under conditions of CoCl2-stimulated hypoxia.
Demethylation of the CLU promoter in LNCaP cells enhances nCLU expression and induces apoptotic cell death
The methylation status of the CLU promoter in the prostate cancer cell lines, PC3M, DU145 and LNCaP, was established using a methylation-specific PCR-based approach that detects restriction-sensitive sites. In PC3M and DU145 cell lines, the CLU promoter was found to be unmethylated, as evidenced by the presence of a single specific PCR band at the bottom. However, LNCaP cells exhibited two PCR bands, indicative of CLU promoter methylation (Fig. 6A). If methylation plays a role in nCLU silencing, demethylation of the CLU promoter should result in reactivation of gene expression. Thus, we examined the effect of the demethylating agent, 5-aza-2′-deoxycytidine (AZA), on nCLU expression and subsequent cell death. LNCaP exhibited no nCLU induction in response to treatment with CoCl2 or exposure to 1% O2 alone and were resistant to the cytotoxic effects of hypoxic stress compared to PC3M cells (Figs. 3C and 6B). However, treatment with 3 μM AZA for 3 days followed by treatment with 400 μM CoCl2 for 6 h led to enhanced expression of nCLU mRNA (Fig. 6C) and protein (Fig. 6D). Moreover, co-treatment with AZA and CoCl2 led to a decrease in the viability of LNCaP cells (Fig. 6E). CLU immunostaining and TUNEL assays demonstrated that LNCaP cells treated with AZA and CoCl2 showed a dramatic induction of CLU expression and these CLU-expressing cells overlapped with TUNEL-positive cells (Fig. 6F). Collectively, these data indicate that demethylation of the CLU promoter reactivates nCLU expression and promotes apoptotic cell death.
Fig. 6.
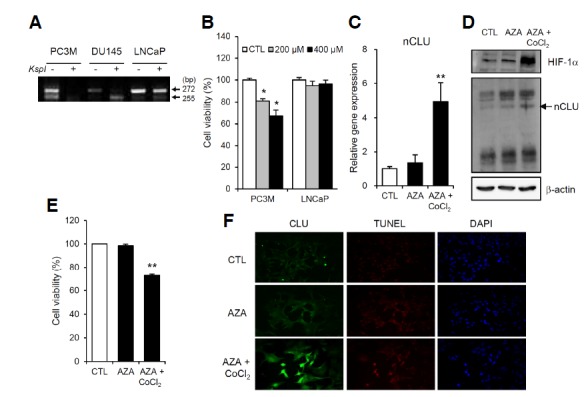
Demethylation of the CLU promoter in LNCaP cells enables CoCl2-induced nCLU expression and apoptotic cell death. (A) Genomic DNA from the prostate cell lines, PC3M, DU145 and LNCaP, was digested with the methylation-sensitive restriction enzyme KspI. Two bands, at 272 and 155 bp, were observed after KspI treatment in LNCaP cells, whereas only a single band at 155 bp was detected in PC3M and DU145 cells. (B) The viability of LNCaP cells was sustained after treatment with CoCl2 treatment compared to that of PC3M cells. (C, D) Treatment with 3 μM AZA for 3 days followed by exposure to 400 μM CoCl2 for 6 h enhanced the mRNA and protein levels of nCLU. (E, F) Co-treatment with AZA and CoCl2 decreased cell viability (%) and induced strong nCLU and TUNEL staining in LNCaP cells. **P < 0.005, *P < 0.01, compared to controls.
DISCUSSION
The present study showed that nCLU expression is transcriptionally regulated by HIF-1α. Three putative HRE sites in the CLU promoter were identified and shown to be functional in luciferase reporter, EMSA, and ChIP assays. Treatment of PC3M cells with 400 μM CoCl2 for 6 h was sufficient to stabilize HIF-1α, as previously reported (Piret et al., 2002), and enhance expression of its downstream target genes, VEGF and GAPDH (Forsythe et al., 1996; Higashimura et al., 2011; Yamaji et al., 2003). We additionally observed an increase in nCLU expression, providing support for nCLU as a HIF-1α target gene.
CoCl2 has been proposed to act by depleting intracellular ascorbate, which, by itself, is sufficient for the inactivation of HIF prolyl hydroxylases and the induction of hypoxia-like stress in cells (Salnikow et al., 2004). It has also been shown that CLU is induced in CoCl2-treated, but not in low-oxygenated, human diploid fibroblasts (HDFs), suggesting that distinct cellular components of HDFs may respond differentially to hypoxic stress (Poulios et al., 2006). Here, we administered short-term, low-dose CoCl2 as an HIF-1α-inducing stimulus to avoid the side effect of higher concentrations of CoCl2 or longer exposure regimens. For physiological relevance, we further performed experiments under low oxygen (1% O2) and established nCLU as a truly hypoxia-responsive gene.
