

Abstract
The Abelson (ABL) family of nonreceptor tyrosine kinases, ABL1 and ABL2, transduces diverse extracellular signals to protein networks that control proliferation, survival, migration, and invasion. ABL1 was first identified as an oncogene required for the development of leukemias initiated by retroviruses or chromosome translocations. The demonstration that small molecule ABL kinase inhibitors effectively treat chronic myeloid leukemia opened the door to the era of targeted cancer therapies. Recent reports have uncovered roles for ABL kinases in solid tumors. Enhanced ABL expression and activation in some solid tumors, together with altered cell polarity, invasion or growth induced by activated ABL kinases, suggest that drugs targeting these kinases may have utility in treating selected solid tumors.
Introduction
Tyrosine kinase signaling networks are required for the regulation of multiple cellular processes including growth, survival, invasion and angiogenesis during tumor initiation and progression. Drug discovery efforts in the past two decades have focused on developing tyrosine kinase inhibitors (TKIs) for cancer therapy. To date, the most successful of these efforts has been the development of ABL TKIs for the treatment of chronic myeloid leukemia (CML), which expresses the fusion oncoprotein BCR-ABL1. ABL1 was first discovered over 30 years ago as the oncogene in the Abelson murine leukemia virus1 and later identified as an oncogene associated with chromosome translocations in human leukemias2. These ABL1 fusion genes encode constitutively activated forms of the ABL1 tyrosine kinase required for cellular transformation. The ABL family of protein kinases comprised of ABL1 and ABL2 (ARG), function to link diverse extracellular stimuli to signaling pathways controlling cell growth, survival, invasion, adhesion, and migration3-6. In recent years, activation of ABL1 and ABL2 has been detected in solid tumors, including breast, colon, lung, and kidney carcinoma, as well as melanoma7-16. Activation of ABL kinases in solid tumors is not linked to chromosome translocation events, but rather is driven by enhanced ABL1 or ABL2 expression and/or by oncogenic tyrosine kinases, chemokine receptors, oxidative stress, and/or inactivation of negative regulatory proteins (cbioportal.org; 3, 16-21). Here we compare the modes of ABL family kinase activation in leukemias and solid tumors. Further, we dissect the role of the ABL kinases in the regulation of cellular processes critical for the progression of solid tumors including cell polarity, invasion, proliferation and survival. Several FDA-approved drugs targeting ABL kinases including imatinib and dasatinib have been tested for the treatment of solid tumors with mixed effectiveness; potentially due to the complexity of signaling pathways activated in solid tumors, the emergence of therapy resistance, and the fact these inhibitors have multiple cellular targets. Thus, these results highlight the necessity both to develop ABL kinase inhibitors with greater specificity and to identify biomarkers for patient stratification.
Function, Structure, and Enzymatic Regulation of ABL Kinases
Physiological Roles of Murine ABL kinases
The two mammalian ABL kinases, ABL1 and ABL2, have both unique and overlapping physiological roles as revealed by genetic inactivation in mice. Disruption of murine Abl1 on a mixed (129/SvEv and C57BL/6J) genetic background resulted in neonatal lethality in about 50% of the mice, a phenotype that was more severe on the C57BL/6J genetic background22, 23. The Abl1 knockout mice exhibit splenic and thymic atrophy, reduced numbers of B and T cells, cardiac abnormalities, and osteoporosis linked to defective osteoblast proliferation and premature senescence22-26. In contrast, Abl2 (Arg) knockout mice are viable and exhibit neuronal defects that include age-related dendrite destabilization and regression27, 28. Functional overlap by Abl1 and Abl2 kinases is supported by the embryonic lethality of Abl1−/− Abl2−/− mice27.
