

Abstract
Cancer is unique amongst human diseases in that its cellular manifestations arise and evolve through the acquisition of somatic alterations in the genome. In particular, instability in the number and structure of chromosomes is a near-universal feature of the genomic alterations associated with epithelial cancers, and is triggered by the inactivation of tumour suppressor mechanisms that preserve chromosome integrity in normal cells. The nature of these mechanisms, and how their inactivation promotes carcinogenesis, remains enigmatic. I will review recent work from our laboratory on the tumour suppressor BRCA2 that addresses these issues, focusing on new insights into cancer pathogenesis and therapy that are emerging from improved understanding of the molecular basis of chromosomal instability in BRCA2-deficient cancer cells.
Keywords: BRCA2, cancer, chromosomal aberrations, DNA repair, genome instability, mitosis
INTRODUCTION
It has been clear for many years that somatic alterations of the genome occur at a high frequency in the majority of cells isolated from most common forms of cancer. Recent studies using next-generation sequencing (NGS) have provided a catalogue of such changes at much higher resolution than has previously been possible. Besides small sequence changes (such as point mutations, and small insertions or deletions of nucleotides), NGS studies highlight the abundance of chromosomal aberrations in cancer genomes. These chromosomal aberrations include different forms of structural abnormalities, such as gross chromosomal rearrangements (eg., large insertions or deletions, rearrangement between chromosomes, chromosome fragmentation etc) and regions of amplification, as well as numerical aberrations associated with the gain or loss of whole chromosomes (or aneuploidy). Neither the mechanisms underlying chromosomal instability in cancer cells, not its potential significance to carcinogenesis and cancer evolution, is well understood.
Inherited mutations affecting the BRCA2 tumour suppressor provide a powerful model in which to study these issues. BRCA2 was identified in the 1990s through the analysis of families at high risk from breast and ovarian cancer. It encodes a large protein, bearing little resemblance to molecules of known function, which is widely expressed in the dividing cells of many tissues, and localises to the cell nucleus. It has become clear over the past decade that BRCA2 plays an essential role in preserving the structural and numerical stability of chromosomes during cell division. This review will focus on recent work from our laboratory that provides insights into the pathogenesis and treatment of epithelial cancers associated with BRCA2 mutations.
GERMLINE BRCA2 MUTATIONS AND CANCER SUSCEPTIBILITY
Germline mutations affecting one allele of BRCA2 were first shown to confer susceptibility to breast and ovarian cancers with a high penetrance, which, by the age of 70 years approaches 40–50% for breast cancer, and 10–15% for ovarian. Individuals who carry BRCA2 mutations also have an increased risk of cancers of the male breast (∼75-fold relative risk), pancreas (4-8-fold), prostate (2-4-fold) or other tissues (Breast Cancer Linkage Consortium, 1999; Thompson and Easton, 2002). It has been proposed that germline heterozygosity for BRCA2 mutations suffices for cancer predisposition, and there is evidence that the second BRCA2 allele is inactivated by somatic alterations during tumour progression, consistent with the expectation for a classical ‘two hit’ tumour suppressor gene described by Knudson.
BRCA2 PERFORMS MULTIPLE CELLULAR FUNCTIONS IN TUMOUR SUPPRESSION
A few years after the discovery of the human BRCA2 gene, we reported that cells carrying a genetically engineered truncation in murine BRCA2 spontaneously develop instability in chromosome structure and number during division (Fig. 1). These aberrations offer important clues to the cellular functions of BRCA2 in tumour suppression. For instance, structural aberrations include many chromatid-type lesions, such as breaks that affect a single sister chromatid, as well as tri-radial and quadri-radial structures. Such aberrations are also diagnostic of Fanconi Anaemia, providing a first connection between this syndrome and cancers associated with BRCA2 deficiency (Patel et al., 1998). Similar abnormalities were afterwards shown to occur in murine cells lacking BRCA1 (Xu et al., 1999) and in cancer cells from BRCA mutation carriers (Gretarsdottir et al., 1998; Tirkkonen et al., 1997).
Fig. 1.
