

Abstract
Retinoic acid inducible gene-I (RIG-I) is critical in the activation of the type I IFN-dependent antiviral innate immune response to hepatitis C virus (HCV) infection. We examined whether hepatic stellate cells (HSC; LX-2) possess a functional RIG-I signaling pathway and produce antiviral factors that can inhibit HCV. We showed that LX-2 cells treated with the RIG-I ligand (5′ppp-dsRNA) expressed significantly higher levels of IFN-β and IFN-λ than the control cells. The RIG-I activation in LX-2 cells also induced the expression of Toll-like receptor 3 (TLR3) and IFN regulatory factor-7 (IRF-7), the key regulators of the IFN signaling pathway. When HCV Japanese fulminant hepatitis (JFH)-1-infected hepatocytes were co-cultured with LX-2 cells stimulated with 5′ppp-dsRNA or incubated in media conditioned with supernatant (SN) from 5′ppp-dsRNA-stimulated LX-2 cells, HCV replication in hepatocytes was suppressed significantly. This LX-2 cell action on HCV replication was mediated through both IFN-β and IFN-λ, as Abs to IFN-α/β or IFN-λ receptors could neutralize the LX-2 SN-mediated anti-HCV effect. The role of IFNs in LX-2 cell-mediated anti-HCV activity is further supported by the observation that LX-2 SN treatment induced the expression of IFN stimulated genes, 2′-5′-oligoadenylate synthase-1 (OAS-1) and myxovirus resistance A (MxA), in HCV-infected Huh7 cells. These observations highlight the importance of HSC in liver innate immunity against HCV infection via a RIG-I-mediated signaling pathway.
Keywords: Hepatic stellate cells, hepatitis C virus, interferon, interferon stimulated genes, retinoic acid inducible gene-I
Introduction
Hepatic stellate cells (HSC) are liver pericytes that reside in the space between parenchymal cells and sinusoidal endothelial cells of the human liver. HSC are rich in vitamin A and store nearly 80% of retinoids of the whole body in its lipid droplets in the cytoplasm. 1,2 In normal liver, HSC are in a quiescent state and represent 5–8% of the total number of liver cells.2 HSC become activated following liver injury, and activated HSC enhanced migration and deposition of extracellular matrix components, resulting in liver fibrosis.3,4 Recent evidence indicated that HSC play a role in the innate immune system of the liver. It was reported that HSC could function as liver-resident antigen- presenting cell (APC) that present lipid antigens to Natural killer T (NKT) cells, inducing cell proliferation. 5 HSC can act as regulatory bystanders, enhancing differentiation and accumulation of regulatory T cells (Tregs), which may be the basis of the tolerogenic nature of the liver.6
The interaction between hepatitis C virus (HCV) and the innate immunity system in liver plays a key role in the immunopathogenesis of HCV disease. Toll-like receptors (TLRs) and the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are major cellular receptors that recognize viral pathogen-associated molecular patterns (PAMPs) during viral infection.7 Several TLR members play a critical role in the recognition of viral nucleic acids.8 Among these TLRs, TLR3 has a crucial role in virus-mediated innate immune responses9–11 as it recognizes dsRNA that either constitutes the genome of one class of viruses or is generated during the lifecycle of viruses.12 IFN-α/β have been recognized as the first line of the TLR3 activation-mediated antiviral response.13 In addition to TLR3, the RLR family has been reported as an important mediator of antiviral immunity. RIG-I can detect viral genomic RNA during negative-strand RNA virus infection14 and trigger a type I IFN-mediated immune response that protects the host against viral infection.15 RIG-I plays an important role in HCV genome recognition, resulting in activation of the type I IFN-dependent antiviral innate immune response to restrict HCV infection.16
Because the majority of HCV-infected subjects develop chronic infection, it is likely that HCV uses complex and unique mechanisms to evade or subvert host innate immunity to establish persistent infection. In order to counteract the host cell innate immunity viruses use mechanisms to block recognition and signaling of TLRs and RIG-I. Studies of HCV–host interactions have revealed that HCV protease NS3/4A ablates TLR3 signaling by cleaving the TLR3 adaptor protein, Toll-IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF),17 and blocks RIG-I signaling by cleaving mitochondrial antiviral signaling protein (MAVS) off the mitochondria.18 Thus, to activate TLRs and/or RIG-I by their ligands represents a promising approach for the treatment of HCV infection. Whilst most studies have focused on the interactions between liver immune cells and HCV, there is limited information about whether other residential cells in the liver participate in innate immune responses to HCV infection of hepatocytes. Specifically, we know little about whether HSC possess functional RIG-I signaling pathway and produce anti-HCV factors. Therefore, this study examined whether HSC have the ability to mount a RIG-I-mediated innate immunity that is effective in the control of HCV infection of human hepatocytes.