Structure and Regulation of ABL Enzymatic Activity
ABL1 and ABL2 function redundantly in some cellular contexts, but also have unique roles, owing to distinct and conserved sequences and structural domains (Fig. 1). ABL1 and ABL2 share amino (N)-terminal regulatory and catalytic domains that are over 90% identical and include the Src homology 3 (SH3), SH2 and SH1 (tyrosine kinase) domains (Fig. 1A). The carboxy (C)-terminus of both ABL kinases contains a conserved filamentous (F) actin-binding domain29, 30 (and reviewed in 6) (Fig. 1A). ABL1 contains a G-actin (globular actin)-binding domain upstream of the F-actin-binding domain, and ABL2 has a second internal F-actin binding domain characterized by an I/LWEQ motif, which is required to promote the formation of F-actin-rich structures at the cell periphery30. The ABL kinases share conserved PXXP motifs that mediate binding to SH3 domain-containing proteins. Consistent with its nuclear and cytoplasmic localization, ABL1 has three nuclear localization signal (NLS) motifs and one nuclear export signal (NES) in its C-terminus. In contrast, ABL2, which lacks the NLS motifs, localizes primarily to the cytoplasm and preferentially accumulates at F-actin-rich sites in the cell periphery, focal adhesions, adherens junctions, invadopodia and phagocytic cups31-34. Multiple isoforms of ABL1 and ABL2 have been detected; alternative splicing of the first exons results in isoforms with distinct N-terminal sequences (Fig. 1A). The 1b isoforms of both ABL kinases contain an N-terminal glycine that is myristoylated, while the 1a variants lack this site and the corresponding modification.
Figure 1. Modular domain structure of ABL family kinases.

A. Schematic representation of the modular domains of the ABL kinases. Alternative splicing of ABL1 and ABL2 produces several isoforms, including 1a isoforms (solid line) and the 1b isoforms (jagged line); the later are targeted for N-terminal myristoylation153. The ABL N-termini are comprised of the Src homology 3 (SH3), SH2 and SH1 domains. The ABL C-termini contain a conserved F-actin-binding domain. ABL1 has a G-actin binding domain whereas ABL2 has a second internal F-actin binding domain. ABL1 has three nuclear localization signal (NLS) motifs and one nuclear export signal (NES) in its C-terminus. Both ABL1 and ABL2 have conserved PXXP motifs to mediate protein-protein interactions. Phosphorylation (P) of ABL1 at Y412 within the activation loop (ABL2 Y439) and Y245 in the SH2-kinase domain linker (ABL2 Y272) stabilizes the active conformation. B. Representation of auto-inhibited (closed) and active (open) ABL kinases to depict the role of intramolecular interactions in the regulation of ABL kinase activity. The SH3 domain binds to the linker sequence connecting the SH2 and kinase domains, while the SH2 domain interacts with C-terminal lobe of the kinase (SH1) domain forming a SH3-SH2 clamp structure. The myristoylated residue in the N-terminus of the 1b ABL isoforms binds to a hydrophobic pocket within the C-lobe of the kinase domain, stabilizing the auto-inhibited conformation. The configuration and interactions of the ABL C-terminal sequences are not included in the model. C. Specificity of selected ABL TKIs: imatinib (Gleevec, STI571; Novartis), nilotinib (Tasigna, AMN107; Novartis), dasatinib (Sprycel, BMS-354825; Bristol-Myers Squibb), ponatinib (Iclusig, AP24534; Ariad Pharmaceuticals) and GNF-2, GNF-5 (allosteric inhibitors in preclinical studies). The kinase selectivity profiles for imatinib, nilotinib, and dasatinib were generated based on the binding of cellular kinases to inhibitors immobilized on solid support matrices (reviewed previously in 75). Ponatinib-sensitive kinases were identified by in vitro kinase assays; shown are targets with IC50 values of less than 20 nM152. Kinases sensitive to GNF-2 and GNF-5 were identified by in vitro kinase assays56, 154, 155.
The catalytic activities of the ABL kinases are tightly regulated by inter- and intramolecular interactions and post-translational modifications (Fig. 1B). This topic has been extensively reviewed and thus will only be briefly discussed here5, 6, 35. Among prominent intramolecular interactions required for autoinhibition are those involving the SH3 and SH2 domains35 (Fig. 1B). The SH3 domain binds to the polyproline-containing linker sequence that connects the SH2 and kinase domains, while the SH2 domain interacts with C-terminal lobe of the kinase domain (SH1) forming a SH3-SH2-SH1 clamp structure35. Autoinhibition is also mediated by the binding of the myristoylated residue in the ABL N-terminus to a hydrophobic pocket within the C-lobe of the kinase domain36. This interaction stabilizes the autoinhibited conformation of the myristoylated 1b isoform of the ABL kinase (Fig. 1B). Inhibitory intramolecular interactions that stabilize the 1a isoform of ABL are poorly understood and may involve binding to lipid residues in trans. In this regard, both ABL1 and ABL2 are inhibited by the lipid phosphatidylinositol 4,5-bisphosphate (PIP2) and experimentally decreasing PIP2 levels in the cell stimulates ABL kinase activity37.