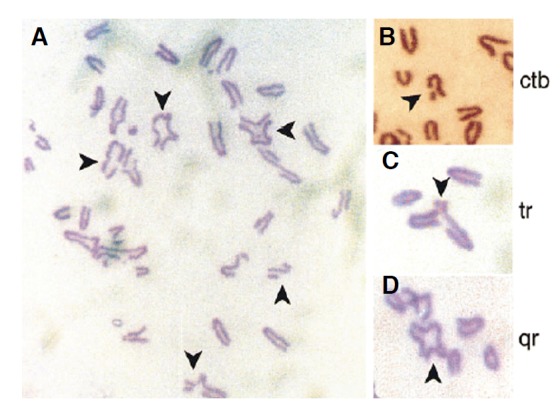
Structural chromosomal aberrations in metaphase spreads from murine embryo fibroblasts homozygous for a targeted truncation in Brca2. (A) shows a typical meta-phase spread, with arrows marking abnormal chromsomes. Chromatid-type aberrations enlarged in (B–D) show breaks affecting a single sister chromatid, tri-radial and quadri-radial chromosomes respectively. Reproduced with the permission of Elsevier from Patel et al. (1998).
Structural chromosomal aberrations following BRCA2 inactivation are believed to arise from its role in DNA repair by homologous recombination (HR). BRCA2 binds directly to the RAD51 recombination enzyme, which has a catalytic activity central to HR. RAD51 coats single-stranded DNA substrates to form a helical nucleoprotein filament that invades and pairs with the homologous DNA duplex, initiating strand exchange between the paired DNA molecules (reviewed in West, 2003). There is compelling evidence that BRCA2 works directly to regulate the availability and activity of RAD51 in these reactions, by controlling the strand-specificity of binding to single-stranded (ss) versus double-stranded (ds) DNA substrates. BRCA2 binds directly to RAD51 through evolutionarily conserved BRC repeat motifs. In human BRCA2, 8 BRC motifs are dispersed in an 1127-residue region encoded by exon 11, in which their spacing exhibits conservation amongst mammalian species. We showed (Carreira et al., 2009; Shivji et al., 2009) that the binding of BRCA2 BRC repeats to RAD51 suffices to promote the stability of RAD51-ssDNA binding, whilst inhibiting the binding of RAD51 to dsDNA substrates. During strand exchange reactions, when homologous ssDNA and dsDNA are both present, BRCA2 stimulates strand exchange by preventing premature and ultimately sterile engagement of RAD51 with dsDNA before formation of the ssDNA-RAD51 filament and the initiation of strand invasion and homologous pairing. Thus, BRCA2 targets RAD51 to ssDNA while inhibiting dsDNA binding and these contrasting activities together bolster one another to stimulate HR (Fig. 2).
Fig. 2.
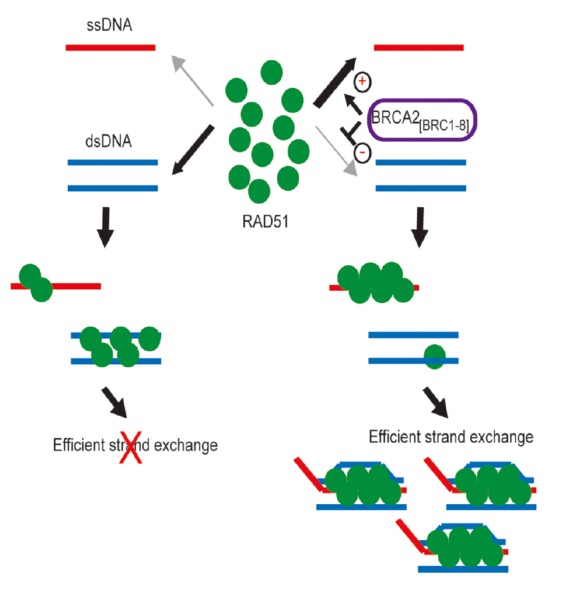
Opposing effects of BRCA2 BRC repeats on RAD51 binding to ssDNA versus dsDNA bolster one another to stimulate strand exchange. When BRCA2 BRC repeats are absent (left-hand side), ssDNA and dsDNA compete for RAD51 binding. Strand exchange is inefficient because premature, stable RAD51 assembly on dsDNA is futile. The BRC repeats of BRCA2 favour the preferential assembly of RAD51 onto ssDNA because their interaction with RAD51 promotes RAD51-ssDNA binding while suppressing RAD51-dsDNA binding (right-hand side). These opposing effects reinforce one another to guide stepwise progression through strand exchange. Reproduced from Shivji et al. (2009).