Materials and methods
Reagents
Mouse Ab against HCV core antigen was purchased from ABR Affinity BioReagents (Thermo Scientific, Rockford, IL, USA). Mouse anti-IFNAR2 Ab was purchased from LifeSpan BioScience (Seattle, WA, USA). Mouse anti-IL-10Rβ Ab was purchased from R&D Systems (Minneapolis, MN, USA). Mouse IgG was from Molecular Probes (Eugene, OR, USA). Hoechst 33 342 was purchased from Molecular Probes (Carlsbad, CA, USA). LyoVec transfection reagent, 5′ppp-dsRNA and 5′ppp-dsRNA control were purchased from Invivogen (San Diego, CA, USA). ELISA kits for IFN-β were from Fujirebio (Tokyo, Japan), ELISA kits for IFN-λ1 were from eBioscience (San Diego, CA, USA) and ELISA kits for IFN-λ2/3 were purchased from Biolegend (San Diego, CA, USA). siRNA against RIG-I and negative control siRNA were purchased from Qiagen (Cambridge, MA, USA).
Cell culture
LX-2, an immortalized human hepatic stellate cell line, was kindly provided by Dr Scott L. Friedman (Mount Sinai School of Medicine, New York, NY, USA). LX-2 cells were cultured in DMEM containing 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml), as described previously.19 Huh7 cells (generously provided by Dr Charles Rice, The Rockefeller University, New York, NY, USA) were maintained in DMEM with 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml).
RIG-I ligand stimulation
LX-2 cells were seeded at a density of 105/well in a 24-well plate. After culturing for 24 h, the cells were stimulated with RIG-I ligand (5′ppp-dsRNA, 1 μg/ml) using LyoVec transfection reagent. The cell cultures were replaced with fresh medium 16 h post-stimulation. Cells were collected for mRNA extraction and culture supernatant (SN) was collected 48 h post-stimulation for HCV inhibition experiments in Japanese fulminant hepatitis (JFH)-1-infected Huh7 cells. As a negative control, cells were stimulated with 5′ppp-dsRNA control. For the blocking experiments using siRNA against RIG-I to block RIG-I signaling pathway, LX-2 cells were transfected with RIG-I siRNA (60 nM) for 24 h prior to 5′ppp-dsRNA stimulation.
HCV JFH-1 infection
The generation of infectious HCV JFH-1 and infection of Huh7 cells (multiplicity of infection of 0.01) were carried out as described previously.20,21 HCV JFH-1 infection of Huh7 cells was analyzed by immunostaining with the mouse anti-HCV core Ab or by real-time RT-PCR for HCV RNA.
Co-culture of Huh7 cells with LX-2 cells and SN from LX-2 cell cultures
For the co-culture experiments, LX-2 cells were first stimulated with 5′ppp-dsRNA (1 μg/ml) for 16 h and then co-cultured with HCV JFH-1-infected Huh7 cells in 0.4 μm-pore transwell tissue culture plates (Costar, Cambridge, MA, USA). LX-2 cells were placed in the lower compartment and Huh7 cells were cultured in the upper compartment. Huh7 cells were then collected for RNA extraction and real-time RT-PCR at 48 h after co-culture. For the experiments using LX-2 SN, HCV JFH-1-infected Huh7 cells were cultured in media with or without SN from LX-2 cells stimulated with 5′ppp-dsRNA (5%, 10% and 20% vol/vol) for 48 h. LX-2 SN was added to Huh7 cells infected with JFH-1 at d 3 post-infection. SN from LX-2 cells stimulated with 5′ppp-dsRNA control was used as a negative control.
RNA extraction and real-time RT-PCR
Total RNA from culture cells was extracted with Tri-Reagent (Molecular Research Center, Cincinnati, OH, USA) as described previously.22 Total RNA (1 μg) was subjected to RT using the RT system (Promega, Madison, WI, USA) with random primers for 1 h at 42°C. The reaction was terminated by incubating the reaction mixture at 99°C for 5 min and the mixture was kept at 4°C. The resulting cDNA was used as a template for the real-time PCR quantification. Real-time PCR was performed with 1/10 of the cDNA with the iQ SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA) as described previously.23 The amplified products were visualized and analyzed using the software MyiQ provided with the thermocycler (iCycler iQ real time PCR detection system; Bio-Rad Laboratories). The oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA, USA) and sequences will be available upon request. The cDNA was amplified by PCR and the products were measured using SYBR green I (Bio-Rad Laboratories). The data were normalized to GAPDH and presented as the change in induction relative to that of untreated control cells.