ABL activity can be negatively or positively regulated by intermolecular interactions with distinct binding partners. In general, intermolecular interactions that disrupt autoinhibitory interactions and stabilize the active (open) conformation of the ABL kinases promote increased enzymatic activity. For example, the catalytic efficiency of the ABL kinases can be enhanced by interactions with adaptor proteins such as RIN1, which interacts with both the ABL SH3 and SH2 domains38. Intermolecular interactions that stabilize the inactive conformation of the ABL kinases inhibit enzymatic activity and downstream signaling. Among the proteins reported to negatively regulate ABL kinase activity are PRDX1 (PAG), FUS, and the PSTPIP1 adaptor protein5, 11, 39, 40.
The enzymatic activity of the ABL kinases is also regulated by tyrosine phosphorylation. Among the tyrosine residues phosphorylated on ABL1 are Y412 in the activation loop (corresponds to ABL2 Y439) and Y245 in the SH2-kinase domain linker (corresponds to ABL2 Y272). Phosphorylation of these tyrosines stabilizes the active conformation of the ABL kinases, and can occur by transphosphorylation between ABL proteins or may be mediated by Src family kinases41. The stability of ABL kinases can be modulated by tyrosine phosphorylation. Phosphorylation of ABL1 on Y245 and Y412 decreases protein stability through ubiquitination and subsequent degradation by the 26S proteasome machinery42.
ABL Activation in Leukemias
Oncogenic activation of the ABL kinases occurs in Philadelphia-positive (Ph+) human leukemias as a consequence of the t(9;22)(q34;q11) chromosome translocation that generates BCR-ABL1 fusion proteins with constitutive tyrosine kinase activity. Three different BCR-ABL1 proteins have been identified that differ in the amount of BCR sequences retained in the fusion protein, and which are associated with distinct types of leukemia: P210 BCR-ABL1 is the molecular hallmark of CML (reviewed in 43); P185 BCR-ABL1 is present in 20-30% of adult and 3-5% of childhood B-cell acute lymphocytic leukemia (B-ALL)44, 45; and P230 BCR-ABL1 has been detected in neutrophilic-CML46 as well as rare cases of CML47. The preferential association of the various BCR-ABL1 forms with distinct leukemias is not absolute as some cases of Ph+ B-ALL express P210 BCR-ABL143. The role of the BCR-ABL1 fusion proteins in the initiation and progression of leukemia has been extensively covered in previous reviews and will not be discussed here48, 49. Oncogenic activation of ABL1 in the BCR-ABL1 chimeric proteins is dependent on the presence of an N-terminal coiled-coil (CC) oligomerization domain in BCR50, 51. Multiple signaling pathways have been identified that function to mediate the oncogenic activity of BCR-ABL1, and include the RAS, NF-κB, PI3K-AKT, JUN, β-catenin, and STAT3 signaling pathways48, 52. Recent proteomic approaches have identified proximal signaling networks linked to BCR-ABL153. Interestingly, some of the adaptors and signaling enzymes identified as components of the BCR-ABL1 network have been functionally linked to the ABL1 and/or ABL2 kinases5, 54.