Another role of BRCA2 whose loss may trigger chromosomal breakage is its essential function in the stabilization of stalled DNA replication forks in the late S and G2 phases of the cell cycle. Our laboratory showed several years ago that replication intermediates formed at stalled forks were broken down in BRCA2-deficient cells into dsDNA breaks (Lomonosov et al., 2003), and this has been subsequently attributed to the inhibition of endonuclease activity at stalled forks in a manner that may be independent of RAD51-mediated DNA repair (Schlacher et al., 2011).
Inactivation of BRCA2 also causes aneuploidy. Murine BRCA2-deficient cells accumulate with > 4N DNA content (Patel et al., 1998), and aneuploidy is found in cancer cells from BRCA2 mutation carriers. The origin of these anomalies is less clear, but may possibly be explained by functions of BRCA2 in mitotic progression at the spindle assembly checkpoint during the transition from metaphase to anaphase (Choi et al., 2012), and/or in the defective completion of cell division by cytokinesis at the stage of daughter cell abscission (Daniels et al., 2004; Lee et al., 2011; Mondal et al., 2012). However, roles for BRCA2 in controlling mitotic entry following activation of the G2 checkpoint for DNA damage have also been proposed (Ayoub et al., 2009; Menzel et al., 2011), although it is currently unclear if their loss contributes to chromosomal instability in BRCA2-deficient cells.
Concerning the spindle assembly checkpoint, recent data suggest (Choi et al., 2012) that BRCA2 recruits the PCAF acetyltransferase to the kinetochores of metaphase chromosomes during mitosis, facilitating the acetylation by PCAF of the kinetochore protein BubR1, increasing its stability. Inactivation of BRCA2 decreases BubR1 acetylation and the level of BubR1 during mitosis, leading to weakened checkpoint activity and aneuploidy.
BRCA2 inactivation also causes an increase in the frequency of cells that fail to complete division, but instead form multi-nucleate structures with ≥ 4N DNA content (Daniels et al., 2004). Although cell cleavage by the actomyosin contractile ring can be initiated, and often reaches near-completion, in many divisions the abscission of the daughter cells does not proceed further. Indeed, BRCA2 is localized to the cytokinetic midbody by filamin A, where it recruits components of the ESCRT (endosomal sorting complex required for transport) protein complex implicated in cytokinetic abscission (Mondal et al., 2012). Moreover, the cytokinetic role of BRCA2 appears to be separable (Lee et al., 2011) from its function in RAD51 regulation during HR (Fig. 3). Thus, BRCA2 binds to the cytokinetic regulator HMG20b via the BRC repeat BRC5, which binds poorly to RAD51. Conversely, the BRC4 repeat binds strongly to RAD51, but not to HMG20b. In cells, BRC5 overexpression inhibits the BRCA2-HMG20b interaction, triggering defects in cytokinesis. In contrast, BRC4 inhibits the BRCA2-RAD51 interaction and the assembly of RAD51 at sites of DNA damage, but has no effect on cytokinesis.
Fig. 3.