Immunofluorescence assay
HCV JFH-1-infected Huh7 cells were cultured at a density of 105/well in a 24-well plate. Huh7 cells were washed with cold 1 x PBS (with Ca2+ and Mg2+) twice. Cells were fixed at 4°C in 4% paraformaldehyde–4% sucrose in PBS for 20 min followed by 0.2% Triton X-100 for an additional 10 min. Cells were blocked in Block Solution (Pierce, Rockford, IL, USA) for 1 h at 18–22°C (room temperature). To examine the expression of HCV core protein, HCV-infected cells were incubated with Ab to HCV core protein (1:500) for 2 h at room temperature. After washing five times with 1 x PBS, the cells were incubated with FITC-conjugated goat anti-mouse IgG Ab (green, 1:100) for 1 h. After washing five times with 1 x PBS, the cells were then mounted on glass coverslips in mounting media (Biomeda, Foster City, CA, USA) and viewed with a fluorescence microscopy (Zeiss, Jena, Germany). Hoechst 33342 was used for nuclei staining.
ELISA
SN collected from 5′ppp-dsRNA stimulated LX-2 cultures were examined for the protein levels of IFN-β, IFN-λ1 and IFN-λ2/3 by ELISA, which was performed according to the manufacturer’s instructions.
Statistical analysis
Student’s t-test was used to evaluate the significance of difference between groups, and multiple comparisons were performed by regression analysis and one-way ANOVA. P-values of less than 0.05 were considered significant. All data are presented as mean ± SD. Statistical analyses were performed with SPSS 11.5 for Windows. Statistical significance was defined as P<0.05.
Results
RIG-I signaling of LX-2 cells inhibits HCV replication in hepatocytes
We first examined whether LX-2 cells or SN from LX-2 cell cultures (LX-2 SN) had a cytotoxicity effect on Huh7 cells. Little cytotoxic effect was observed in Huh7 cells either co-cultured with LX-2 cells stimulated with or without 5′ppp-dsRNA or treated with LX-2 SN (data not shown). We then determined whether LX-2 cells stimulated with 5′ppp-dsRNA release soluble antiviral factor(s) that suppresses HCV replication in Huh7 cells. We demonstrated that 5′ppp-dsRNA had little effect on HCV JFH-1 replication in Huh7 cells (Figure 1A). In contrast, HCV replication was significantly inhibited in Huh7 cells co-cultured with LX-2 cells stimulated with 5′ppp-dsRNA (Figure 1B). In addition, SN from 5′ppp-dsRNA-stimulated LX-2 cell cultures, when added to HCV JFH-1-infected Huh7 cells, inhibited viral RNA expression in a concentration-dependent manner (Figure 1C). We further examined the anti-HCV activity of LX-2 SN under three different conditions: Huh7 cells were incubated with LX-2 SN either 24 h before HCV infection, simultaneously with HCV infection or 8 h after infection. Cells that were pretreated for 24 h with 10% (v/v) of LX-2 SN and then infected had significantly lower levels of HCV RNA than untreated and infected cells (Figure 1D). Similarly, cells treated with LX-2 SN and infected simultaneously or after 8 h HCV JFH-1 infection also had significantly lower levels of HCV RNA than the control cells (Figure 1D). LX-2 SN-mediated inhibition of HCV replication was also confirmed by diminished percentage of HCV core antigen-positive cells in JFH-1-infected Huh7 cells treated with LX-2 SN (Figure 1E).
Figure 1.