Inhibition of P210 BCR-ABL1 with small molecule chemical inhibitors in chronic phase CML is perhaps the most successful example of molecular targeted therapy to date55 (see Box 1). The BCR-ABL1 inhibitor imatinib (Gleevec; STI571) is an effective first-line therapy for the majority of patients diagnosed with chronic phase CML, but it is much less effective when used to treat blast crisis CML and Ph+ B-ALL patients43. Inhibitors of the ABL kinases are classified into three main classes based on their mechanism of action: type 1 inhibitors target the active conformation of the kinase domain (dasatinib, bosutinib), whereas type 2 inhibitors stabilize the inactive conformation of the kinase domain preventing its activation (imatinib, nilotinib, ponatinib). The third category of inhibitors includes allosteric inhibitors which do not compete for ATP binding, but rather bind to regulatory domains to inhibit kinase activity (Figs. 1B,C). Among these are GNF-2 and GNF-5 which target the myristoyl-binding pocket in the C-lobe of the ABL kinase domain56. In contrast to the ATP-competitive inhibitors that target multiple kinases, the allosteric inhibitors are highly selective for the ABL kinases (Fig. 1C). These allosteric inhibitors decrease BCR-ABL1 enzymatic activity and inhibit BCR-ABL1-driven leukemogenesis in mice56; thus underscoring the importance of intramolecular autoinhibitory mechanisms regulating kinase activity of the oncogenic fusion protein.
Box 1. ATP-competitive ABL kinase inhibitors.
Imatinib (Gleevec, STI571; Novartis), was the first targeted ABL inhibitor developed and approved for the treatment of CML. Treatment of CML and other Ph+ leukemias expressing BCR-ABL1 produced durable remissions in adults and children treated in the chronic phase49, 150. Remarkably, treatment of children with Ph+ ALL with imatinib plus chemotherapy has shown improved early event-free survival151. While imatinib has proven an effective drug for patients with CML, up to one-third of CML patients require alternate therapies132. The most common reason for imatinib resistance is the development of mutant forms of BCR-ABL1, including the T315I gatekeeper mutant, which prevent imatinib from docking in the ATP-binding pocket. Second-generation inhibitors such as nilotinib (Tasigna, AMN107; Novartis), which inhibits ABL kinases at 10- to 20- fold lower concentrations than imatinib, are used to treat imatinib-resistant disease131. Other second-generation inhibitors, including dasatinib (Sprycel, BMS-54825; Bristol-Myers Squibb) and bosutinib (Bosulif, SKI-606; Pfizer), inhibit ABL kinases at even lower concentrations than nilotinib, but like nilotinib and imatinib are ineffective against the BCR-ABL1 T315I mutant131. More recently, ponatinib (Iclusig, AP24534; Ariad Pharmaceuticals) was shown to inhibit both wild type and imatinib-resistant BCR-ABL1 mutants, including the T315I mutant, with similar potency. Similar to dasatinib and bosutinib, ponatinib has broad target specificity152 (Fig. 1C).
ABL1 has been identified as a fusion partner with an increasing number of genes in rare chromosome translocations in T cell acute lymphoblastic leukemia (T-ALL), B-ALL and other leukemias (Fig. 2). In addition to BCR, at least eight other ABL1 fusion partners have been identified to date including NUP214, ETV6 (TEL), EML, ZMIZ1, RCSD1, SFPQ, FOXP1 and SNX257-59 (Fig. 2). The BCR-ABL1 and ETV6-ABL1 fusions are associated with several types of leukemia including CML, B-ALL, T-ALL and acute myeloid leukemia (AML), and these ABL1 translocations may occur in hematopoietic pluripotent or committed stem cells (reviewed in 58, 60.
Figure 2. ABL activation by chromosome translocations in leukemia.