Recent reports have shown that CLU overexpression increases drug sensitivity under hypoxic conditions by increasing apoptosis in colorectal cancer (Kevans et al., 2012). Diverse cross-talk between CLU and hypoxia suggests that regulation of CLU expression is important for tumor formation and cancer progression, although a direct relationship between the two has not yet been investigated. To our knowledge, this is the first study to identify HRE sites in the CLU promoter and demonstrate that they directly bind HIF-1α. Although the full-length promoter spans ∼1,500–2,000 bp of the 5′ regulatory region, we cloned only the sequence from −806 to +51 bp for our luciferase constructs because it contains the clusterin response element (CRE), which is critical for CLU induction, as reported in hepatocellular carcinomas (Liao et al., 2009). The functional HRE sites responsible for hypoxia-induced apoptotic cell death were found within this region.
Our study clearly showed the epigenetic regulation of the CLU promoter by methylation, as previously described (Rauhala et al., 2008; Sala et al., 2009). Since the CpG islands in the CLU promoter in PC3M and LNCaP cells have been reported to be differentially methylated, we selected these two cell lines for comparing the levels of nCLU expression and apoptosis. The major difference in methylation is found between −534 and −99 bp within the CLU promoter (Rosemblit and Chen, 1994). Serrano et al. (2009) suggested that the methylation position most sensitive to the KspI enzyme (−100) is of particular interest, because it is close to the TAATA box and is the target sequence for the transcription factor, AP1. Moreover, HRE1 (−85 to −81) and HRE2 (−291 to −287) sites identified in this study are also in close proximity to this site. Thus, it is reasonable to speculate that DNA sequences capable of being methylated, particularly those proximal to the HRE sites, are critical for epigenetic modification-dependent repression of HIF-1α-mediated transcription, which may be an initial step towards tumorigenesis in early prostate cancers.
In this study, we demonstrated that HIF-1α is a major HRE-binding factor in the CLU promoter. However, it should be noted that there are two isoforms of HIF-α - HIF-1α and HIF-2α - that are highly conserved with respect to protein structure; both are also stabilized by hypoxia and bind to the same HRE sites. However, HIF-2α has a distinct, tissue-specific expression pattern and binding partners (Loboda et al., 2010). HIF-1α is expressed ubiquitously, whereas HIF-2α expression is limited to the endothelium, kidney, lung, heart, and small intestine (Wiesener et al., 2003). In neuroblastomas, HIF-2α is known to be associated with a more aggressive metastatic phenotype (Holmquist-Mengelbier et al., 2006). Since our study was performed in prostate cancer cells and focused on the apoptotic function of nCLU at the early stage of cancer, we investigated HIF-1α as the major transcriptional regulator of nCLU expression.
A previous study has reported that von Hippel-Lindau (VHL), a tumor-suppressor protein, regulates CLU expression in a HIF-independent manner in renal carcinomas (Nakamura et al., 2006). However, this study investigated the secretion of sCLU and provided no information about nCLU or methylation status of the promoter. Here, we clearly showed that nCLU expression is HIF-1α dependent and regulated by promoter methylation. As previously shown, the two CLU isoforms are present in various levels in different tissues (Bettuzzi, 2009; Rizzi and Bettuzzi, 2010; Rizzi et al., 2009), and we speculate that each isoform may be differentially expressed according to the abundance of tissue-specific transcription factors and their cis-regulatory elements on the promoter as well as by various epigenetic modifications.
We previously proposed that nCLU sequesters the anti-apoptotic Bcl-1 family member, Bcl-xL, via its putative BH3 motif and releases bound Bax, thereby promoting apoptosis accompanied by activation of caspase-3 and release of cytochrome c (Kim and Choi, 2011; Kim et al., 2011; 2012; Lee et al., 2011). These results suggest a novel, nCLU-mediated apoptosis mechanism in normal cells. We further propose here that nCLU expression may be silenced in the early stage of prostate cancers via promoter methylation, which facilitates the survival of cancer cells (Rizzi and Bettuzzi, 2009; 2010). A recent report showed that HIF-1α directly regulates BCL-xL transcription by binding to a HRE in the BCL-xL promoter in prostate cancer cells (Chen et al., 2009). This suggests that BCL-xL overexpression is an important mechanism by which HIF-1α protects prostate cancer cells from apoptosis. Thus, our results strongly support the concept that CLU reactivation through relief of methylation-mediated repression would be a good strategy for treating drug resistance.