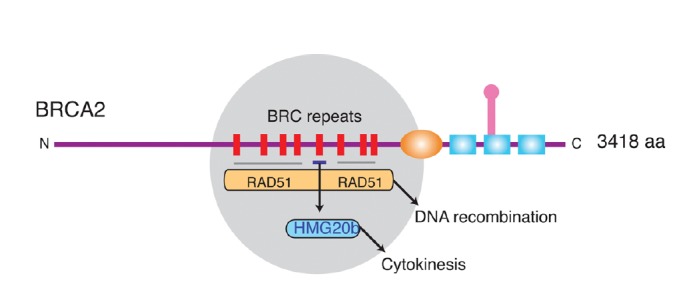
Divergent tumour-suppressive functions of the BRC repeats of BRCA2. The grey circle highlights in a schematic depiction of human BRCA2 the evolutionarily conserved region that contains 8 BRC repeats whose sequence and spacing is preserved amongst mammalian orthologues. Most BRC repeats, including BRC4, are known to bind and control the RAD51 recombinase in reactions that lead to homologous DNA recombination. BRC5 (but not BRC4) binds directly to HMG20b, and this interaction regulates an unrecognized function for HMG20b in the efficient completion of cell division by cytokinesis. Thus, divergent tumour suppressive functions maintaining chromosome segregation as well as chromosome structure may be mediated by the conserved BRC repeats of BRCA2. Reproduced from Lee et al. (2011).
Thus, in summary, multiple functions for BRCA2 in the preservation of chromosome stability may explain its tumour suppressive function. These include functions in the control of RAD51 during DNA repair by homologous recombination, in mitotic entry at the G2/M transition, and in mitotic progression through functions at the spindle assembly checkpoint and during cytokinesis. The loss of these functions may account for the observed aberrations in chromosome structure and number that occur when BRCA2 is inactivated in cancer cells. The occurrence of chromosomal instability, in turn, plausibly drives mutagenesis and neoplastic transformation in cells lacking BRCA2 (Venkitaraman, 2002; 2009).
A GENETICALLY ENGINEERED MOUSE MODEL (GEMM) FOR TISSUE-SPECIFIC CARCINOGENESIS ASSOCIATED WITH BRCA2 DEFICIENCY
Many genetically engineered mouse models (GEMMs) for the role of tumour suppressors in human carcinogenesis fail to recapitulate the complex combinations of germline and somatic alterations typically associated with human cancers, but instead, focus on the constitutive or conditional inactivation of one or both alleles of the tumour suppressor gene in different mouse tissues. In particular, human diseases associated with tumour suppressors such as BRCA2 involve germline BRCA2 alterations in all somatic tissues, with loss of the second allele expected only in the developing cancer cells, where they coexist with a number of additional oncogenic or tumour suppressive alterations. The failure of most available GEMMs to mimic these features means that they are frequently of limited value in modeling human carcinogenesis or cancer therapy.
To address this lacuna, we have created in our laboratory a GEMM for pancreatic cancer associated with inherited BRCA2 mutations (Skoulidis et al., 2010). Pancreatic ductal adenocarcinoma (PDAC) represents the fourth leading cause of cancer mortality worldwide, with an incidence of approximately 217,000 new cases each year associated with ∼213,000 deaths (Parkin et al., 2001). The most frequent genetic events underlying the initiation and progression of human pancreatic cancer (Hezel et al., 2006; Maitra and Hruban, 2008) include activating mutations in the KRAS proto-oncogene, in > 90% of PDAC cases (Caldas and Kern, 1995), besides mutations inactivating the TP53 gene in 50–75% of patients (Redston et al., 1994). Moreover, mutations inactivating BRCA2 are found in an estimated 5–20% of familial PDAC (Couch et al., 2007; Hahn et al., 2003). Conversely, germline inheritance of pathogenic BRCA2 mutations carries an ∼4-8-fold increased lifetime risk of developing PDAC (Breast Cancer Linkage Consortium, 1999).