Schematic representation of ABL1, and the various ABL1 and ABL2 fusion proteins that arise as a consequence of chromosome translocations in various types of human leukemia. The BCR-ABL1 and ETV6-ABL1 fusions are associated with several types of leukemia including chronic myeloid leukemia (CML), B-cell acute lymphoblastic leukemia (B-ALL), T-cell ALL (T-ALL) and acute myeloid leukemia (AML)58. The ETV6-ABL2 fusion protein is present in rare cases of T-ALL and AML60. The NUP214-ABL1 and EML1-ABL1 fusions are primarily linked to T-ALL, while the RCSD1-ABL1, SFPQ-ABL1, ZMIZ1-ABL1, FOXP1-ABL1 and SNX2-ABL1 fusions are associated with B-ALL57, 59. The coiled-coil (CC) and other motifs in the fusion partner promote oligomerization of the resulting chimeric ABL kinases. The BCR-ABL1, NUP214-ABL1, ETV6-ABL1, ETV6-ABL2, EML1-ABL1, and ZMIZ1-ABL1 fusion proteins retain the SH3, SH2 and SH1 domains of the ABL kinase. The RCSD1-ABL1, SFPQ-ABL1, FOXP1-ABL1 and SNX2-ABL1 fusions lack the SH3 but retain part of or the entire SH2 domain. The v-Abl1 oncoprotein fuses Gag sequences of Abelson murine leukemia virus to sequences upstream of the Abl1 SH2 domain 57, 58. The v-Abl1 oncoprotein induces murine lymphosarcoma and acute pre-B cell leukemia156, 157 (HELP = hydrophobic EMAP-like protein domain, RRM = RNA recognition motif)
In contrast, the NUP214-ABL1 and EML1-ABL1 fusions are primarily linked to T-ALL, while the RCSD1-ABL1, SFPQ-ABL1, ZMIZ1-ABL1, FOXP1-ABL1 and SNX2-ABL1 fusions are associated with B-ALL. In general, the fusion partners encode proteins that contain coiled-coil or helix-loop-helix motifs that promote oligomerization of the resulting chimeric ABL kinases. However, the NUP-ABL1 fusion, which is present in about 6% of T-ALL cases, requires localization to the nuclear pore complex rather than oligomerization for enhanced transforming activity61. While the majority of chimeric fusions involve ABL1, the ABL2 gene was identified as a partner of ETV6 in rare cases of T-ALL and AML60 (Fig. 2). In general, the distinct partners fused to the N-terminus of the ABL kinases promote enhanced kinase and transforming activities by disrupting inhibitory intramolecular interactions, providing sequences that facilitate oligomerization, enhancing tyrosine phosphorylation, and/or by recruiting the chimeric kinases to distinct subcellular sites and protein complexes. The effectiveness of the ABL TKIs for the treatment of leukemias associated with the diverse ABL fusion partners other than BCR remains to be established.
ABL Kinases in Solid Tumors
In contrast to the well-established role of BCR-ABL1 in CML, less is known about the role of the ABL kinases in solid tumors. Accumulating data support a role for ABL kinases in solid tumors characterized by enhanced expression and/or activation of these kinases. The Cancer Genome Atlas (TCGA) and other studies have shown that ABL1 and ABL2 are amplified and/or overexpressed in invasive breast carcinoma, ovarian serous cystadenocarcinoma, lung adenocarcinoma, and lung squamous cell carcinoma (17-20) (Table 1). Alterations in ABL1 or ABL2 are also found in skin cutaneous melanoma, bladder urothelial carcinoma, colorectal adenocarcinoma, rhabdoid tumors, as well as renal medullary and clear cell renal carcinoma (cbioportal.org; 20, 21, 62). Recent data generated from large-scale sequencing projects revealed that alterations (amplification, mRNA increase, or somatic mutations) are more common in ABL2 than ABL1, with observed ABL2 alterations in breast invasive carcinoma (18%), uterine endometrioid carcinoma (19%), ovarian serous cystadenocarcinoma (15%), lung adenocarcinoma (16%) and lung squamous cell carcinoma (15%) (cbioportal.org; 17) (Table 1). Prevalent among these alterations is ABL2 amplification, which has been detected in invasive breast carcinomas (12%)17 and non-small cell lung adenocarcinomas (9%) (cbioportal.org; 18, 19). These findings are consistent with earlier reports of elevated ABL2 expression in advanced high-grade colorectal, pancreatic, renal and gastric tumors, implicating an association between ABL2 activity and tumor progression21, 63-65. The prevalence of ABL2 amplification and enhanced gene expression are comparable to those reported for several kinases previously implicated in breast invasive carcinoma (TCGA; 17). Somatic mutations of ABL1 and ABL2 in solid tumors are rare, but have been reported in selected solid tumors, including lung cancer and uterine corpus endometroid carcinoma (66-69, cbioportal.org). The role of these mutations in regulating ABL oncogenic activity remains to be determined.