5′ppp-dsRNA-stimulated LX-2 cells or SN from cell cultures suppresses HCV replication in Huh7 cells. (A) Effect of 5′ppp-dsRNA on HCV JFH-1 replication in Huh7 cells. Huh7 cells were infected with HCV JFH-1 for 48 h, and then stimulated with 5′ppp-dsRNA (1 μg/ml) and total cellular RNA was extracted from cells for the HCV real-time RT-PCR. (B) Co-culture of HCV JFH-1-infected Huh7 cells with 5′ppp-dsRNA-stimulated LX-2 cells. LX-2 cells were plated in the lower compartment of a 24-well plate and stimulated with 5′ppp-dsRNA (1 μg/ml) for 16 h, whilst HCV JFH-1-infected Huh7 cells (d 3 post-infection) were plated in the upper compartment for co-culture. After 48 h co-culture, total cellular RNA extracted from Huh7 cells was subjected to real-time RT-PCR for HCV and GAPDH RNA quantification. (C) Effect of SN of LX-2 cell culture stimulated with 5′ppp-dsRNA (1 μg/ml) on HCV replication in Huh7 cells. HCV JFH-1-infected Huh7 cells (d 3 post-infection) were cultured in the presence or absence of the SN of LX-2 stimulated with 5′ppp-dsRNA at the indicated concentration for 48 h. Total cellular RNA extracted from Huh7 cells was subjected to real-time RT-PCR for HCV and GAPDH RNA quantification. (D) Suppression of HCV RNA expression by LX-2 SN under three different conditions. Huh7 cells were cultured in media conditioned with or without LX-2 SN for either 24 h prior to HCV infection, simultaneously or 8 h post-infection. The cells were then washed five times to remove input HCV after 6 h incubation with HCV JFH1 and then cultured in the presence or absence of LX-2 SN for 72 h. Intracellular RNA extracted from hepatocytes at 72 h post-infection was subjected to real-time RT-PCR for HCV and GAPDH RNA quantification. The data are expressed as HCV RNA levels relative (%) to control (LyoVec only, without co-culture, without stimulation or without LX-2 SN treatment, which is defined as 100%). The results are mean ± SD of triplicate cultures, representative of three experiments (*P<0.05, **P<0.01, 5′ppp-dsRNA vs 5′ppp-dsRNA control). (E) Effect of LX-2 SN on HCV core protein expression in Huh7 cells. HCV JFH-1-infected Huh7 cells (d 3 post-infection) were cultured in the presence or absence of 5′ppp-dsRNA stimulated LX-2 cell culture SN at the indicated concentrations for 48 h. HCV core protein expression was determined by immunofluorescence staining with Ab against HCV core protein (green). The nuclei were stained with Hoechst 33342 (blue). One representative experiment is shown (original magnification 200×).
RIG-I activation induces IFN expression in LX-2 cells
RIG-I is a key regulator in the IFN signaling pathway. Thus, we examined whether 5′ppp-dsRNA can trigger IFN production in HCV JFH-1-infected Huh7 cells. We demonstrated that 5′ppp-dsRNA stimulation failed to induce IFN expression in HCV JFH-1-infected Huh7 cells (Figure 2A, B, C). We then examined whether RIG-I activation can induce IFN expression in LX-2 cells. We showed that LX-2 cells stimulated with 5′ppp-dsRNA expressed higher levels of IFN-β than unstimulated cells at both mRNA and protein levels (Figure 2D, G). In addition, 5′ppp-dsRNA induced the expression of IFN-λ1 and IFN-λ2/3 in LX-2 cells at both mRNA (Figure 2E, H) and protein levels (Figure 2F, I). This induction of IFNs in 5′ppp-dsRNA-stimulated LX-2 cells was not affected by the exposure of the cells to HCV (Figure 2J, K, L).
Figure 2.

Effect of RIG-1 activation on IFN-β and IFN-λ expression. (A–C) HCV JFH-1-infected Huh7 cells (d 3 post-infection) were stimulated with 5′ppp-dsRNA (1 μg/ml) for 48 h. (D–I) LX-2 cells were tranfected with 5′ppp-dsRNA (1 μg/ml) for 48 h. Total RNA extracted from cells was subjected to real-time RT-PCR for the mRNA levels of IFN-β (A, D), IFN-λ1 (B, E) and λ2/3 (C, F). SN was collected from the cell cultures described for ELISA to measure the protein levels of IFN-β (G), IFN-λ1 (H) and λ2/3 (I). The data (A, B, C) are expressed as mRNA levels relative (fold) to the control (without stimulation, which is defined as 1). (J–L) Effect of HCV exposure on 5′ppp-dsRNA-mediated IFN expression in LX-2 cells. LX-2 cells were plated in the lower compartment of a 24-well plate and stimulated with 5′ppp-dsRNA (1 μg/ml), whilst HCV JFH-1-infected Huh7 cells (d 3 post-infection) were plated in the upper compartment for co-culture. After 48 h co-culture, total cellular RNA extracted from LX-2 cells was subjected to real-time RT-PCR for IFN RNA quantification. The results are mean ± SD of three different experiments (*P<0.05, **P<0.01).