We conclude that nCLU acts as a tumor-suppressor protein and that the methylation status of the CLU promoter could serve as a diagnostic indicator of the early stages of prostate cancer. Moreover, modulating CLU promoter methylation could be a promising therapeutic strategy for prostate cancer treatment.
Acknowledgments
We thank Dr. Jae Yong Park at Gyeongsang National University for providing the HIF-1α expression vector. This research was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea (No. 2005-0049415).
Note:
Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Andersen C.L., Schepeler T., Thorsen K., Birkenkamp-Demtroder K., Mansilla F., Aaltonen L.A., Laurberg S., Orntoft T.F. Clusterin expression in normal mucosa and colorectal cancer. Mol. Cell Proteomics. 2007;6:1039–1048. doi: 10.1074/mcp.M600261-MCP200. [DOI] [PubMed] [Google Scholar]
- Bettuzzi S. Chapter 1: Introduction. Adv. Cancer Res. 2009;104:1–8. doi: 10.1016/S0065-230X(09)04001-9. [DOI] [PubMed] [Google Scholar]
- Bettuzzi S., Scorcioni F., Astancolle S., Davalli P., Scaltriti M., Corti A. Clusterin (SGP-2) transient overexpression decreases proliferation rate of SV40-immortalized human prostate epithelial cells by slowing down cell cycle progression. Oncogene. 2002;21:4328–4334. doi: 10.1038/sj.onc.1205594. [DOI] [PubMed] [Google Scholar]
- Chen N., Chen X., Huang R., Zeng H., Gong J., Meng W., Lu Y., Zhao F., Wang L., Zhou Q. BCL-xL is a target gene regulated by hypoxia-inducible factor-1{ alpha} J. Biol. Chem. 2009;284:10004–10012. doi: 10.1074/jbc.M805997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane D.R., Wang Z., Muramaki M., Gleave M.E., Nelson C.C. Differential regulation of clusterin and its isoforms by androgens in prostate cells. J. Biol. Chem. 2007;282:2278–2287. doi: 10.1074/jbc.M608162200. [DOI] [PubMed] [Google Scholar]
- de Silva H.V., Harmony J.A., Stuart W.D., Gil C.M., Robbins J. Apolipoprotein J: structure and tissue distribution. Biochemistry. 1990;29:5380–5389. doi: 10.1021/bi00474a025. [DOI] [PubMed] [Google Scholar]
- Forsythe J.A., Jiang B.H., Iyer N.V., Agani F., Leung S.W., Koos R.D., Semenza G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave M., Miyake H. Use of antisense oligonucleotides targeting the cytoprotective gene, clusterin, to enhance androgen- and chemo-sensitivity in prostate cancer. World J. Urol. 2005;23:38–46. doi: 10.1007/s00345-004-0474-0. [DOI] [PubMed] [Google Scholar]
- Higashimura Y., Nakajima Y., Yamaji R., Harada N., Shibasaki F., Nakano Y., Inui H. Up-regulation of glyceraldehyde-3-phosphate dehydrogenase gene expression by HIF-1 activity depending on Sp1 in hypoxic breast cancer cells. Arch. Biochem. Biophys. 2011;509:1–8. doi: 10.1016/j.abb.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Holmquist-Mengelbier L., Fredlund E., Lofstedt T., Noguera R., Navarro S., Nilsson H., Pietras A., Vallon-Christersson J., Borg A., Gradin K., et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10:413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Kase S., He S., Sonoda S., Kitamura M., Spee C., Wawrousek E., Ryan S.J., Kannan R., Hinton D.R. alphaB-crystallin regulation of angiogenesis by modulation of VEGF. Blood. 2010;115:3398–3406. doi: 10.1182/blood-2009-01-197095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevans D., Gorman S., Tosetto M., Sheahan K., O’Donoghue D., Mulcahy H., O’Sullivan J. Clusterin and chemotherapy sensitivity under normoxic and graded hypoxic conditions in colorectal cancer. J. Gastrointest Cancer. 2012;43:305–313. doi: 10.1007/s12029-011-9277-x. [DOI] [PubMed] [Google Scholar]