Table 1. ABL Kinases in Solid Tumors.
| Cancer Type | Gene | Somatic Mutation |
Amplification | Deletion | mRNA up |
mRNA down |
|---|---|---|---|---|---|---|
| Breast
Invasive Carcinoma (482) |
ABL1 | <1% | <1% | <1% | 5% | 3% |
| ABL2 | <1% | 12% | <1% | 8% | 0 | |
| Uterine
Corpus Endometrioid Carcinoma (233) |
ABL1 | 3% | 0 | <1% | 4% | 1% |
| ABL2 | 6% | 4% | 0 | 9% | 0 | |
| Ovarian
Serous Cystadenocarcinoma (213) |
ABL1 | <1% | 1% | 1% | <1% | 0 |
| ABL2 | 0 | 7% | 0 | 11% | 0 | |
| Lung Adenocarcinoma (129) |
ABL1 | <1% | <1% | 0 | 2% | 7% |
| ABL2 | 4% | 9% | 0 | 4% | 0 | |
| Lung Squamous
Cell Carcinoma (177) |
ABL1 | 2% | 0 | 0 | 6% | <1% |
| ABL2 | 4% | <1% | <1% | 10% | 0 |
Data compiled from The Cancer Genome Atlas18 (cbioportal.org). mRNA z-score threshold +/−: 2.0. For each cancer type, the number of cases is shown in parenthesis.
In addition to genomic amplification and changes in ABL mRNA expression in tumors, increased ABL kinase activity has been reported in several solid tumors. Quantitative proteomic analysis in cancer cells expressing the ERBB receptors showed that ABL1 was rapidly activated in response to epidermal growth factor (EGF) stimulation7. This is consistent with previous findings that showed ABL kinases are activated downstream of the receptor tyrosine kinases (RTKs) platelet-derived growth factor receptor (PDGFR) and EGF receptor (EGFR, also known as ERBB1) in fibroblasts70. Subsequent studies demonstrated that ABL kinases are tyrosine phosphorylated and activated in breast, lung, colorectal, gastric, and prostate cancer cells as well as in melanoma8-16. Analysis of global tyrosine phosphorylation profiles of lung cancer cells showed higher levels of ABL tyrosine phosphorylation, but the identity of the upstream signals promoting tyrosine phosphorylation of the ABL kinases, and their role in the initiation and progression of lung tumors is largely unknown10-13.
The mechanisms regulating ABL activation in solid tumors differs from the constitutive kinase activation observed in BCR-ABL1+ disease (Table 2). In solid tumor cells, ABL activation has often been linked to stimulation by hyperactive RTKs and chemokine receptors8, 33. Indeed, activation of ABL kinases in breast cancer cells has been shown to occur downstream of the EGFR, Her2 (ERBB2), and insulin-like growth factor receptor (IGFR) as well as the chemokine receptor, CXCR48, 33, 71-73. Activation of ABL1 in GTL-16 gastric carcinoma cells and HepG2 hepatocellular carcinoma cells was induced by ligand-dependent activation of MET15, while ABL1 activation in HT-29 colon cancer cells was stimulated by PDGF9. Enhanced ABL1 kinase activity in human anaplastic thyroid carcinoma cells was induced by the RET-PTC3 fusion protein, a constitutively active form of the RET RTK74. Thus, the catalytic activity of the ABL kinases can be up-regulated by ligand-dependent and ligand-independent activation of RTKs. In addition to activation by upstream kinases, ABL kinases can also be activated through the inactivation of negative regulatory networks including interactions with inhibitory proteins. In this regard, activation of ABL1 in two lung adenocarcinoma cell lines correlated with loss of the tumor suppressor protein FUS11.