RIG-I activation induces RIG-I, TLR3 and IRF expression in LX-2 cells
The investigation of the mechanism(s) of 5′ppp-dsRNA- mediated induction of IFN showed that 5′ppp-dsRNA significantly increased RIG-I (Figure 3A) and TLR3 (Figure 3B) expression in LX-2 cells. Although 5′ppp-dsRNA had little effect on IFN regulatory factor (IRF)-3 expression (Figure 3C), it significantly induced IRF-7 expression in LX-2 cells (Figure 3D).
Figure 3.

Effect of 5′ppp-dsRNA stimulation on RIG-1, TLR3 and IRF expression. LX-2 cells were stimulated with 5′ppp-dsRNA (1 μg/ml) for 48 h. Total RNA extracted from cells was subjected to real-time RT-PCR for the mRNA levels of RIG-I (A), TLR3 (B), IRF-3 (C) and IRF-7 (D). The data are expressed as mRNA levels relative (fold) to the control (without stimulation, which is defined as 1). The results are mean ± SD of three different experiments (**P<0.01, 5′ppp-dsRNA vs control).
RIG-I siRNA blocks RIG-I activation-mediated IFN expression
To further determine whether RIG-I/IFN signaling pathway is critical in the LX-2 SN-mediated anti-HCV effect, siRNA against RIG-I was used to knockdown RIG-I expression in LX-2 cells. As shown in Figure 4A, the knockdown efficiency of RIG-I expression by RIG-I siRNA was approximately 70% and 5′ppp-dsRNA-induced RIG-I expression was significantly decreased in LX-2 cells transfected with RIG-I siRNA. In addition, 5′ppp-dsRNA-induced expression of IFN-β (Figure 4B) and IFN-λ (Figure 4C, D) was largely blocked in the cells transfected with RIG-I siRNA.
Figure 4.

Effect of siRNA of RIG-1 on 5′ppp-dsRNA-mediated IFN expression. LX-2 cells were transfected with or without siRNA against RIG-I (60 nM) for 24 h prior to 5′ ppp-dsRNA stimulation (1 μg/ml). Negative control siRNA was used as a negative control in siRNA transfection experiment. Total RNA extracted from cells was subjected to real-time RT-PCR for mRNA levels of RIG-I (A), IFN-β (B), IFN-λ1 (C) and λ2/3 (D). The data are expressed as mRNA levels relative (fold) to the control (stimulated with 5′ppp-dsRNA control, which is defined as 1). The results are mean ± SD of three different experiments (*P<0.05, **P<0.01).
IFN-β and IFN-λ are involved in the anti-HCV activity of HSC
To investigate whether RIG-I activation-induced IFNs are responsible for LX-2 SN-mediated anti-HCV activity, we incubated HCV JFH-1-infected Huh7 cells with Ab to IFNAR2 prior to LX-2 SN treatment. As shown in Figure 5A, Ab to IFNAR2 partially blocked the ability of LX-2 SN to inhibit HCV replication in Huh7 cells. In addition, we also incubated HCV JFH-1-infected Huh7 cells with Ab to the extracellular domain of IL-10Rβ (IFN-λ receptor) prior to LX-2 SN treatment. As shown in Figure 5B, Ab to IL-10Rβ partially compromised LX-2 SN-mediated HCV inhibition in Huh7 cells.
Figure 5.
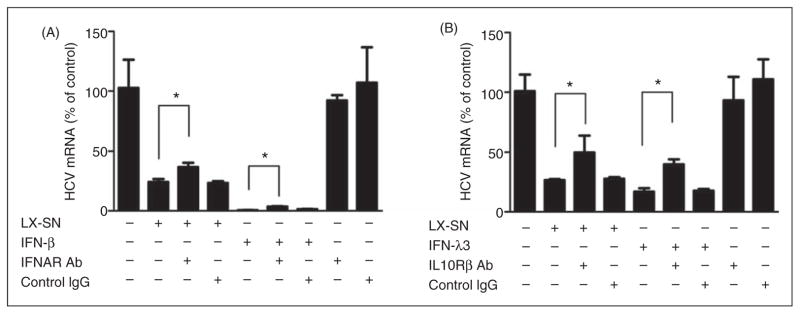
Effect of Abs against to IFNAR2 and IL-10Rβ on LX-2 SN-mediated anti-HCV activity. HCV JFH-1-infected Huh7 cells (d 3 post-infection) were pretreated with Ab to IFNAR2 (A) or IL-10Rβ (B), or control IgG at the concentration of 5 μg/ml for 1 h followed with SN from 5′ppp-dsRNA-stimulated (10%, v/v) LX-2 cell cultures for 48 h. Recombinant human IFN-β (400 IU/ml) or IFN-λ3 (10 ng/ml) was used as a positive control for (A) or (B) respectively. HCV JFH-1 replication was measured by real-time RT-PCR for HCV RNA 48 h post-treatment. Value is mean ± SD of three different cultures (*P<0.05).