- Kim N., Choi W.S. Proapoptotic role of nuclear clusterin in brain. Anat. Cell Biol. 2011;44:169–175. doi: 10.5115/acb.2011.44.3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N., Yoo J.C., Han J.Y., Hwang E.M., Kim Y.S., Jeong E.Y., Sun C.H., Yi G.S., Roh G.S., Kim H.J., et al. Human nuclear clusterin mediates apoptosis by interacting with Bcl-XL through C-terminal coiled coil domain. J. Cell Physiol. 2011;227:1157–1167. doi: 10.1002/jcp.22836. [DOI] [PubMed] [Google Scholar]
- Kim N., Han J.Y., Roh G.S., Kim H.J., Kang S.S., Cho G.J., Park J.Y., Choi W.S. Nuclear clusterin is associated with neuronal apoptosis in the developing rat brain upon ethanol exposure. Alcohol Clin. Exp. Res. 2012;36:72–82. doi: 10.1111/j.1530-0277.2011.01588.x. [DOI] [PubMed] [Google Scholar]
- Lee D.H., Ha J.H., Kim Y., Bae K.H., Park J.Y., Choi W.S., Yoon H.S., Park S.G., Park B.C., Yi G.S., et al. Interaction of a putative BH3 domain of clusterin with anti-apoptotic Bcl-2 family proteins as revealed by NMR spectroscopy. Biochem. Biophys. Res. Commun. 2011;408:541–547. doi: 10.1016/j.bbrc.2011.04.054. [DOI] [PubMed] [Google Scholar]
- Leskov K.S., Klokov D.Y., Li J., Kinsella T.J., Boothman D.A. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 2003;278:11590–11600. doi: 10.1074/jbc.M209233200. [DOI] [PubMed] [Google Scholar]
- Liao F.T., Lee Y.J., Ko J.L., Tsai C.C., Tseng C.J., Sheu G.T. Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J. Gen. Virol. 2009;90:1124–1134. doi: 10.1099/vir.0.007211-0. [DOI] [PubMed] [Google Scholar]
- Loboda A., Jozkowicz A., Dulak J. HIF-1 and HIF-2 transcription factors--similar but not identical. Mol Cells. 2010;29:435–442. doi: 10.1007/s10059-010-0067-2. [DOI] [PubMed] [Google Scholar]
- Lund P., Weisshaupt K., Mikeska T., Jammas D., Chen X., Kuban R.J., Ungethum U., Krapfenbauer U., Herzel H.P., Schafer R., et al. Oncogenic HRAS suppresses clusterin expression through promoter hypermethylation. Oncogene. 2006;25:4890–4903. doi: 10.1038/sj.onc.1209502. [DOI] [PubMed] [Google Scholar]
- Millar N.L., Reilly J.H., Kerr S.C., Campbell A.L., Little K.J., Leach W.J., Rooney B.P., Murrell G.A., McInnes I.B. Hypoxia: a critical regulator of early human tendinopathy. Ann. Rheum. Dis. 2012;71:302–310. doi: 10.1136/ard.2011.154229. [DOI] [PubMed] [Google Scholar]
- Nakamura E., Abreu-e-Lima P., Awakura Y., Inoue T., Kamoto T., Ogawa O., Kotani H., Manabe T., Zhang G.J., Kondo K., et al. Clusterin is a secreted marker for a hypoxia-inducible factor-independent function of the von Hippel-Lindau tumor suppressor protein. Am. J. Pathol. 2006;168:574–584. doi: 10.2353/ajpath.2006.050867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otrock Z.K., Hatoum H.A., Awada A.H., Ishak R.S., Shamseddine A.I. Hypoxia-inducible factor in cancer angiogenesis: structure, regulation and clinical perspectives. Crit. Rev. Oncol. Hematol. 2009;70:93–102. doi: 10.1016/j.critrevonc.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Park S.Y., Kim J.B., Han Y.M. REST is a key regulator in brain-specific homeobox gene expression during neuronal differentiation. J. Neurochem. 2007;103:2565–2574. doi: 10.1111/j.1471-4159.2007.04947.x. [DOI] [PubMed] [Google Scholar]
- Piret J.P., Mottet D., Raes M., Michiels C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann. N Y Acad. Sci. 2002;973:443–447. doi: 10.1111/j.1749-6632.2002.tb04680.x. [DOI] [PubMed] [Google Scholar]
- Poulios E., Trougakos I.P., Gonos E.S. Comparative effects of hypoxia on normal and immortalized human diploid fibroblasts. Anticancer Res. 2006;26:2165–2168. [PubMed] [Google Scholar]
- Pucci S., Bonanno E., Pichiorri F., Angeloni C., Spagnoli L.G. Modulation of different clusterin isoforms in human colon tumorigenesis. Oncogene. 2004;23:2298–2304. doi: 10.1038/sj.onc.1207404. [DOI] [PubMed] [Google Scholar]
- Rauhala H.E., Porkka K.P., Saramaki O.R., Tammela T.L., Visakorpi T. Clusterin is epigenetically regulated in prostate cancer. Int. J Cancer. 2008;123:1601–1609. doi: 10.1002/ijc.23658. [DOI] [PubMed] [Google Scholar]