In patients with germline mutations in BRCA2, all somatic tissues contain the mutant allele, whereas the second BRCA2 allele is wildtype (Wooster et al., 1994). Loss of this second, wild-type BRCA2 allele in nascent cancer cells (termed ‘loss of heterozygosity’ or LOH) is widely believed to be essential for carcinogenesis in these patients. The pancreatic cancer model developed in our laboratory (Skoulidis et al., 2010) recapitulates these events (Fig. 4). A truncated allele of murine Brca2 (Brca2Tr), resembling deleterious human germline mutations, which truncates the gene in an evolutionarily conserved and functionally critical region encoded by exon 11, is present in all somatic tissues (Friedman et al., 1998). The second Brca2 allele, Brca2F11 (Jonkers et al., 2001) can be conditionally disrupted to remove exon 11 by loxP-CRE recombination in the pancreas when PDX1-CRE is expressed there, mimicking LOH in human cancers from mutation carriers. Notably, tissue-specific activation of oncogenic KrasG12D is driven on a single allele by loxP-CRE mediated recombination in the pancreas, mimicking a genetic event that frequently triggers Kras activation in human cancers. In addition, in some strains, the mutant p53 allele mutant Tp53R270H can also be activated in the pancreas via CRE-mediated recombination. Thus, this mouse model not only faithfully recapitulates several of the key genetic alterations that have been implicated in PDAC pathogenesis, but also mimics the combination of mutant BRCA2 alleles found in the somatic tissues and cancer tissues from BRCA2 mutation carriers.
Unexpectedly, our studies reveal that BRCA2 heterozygosity is sufficient to promote pancreatic cancer development in mice and men; inactivation of the second wild-type allele by LOH is not necessary. Irrespective of p53 mutations, heterozygosity for Brca2Tr acts with KrasG12D to accelerate the progression and development of PDAC. Similarly, in a small number of human pancreatic cancer samples from carriers of the Icelandic founder mutation in BRCA2 (BRCA2999Del5) (Mikaelsdottir et al., 2004), the majority (75%) of the cases tested do not exhibit LOH. These findings suggest that BRCA2 - contrary to belief - does not act as a typical Knudson ‘two hit’ tumour suppressor.
It is not clear how heterozygosity for Brca2Tr can promote tumourigenesis. One possibility is that this genotype causes defects in DNA repair, but this is not convincingly borne out by studies on cells derived from Brca2Tr-heterozygous mice (Patel et al., 1998; Yu et al., 2000), or from a similar transgenic model carrying another truncating allele in Brca2 (Tutt et al., 2002). Thus, the evidence that heterozygosity for the Brca2Tr mutant allele creates a DNA repair defect is not convincing, although the possibility has not yet been fully excluded.
Our laboratory has recently identified (Jeyasekharan et al., 2013) a protein-targeting mechanism that is disrupted even by heterozygosity for the human mutation BRCA2D2723H, which is associated with cancer. We find that in the mutant protein BRCA2D2723H a nuclear export signal in BRCA2 that is normally masked by its interaction with a partner protein, DSS1, is exposed, rendering the mutant BRCA2D2723H into a substrate for binding by the nuclear exportin CRM1. This mislocalizes mutant BRCA2D2723H into the cytoplasm, in turn decreasing RAD51 nuclear retention even when wild-type BRCA2 is also present. Thus, these findings suggest that a mechanism regulating the normal nucleo-cytoplasmic distribution of BRCA2 and RAD51 can be impaired by a heterozygous cancer-associated mutation.
BRCA2 HETEROZYGOSITY SUFFICES FOR CARCINOGENESIS: IMPLICATIONS FOR CANCER THERAPY
Our results suggesting that Brca2 heterozygosity is sufficient for pancreatic carcinogenesis driven by mutant Kras in mice and men have important implications for cancer therapy. This is because the rationale for the use of targeted agents such as PARP1 inhibitors (PARP1i) in BRCA2-deficient cancers requires bi-allelic BRCA2 inactivation in the tumour cells (Bryant et al., 2005; Farmer et al., 2005). Indeed, we have shown in our work (Skoulidis et al., 2010) that PDAC cells that retain a functional Brca2 allele are resistant to PARPi such as the AstraZeneca compound Olaparib. Our findings argue that in humans, PARP1 inhibitors should only be used in patients when BRCA2 LOH can be verified in the tumour. Assessment of LOH therefore becomes a critical requirement in the design of human clinical trials for the treatment of BRCA2-deficient cancers.
There is mounting evidence that heterozygosity for BRCA2 is sufficient for human carcinogenesis in tissues other than the pancreas, with the attendant implications for therapy using PARP1i. Lack of LOH has been documented using different methods in up to ∼30% of breast cancers (King et al., 2007), by NGS in ∼25% high-grade serous ovarian carcinomas (Cancer Genome Atlas, 2011), and in up to 55% of prostatic cancers associated with BRCA2 mutation carriers (Willems-Jones et al., 2012). Thus, it will be essential in future clinical trial designs to test for LOH in tumour samples to help stratify likely responders to PARP1i or similar targeted therapies.