LX-2 SN induce ISG expression in HCV-infected Huh7 cells
The action of IFN on virus-infected cells is to elicit an antiviral state, which is characterized by the induction of IFN-stimulated genes (ISGs).24 We thus examined the expression of several key IFN-inducible genes in HCV JFH-1-infected Huh7 cells incubated with SN from LX-2 cell cultures stimulated with 5′ppp-dsRNA. As shown in Figure 6, although IFN-stimulated gene 56 (ISG56) and protein kinase R (PKR) were not affected, SN from RIG-I-activated LX-2 cells induced 2′-5′-oligoadenylate synthase-1 (OAS-1) and myxovirus resistance A (MxA) gene expression dose-dependently in HCV-infected hepatocytes.
Figure 6.
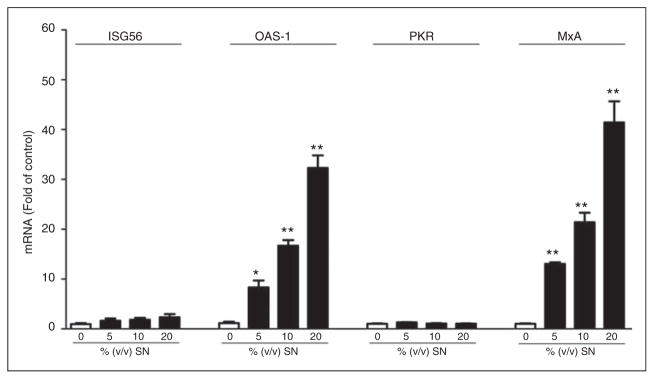
Effect of LX-2 SN on the expression of ISG56, OAS-1, protein kinase R (PKR) and MxA in HCV JFH-1-infected Huh7 cells. HCV JFH-1-infected Huh7 cells (d 3 post-infection) were cultured in the presence or absence of SN from 5′ppp-dsRNA-stimulated LX-2 cell cultures at the indicated concentrations for 48 h. Total cellular RNA extracted from Huh7 cells was subjected to real-time RT-PCR for ISG56, OAS-1, PKR and MxA mRNA quantification. The data are expressed as RNA levels relative (fold) to control (without SN treatment, which is defined as 1). The results are mean ± SD of three different experiments (*P<0.05, **P<0.01).
Discussion
The interactions between HCV and the innate immunity system in liver play a key role in the consequences of HCV infection. HCV persistent infection is associated with its ability to evade the host immune response and counteract antiviral defenses. RIG-I as a pattern recognition receptor plays an important role in liver innate immunity against HCV infection. RIG-I recognizes viral dsRNA and induces downstream signals, leading to IFN-α/β production.15 Although RIG-I operates independently of the TLRs,25 RIG-I signaling culminates in the induction of the type I IFN response, one of the earliest alarm bells of the immune system, which inhibits viral replication without killing infected cells.26 It has been demonstrated that the HCV PAMP RNA-stimulated RIG-I-dependent signaling induced a hepatic innate immune response in vivo, leading to IFN and ISG expression that suppresses HCV infection.16 This RIG-I signaling, however, could be blocked by HCV protease NS3/4 A, which has the ability to cleave MAVS off the mitochondria.18
We demonstrated that HSC possess functional RIG-I that can be activated by the RIG-I ligand, resulting in the induction of IFNs and inhibition of HCV replication in hepatocytes. This RIG-I signaling-mediated anti-HCV activity is potent, as SN from RIG-I-activated HSC cell cultures showed anti-HCV activity under three different conditions (before, during and after infection) in HCV JFH-1-infected Huh7 cells (Figure 1D). In contrast, 5′ppp-dsRNA direct stimulation of HCV-infected Huh7 cells failed to inhibit virus replication (Figure 1A), which supports the report that HCV has the ability to inhibit RIG-I signaling in infected hepaotcytes.18 These data suggest that RIG-I activation-based therapeutic approaches may not be effective in the control of HCV in infected hepatocytes. Therefore, RIG-I signaling of liver bystander cells that can produce antiviral factors is now becoming the focus of research. We demonstrated that RIG-I-activated LX-2 cells produced IFN-β. The role of IFN-β in RIG-I-mediated HCV inhibition was evidenced by the observation that Abs to IFN-α/β receptor could compromise the LX-2-SN-mediated anti-HCV effect in Huh7 cells (Figure 5A). The importance of the RIG-I-activated IFN signaling pathway in LX-2 cell-mediated anti-HCV activity was further demonstrated in the experiments, which showed that to knockdown RIG-I by specific siRNA could block the IFN induction of 5′ppp-dsRNA (Figure 4).