- Rizzi F., Bettuzzi S. Clusterin (CLU) and prostate cancer. Adv. Cancer Res. 2009;105:1–19. doi: 10.1016/S0065-230X(09)05001-5. [DOI] [PubMed] [Google Scholar]
- Rizzi F., Bettuzzi S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr. Relat Cancer. 2010;17:R1–17. doi: 10.1677/ERC-09-0140. [DOI] [PubMed] [Google Scholar]
- Rizzi F., Coletta M., Bettuzzi S. Chapter 2: Clusterin (CLU): From one gene and two transcripts to many proteins. Adv. Cancer Res. 2009;104:9–23. doi: 10.1016/S0065-230X(09)04002-0. [DOI] [PubMed] [Google Scholar]
- Rosemblit N., Chen C.L. Regulators for the rat clusterin gene: DNA methylation and cis-acting regulatory elements. J. Mol. Endocrinol. 1994;13:69–76. doi: 10.1677/jme.0.0130069. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.E., Silkensen J. Clusterin: physiologic and pathophysiologic considerations. Int. J. Biochem. Cell Biol. 1995;27:633–645. doi: 10.1016/1357-2725(95)00027-m. [DOI] [PubMed] [Google Scholar]
- Sala A., Bettuzzi S., Pucci S., Chayka O., Dews M., Thomas-Tikhonenko A. Regulation of CLU gene expression by oncogenes and epigenetic factors implications for tumorigenesis. Adv. Cancer Res. 2009;105:115–132. doi: 10.1016/S0065-230X(09)05007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K., Donald S.P., Bruick R.K., Zhitkovich A., Phang J.M., Kasprzak K.S. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J. Biol. Chem. 2004;279:40337–40344. doi: 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- Schepeler T., Mansilla F., Christensen L.L., Orntoft T.F., Andersen C.L. Clusterin expression can be modulated by changes in TCF1-mediated Wnt signaling. J. Mol. Signal. 2007;2:6. doi: 10.1186/1750-2187-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G.L. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998;8:588–594. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- Semenza G.L. HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- Serrano A., Redondo M., Tellez T., Castro-Vega I., Roldan M.J., Mendez R., Rueda A., Jimenez E. Regulation of clusterin expression in human cancer via DNA methylation. Tumour. Biol. 2009;30:286–291. doi: 10.1159/000259912. [DOI] [PubMed] [Google Scholar]
- Shannan B., Seifert M., Leskov K., Willis J., Boothman D., Tilgen W., Reichrath J. Challenge and promise: roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006;13:12–19. doi: 10.1038/sj.cdd.4401779. [DOI] [PubMed] [Google Scholar]
- Trougakos I.P., Gonos E.S. Clusterin/apolipoprotein J in human aging and cancer. Int. J. Biochem. Cell Biol. 2002;34:1430–1448. doi: 10.1016/s1357-2725(02)00041-9. [DOI] [PubMed] [Google Scholar]
- Trougakos I.P., So A., Jansen B., Gleave M.E., Gonos E.S. Silencing expression of the clusterin/apolipoprotein j gene in human cancer cells using small interfering RNA induces spontaneous apoptosis, reduced growth ability, and cell sensitization to genotoxic and oxidative stress. Cancer Res. 2004;64:1834–1842. doi: 10.1158/0008-5472.can-03-2664. [DOI] [PubMed] [Google Scholar]
- Wiesener M.S., Jurgensen J.S., Rosenberger C., Scholze C.K., Horstrup J.H., Warnecke C., Mandriota S., Bechmann I., Frei U.A., Pugh C.W., et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17:271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- Wilson M.R., Easterbrook-Smith S.B. Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 2000;25:95–98. doi: 10.1016/s0968-0004(99)01534-0. [DOI] [PubMed] [Google Scholar]
- Yamaji R., Fujita K., Takahashi S., Yoneda H., Nagao K., Masuda W., Naito M., Tsuruo T., Miyatake K., Inui H., et al. Hypoxia up-regulates glyceraldehyde-3-phosphate dehydrogenase in mouse brain capillary endothelial cells: involvement of Na+/Ca2+ exchanger. Biochim. Biophys. Acta. 2003;1593:269–276. doi: 10.1016/s0167-4889(02)00397-x. [DOI] [PubMed] [Google Scholar]