REFERENCES
- Ayoub N., Rajendra E., Su X., Jeyasekharan A.D., Mahen R., Venkitaraman A.R. The carboxyl terminus of Brca2 links the disassembly of Rad51 complexes to mitotic entry. Curr. Biol. 2009;19:1075–1085. doi: 10.1016/j.cub.2009.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breast Cancer Linkage Consortium The breast cancer linkage consortium: cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999;91:1310–1316. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Caldas C., Kern S.E. K-ras mutation and pancreatic adenocarcinoma. Int. J. Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Consortium Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira A., Hilario J., Amitani I., Baskin R.J., Shivji M.K., Venkitaraman A.R., Kowalczykowski S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136:1032–1043. doi: 10.1016/j.cell.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E., Park P.G., Lee H.O., Lee Y.K., Kang G.H., Lee J.W., Han W., Lee H.C., Noh D.Y., Lekomtsev S., et al. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Dev Cell. 2012;22:295–308. doi: 10.1016/j.devcel.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Couch F.J., Johnson M.R., Rabe K.G., Brune K., de Andrade M., Goggins M., Rothenmund H., Gallinger S., Klein A., Petersen G.M., et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 2007;16:342–346. doi: 10.1158/1055-9965.EPI-06-0783. [DOI] [PubMed] [Google Scholar]
- Daniels M.J., Wang Y., Lee M., Venkitaraman A.R. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–879. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C., et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Friedman L.S., Thistlethwaite F.C., Patel K.J., Yu V.P., Lee H., Venkitaraman A.R., Abel K.J., Carlton M.B., Hunter S.M., Colledge W.H., et al. Thymic lymphomas in mice with a truncating mutation in Brca2. Cancer Res. 1998;58:1338–1343. [PubMed] [Google Scholar]
- Gretarsdottir S., Thorlacius S., Valgardsdottir R., Gudlaugsdottir S., Sigurdsson S., Steinarsdottir M., Jonasson J.G., Anamthawat-Jonsson K., Eyfjörd J.E. BRCA2 and p53 mutations in primary breast cancer in relation to genetic instability. Cancer Res. 1998;58:859–862. [PubMed] [Google Scholar]
- Hahn S.A., Greenhalf B., Ellis I., Sina-Frey M., Rieder H., Korte B., Gerdes B., Kress R., Ziegler A., Raeburn J.A., et al. BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst. 2003;95:214–221. doi: 10.1093/jnci/95.3.214. [DOI] [PubMed] [Google Scholar]
- Hezel A.F., Kimmelman A.C., Stanger B.Z., Bardeesy N., Depinho R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- Hruban R., Pitman M., Klimstra D.S. 2007. Tumors of the Pancreas, AFIP ATLAS of Tumour Pathology, Series IV, (American Registry of Pathology)
- Jeyasekharan A.D., Liu Y., Hattori H., Pisupati V., Jonsdottir A.B., Rajendra E., Lee M., Sundaramoorthy E., Schlachter S., Kaminski C.F., et al. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol. 2013;20:1191–1198. doi: 10.1038/nsmb.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J., Meuwissen R., van der Gulden H., Peterse H., van der Valk M., Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- King T.A., Li W., Brogi E., Yee C.J., Gemignani M.L., Olvera N., Levine D.A., Norton L., Robson M.E., Offit K., et al. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann. Surg. Oncol. 2007;14:2510–2518. doi: 10.1245/s10434-007-9372-1. [DOI] [PubMed] [Google Scholar]