In addition to the induction of IFN-β, the RIG-I signaling of LX-2 cells also induced the expression of IFN-λ. The induced IFN-λ is responsible, at least partially, for the RIG-I-mediated anti-HCV effect, as the Ab to IFN-λ receptor could block the LX-2 SN action on HCV (Figure 5B). IFN-λ has been shown to have antiviral activities against a number of viruses,27–30 including HCV.31–33 IFN-λ exhibits potent antiviral action on HCV replication in both replicon and the JFH-1 infectious cell system.28,31,32,34,35 Recent studies 36–39 showed that IFN-λ may play a crucial role in HCV clearance in clinical IFN-α treatment. These studies reported that both spontaneous HCV clearance and a sustained viral response (SVR) after pegylated (PEG) IFN-α and ribavirin combination treatment correlated with single nucleotide polymorphisms (SNPs) in the IL28B gene locus, which encodes the IFN-λ3 protein. 36–39 Clinically, IFN-λ-based therapy for genotype 1 chronic HCV infection has been investigated.40,41 Therefore, the demonstration of intracellular IFN-λ induction by the RIG-I signaling of HSC is highly significant, as it supports the notion of developing RIG-I ligand-based therapy for the treatment of HCV-infected patients.
HSC express HCV receptors (CD81, LDL receptor and C1q),42,43 suggesting that HSC may be a potential target for HCV. It has been demonstrated that the HCV E2 protein could bind directly to CD81 on the HSC surface.44 However, there have been no reports showing that HCV can infect HSC productively. We did not observe HCV JFH-1 infection of LX-2 cells (data not shown). This lack of infection by HCV suggests the possibility that HSC possess immunologic mechanisms to protect them. Our data showed that the induction of IFNs in RIG-I-activated LX-2 cells was not affected by the exposure of the cells to HCV. Recent studies have suggested that HSC are involved in liver immunity. It was reported that HSC can induce vigorous NKT cell responses in vitro and in vivo, promoting the homeostatic proliferation of NKT cells.5 HSC could process protein antigens and present peptides to CD4+ and CD8+ T cells.5 HSC could elicit antigen-specific T cells and protected against bacterial infection in a Listeria monocytogenes infection model.5 A recent study indicated that HSC had the ability to enhance the differentiation and accumulation of Tregs.6 HSC can express retinoic acid early inducible-1 (RAE1), CD1d, and MHC I and II, and directly interact with immune cells, such as NK cells, NKT cells and T cells.5,45,46 Our data indicate that HSC have a functional RIG-I signaling system and produce IFNs that inhibit HCV replication in hepatocytes provide additional evidence to support the notion that the activation of RIG-I signaling in HSC can help with the control of HCV infection/replication in the liver. This notion, however, requires future ex vivo and in vivo studies to further define the role of HSC in liver innate immunity against HCV infection.
Acknowledgments
Funding
This work was supported by the grants (DA12815, DA22177 and DA27550) from the National Institutes of Health.
We are grateful to Dr Friedman (Mount Sinai School of Medicine, New York, NY, USA) for providing us with the LX-2 cell line, which was critical for this study.
Footnotes
Reprints and permissions: sagepub.co.uk/journalPermissions.nav
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
References
- 1.Senoo H. Structure and function of hepatic stellate cells. Med Electron Microsc. 2004;37:3–15. doi: 10.1007/s00795-003-0230-3. [DOI] [PubMed] [Google Scholar]
- 2.Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 3.Reynaert H, Thompson MG, Thomas T, Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut. 2002;50:571–581. doi: 10.1136/gut.50.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 6.Ichikawa S, Mucida D, Tyznik AJ, Kronenberg M, Cheroutre H. Hepatic stellate cells function as regulatory bystanders. J Immunol. 2011;186:5549–5555. doi: 10.4049/jimmunol.1003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 9.Kumar A, Zhang J, Yu FS. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology. 2006;117:11–21. doi: 10.1111/j.1365-2567.2005.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starace D, Galli R, Paone A, De Cesaris P, Filippini A, Ziparo E, et al. Toll-like receptor 3 activation induces antiviral immune responses in mouse sertoli cells. Biol Reprod. 2008;79:766–775. doi: 10.1095/biolreprod.108.068619. [DOI] [PubMed] [Google Scholar]
- 11.West J, Damania B. Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J Virol. 2008;82:5440–5449. doi: 10.1128/JVI.02590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 13.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 14.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell. 2010;140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 15.Fujita T, Onoguchi K, Onomoto K, Hirai R, Yoneyama M. Triggering antiviral response by RIG-I-related RNA helicases. Biochimie. 2007;89:754–760. doi: 10.1016/j.biochi.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye L, Wang X, Wang S, Wang Y, Song L, Hou W, et al. CD56+ T cells inhibit hepatitis C virus replication in human hepatocytes. Hepatology. 2009;49:753–762. doi: 10.1002/hep.22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang JH, Lai JP, Douglas SD, Metzger D, Zhu XH, Ho WZ. Real-time RT-PCR for quantitation of hepatitis C virus RNA. J Virol Methods. 2002;102:119–128. doi: 10.1016/s0166-0934(02)00007-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang T, Lin RT, Li Y, Douglas SD, Maxcey C, Ho C, et al. Hepatitis C virus inhibits intracellular interferon alpha expression in human hepatic cell lines. Hepatology. 2005;42:819–827. doi: 10.1002/hep.20854. [DOI] [PubMed] [Google Scholar]
- 24.Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 25.Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benedict CA, Ware CF. RIGing a virus trap. Nat Med. 2005;11:929–930. doi: 10.1038/nm0905-929. [DOI] [PubMed] [Google Scholar]
- 27.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 29.Hou W, Wang X, Ye L, Zhou L, Yang ZQ, Riedel E, et al. Lambda interferon inhibits human immunodeficiency virus type 1 infection of macrophages. J Virol. 2009;83:3834–3842. doi: 10.1128/JVI.01773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 31.Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 32.Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Jilg N, Shao RX, Lin W, Fusco DN, Zhao H, et al. IL28B inhibits hepatitis C virus replication through the JAK-STAT pathway. J Hepatol. 2011;55:289–298. doi: 10.1016/j.jhep.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pagliaccetti NE, Eduardo R, Kleinstein SH, Mu XJ, Bandi P, Robek MD. Interleukin-29 functions cooperatively with interferon to induce antiviral gene expression and inhibit hepatitis C virus replication. J Biol Chem. 2008;283:30079–30089. doi: 10.1074/jbc.M804296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu H, Butera M, Nelson DR, Liu C. Novel type I interferon IL-28A suppresses hepatitis C viral RNA replication. Virol J. 2005;2:80. doi: 10.1186/1743-422X-2-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 37.Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 39.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller DM, Klucher KM, Freeman JA, Hausman DF, Fontana D, Williams DE. Interferon lambda as a potential new therapeutic for hepatitis C. Ann N Y Acad Sci. 2009;1182:80–87. doi: 10.1111/j.1749-6632.2009.05241.x. [DOI] [PubMed] [Google Scholar]
- 41.Muir AJ, Shiffman ML, Zaman A, Yoffe B, de la Torre A, Flamm S, et al. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology. 52:822–832. doi: 10.1002/hep.23743. [DOI] [PubMed] [Google Scholar]
- 42.Bataller R, Paik YH, Lindquist JN, Lemasters JJ, Brenner DA. Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology. 2004;126:529–540. doi: 10.1053/j.gastro.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 43.Mazzocca A, Carloni V, Sciammetta S, Cordella C, Pantaleo P, Caldini A, et al. Expression of transmembrane 4 superfamily (TM4SF) proteins and their role in hepatic stellate cell motility and wound healing migration. J Hepatol. 2002;37:322–330. doi: 10.1016/s0168-8278(02)00175-7. [DOI] [PubMed] [Google Scholar]
- 44.Mazzocca A, Sciammetta SC, Carloni V, Cosmi L, Annunziato F, Harada T, et al. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J Biol Chem. 2005;280:11329–11339. doi: 10.1074/jbc.M410161200. [DOI] [PubMed] [Google Scholar]
- 45.Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, et al. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology. 2009;49:1683–1694. doi: 10.1002/hep.22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809–G816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]