- Lee M., Daniels M.J., Garnett M.J., Venkitaraman A.R. A mitotic function for the high-mobility group protein HMG20b regulated by its interaction with the BRC repeats of the BRCA2 tumor suppressor. Oncogene. 2011;30:3360–3369. doi: 10.1038/onc.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonosov M., Anand S., Sangrithi M., Davies R., Venkitaraman A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003;17:3017–3022. doi: 10.1101/gad.279003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A., Hruban R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel T., Nähse-Kumpf V., Kousholt A.N., Klein D.K., Lund-Andersen C., Lees M., Johansen J.V., Syljuåsen R.G., Sørensen C.S. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep. 2011;12:705–712. doi: 10.1038/embor.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikaelsdottir E.K., Valgeirsdottir S., Eyfjord J.E., Rafnar T. The Icelandic founder mutation BRCA2 999del5: analysis of expression. Breast Cancer Res. 2004;6:R284–290. doi: 10.1186/bcr785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal G., Rowley M., Guidugli L., Wu J., Pankratz V.S., Couch F.J. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev. Cell. 2012;23:137–152. doi: 10.1016/j.devcel.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin D.M., Bray F.I., Devesa S.S. Cancer burden in the year 2000. The global picture. Eur. J. Cancer. 2001;37:S4–66. doi: 10.1016/s0959-8049(01)00267-2. [DOI] [PubMed] [Google Scholar]
- Patel K.J., Yu V.P., Lee H., Corcoran A., Thistlethwaite F.C., Evans M.J., Colledge W.H., Friedman L.S., Ponder B.A., Venkitaraman A.R. Involvement of Brca2 in DNA repair. Mol. Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- Redston M.S., Caldas C., Seymour A.B., Hruban R.H., da Costa L., Yeo C.J., Kern S.E. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025–3033. [PubMed] [Google Scholar]
- Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji M.K., Mukund S.R., Rajendra E., Chen S., Short J.M., Savill J., Klenerman D., Venkitaraman A.R. The BRC repeats of human BRCA2 differentially regulate RAD51 binding on single- versus double-stranded DNA to stimulate strand exchange. Proc. Natl. Acad. Sci. USA. 2009;106:13254–13259. doi: 10.1073/pnas.0906208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F., Cassidy L.D., Pisupati V., Jonasson J.G., Bjarnason H., Eyfjord J.E., Karreth F.A., Lim M., Barber L.M., Clatworthy S.A., et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 2010;18:499–509. doi: 10.1016/j.ccr.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Thompson D., Easton D.F. Cancer incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002;94:1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- Tirkkonen M., Johannsson O., Agnarsson B.A., Olsson H., Ingvarsson S., Karhu R., Tanner M., Isola J., Barkardottir R.B., Borg A., et al. Distinct somatic genetic changes associated with tumor progression in carriers of BRCA1 and BRCA2 germ-line mutations. Cancer Res. 1997;57:1222–1227. [PubMed] [Google Scholar]
- Tutt A.N., van Oostrom C.T., Ross G.M., van Steeg H., Ashworth A. Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation. EMBO Rep. 2002;3:255–260. doi: 10.1093/embo-reports/kvf037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Venkitaraman A.R. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu. Rev. Pathol. 2009;4:461–487. doi: 10.1146/annurev.pathol.3.121806.151422. [DOI] [PubMed] [Google Scholar]
- West S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- Willems-Jones A., Kavanagh L., Clouston D., Bolton D., kCon-Fab Investigators. Fox S., Thorne H. High grade prostatic intraepithelial neoplasia does not display loss of heterozygosity at the mutation locus in BRCA2 mutation carriers with aggressive prostate cancer. BJU Int. 2012;110:E1181–E1186. doi: 10.1111/j.1464-410X.2012.11519.x. [DOI] [PubMed] [Google Scholar]
- Wooster R., Neuhausen S.L., Mangion J., Quirk Y., Ford D., Collins N., Nguyen K., Seal S., Tran T., Averill D., et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12–13. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- Xu X., Weaver Z., Linke S.P., Li C., Gotay J., Wang X.W., Harris C.C., Ried T., Deng C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- Yu V.P., Koehler M., Steinlein C., Schmid M., Hanakahi L.A., van Gool A.J., West S.C., Venkitaraman A.R. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev. 2000;14:1400–1406. [PMC free article] [PubMed] [Google Scholar]