Table 2. Comparison of and ABL1, ABL2, and BCR-ABL1 Kinases: Activation & Signaling.
| Protein Kinase |
ABL1 | ABL2 | BCR-ABL1 |
|---|---|---|---|
| Subcellular localization |
|
|
|
| Mechanisms of Activation |
|
|
|
| Upstream Signals Leading to Reversible Activation |
|
|
NA |
| Cellular Processes Regulated Downstream of ABL Kinase Activation |
|
|
|
Regulation of cell proliferation and survival by ABL kinases
Activation of ABL kinases in solid tumor types has been linked to alterations in cell growth and survival. Several studies have reported inhibitory effects of imatinib, nilotinib and related ABL TKIs (Box 1) on cancer cell proliferation and survival in vitro and in xenograft studies, but the cellular responses to these compounds cannot be solely attributed to inhibition of the ABL kinases as these compounds have numerous kinase and some non-kinase targets75, 76 (Fig. 1C). Depletion of either ABL1 or ABL2 with specific siRNAs in breast cancer cells that express elevated levels of one or both proteins showed that both kinases were required for anchorage-independent growth72. Both ABL kinases were also required for maximal proliferation of melanoma cell lines16. In contrast, growth and survival under nutrient-deprivation required ABL2 but not ABL1 expression in some breast cancer cells (MDA-MB-435s and BT-549)72. Additionally, depletion of ABL2 alone in non-small cell lung cancer lines decreased cell growth77, and ABL1 alone was required for survival and anchorage-independent growth of gastric and hepatocellular carcinoma cells15. Among pathways mediating ABL1-dependent proliferative pathways are the Rac, p38, and ERK5 signaling pathways (reviewed in 78). Thus, cancer cells expressing high levels of ABL1 and/or ABL2 can become reliant on one or both kinases for cell growth and viability.
Accumulating data show that a growing number of signaling networks (such as the transforming growth factor β (TGFβ), WNT, and Hippo pathways) exhibit context-dependent activities in solid tumors and can promote tumorigenesis or tumor suppression under different circumstances79-81. While ABL kinases promote cell proliferation and survival in leukemias and some solid tumor cell lines, three reports have shown that ABL kinases suppress the growth of breast cancer xenografts82-84. Stimulation of the EphB4 receptor with ephrin-B2 in breast cancer cells decreased cell growth through an ABL-CRK pathway, and imatinib blocked the inhibitory effect of ephrin-B2 on the growth of these breast cancer xenografts82. Further, expression of a constitutively active form of ABL1 in the 4T1 murine mammary tumor cell line enhanced the growth suppressive effects of TGF-β in 3-dimensional (3D) cultures, and inhibited growth of 4T1 tumor xenografts83. A recent report showed that knockdown of ABL2 in the MDA-MB-231 breast cancer cell line enhanced the growth of MDA-MB-231 xenograft tumors due to increased cell proliferation 84. ABL1 and ABL2 have overlapping functions during cell proliferation in some cell types, but the consequences of depleting both kinases on the growth of the MDA-MB-231 xenograft tumors was not reported in this study84. In contrast to the growth inhibitory role of ABL kinases in the 4T1 and MDA-MB-231 xenografts, depletion of both ABL1 and ABL2 in the MCF7 breast cancer cell line markedly impaired the growth of MCF7 xenograft tumors, supporting a tumor-promoting function for ABL kinases in this cell line85. A role for ABL kinases in the growth of mammary tumors may depend on the cellular context, the repertoire of oncogenes and tumor suppressors expressed in the tumor, redundancy in the function of ABL1 and ABL2 kinases, and upstream signals that regulate tumor suppressive versus promoting pathways in distinct cell lines.
ABL kinases regulate cellular stress responses
ABL kinases can be activated in response to distinct types of cellular stress including those mediated by reactive oxygen species (ROS)86-89. Elevated levels of ROS are a feature characteristic of many solid tumors90, 91 and are also an inevitable by-product of cellular metabolism. High levels of ROS can induce cell death, whereas lower chronic levels of ROS may induce inflammation, promote tumorigenesis, and other disease states92. Oxidative stress results when the level of oxidants within the cells supersedes the antioxidant defense and may be induced by exogenous as well as endogenous sources93. Due to increased cellular metabolism, cancer cells are vulnerable to oxidative stress as the antioxidant defense system may be operating at its maximum threshold94. ABL kinase activation and signaling appear to be differentially affected by ROS levels. ABL2 was reported to promote cell survival in response to low levels of oxidative stress, but high levels of oxidative stress correlated with ABL2 degradation and apoptosis95, 96. In this regard, increased tyrosine phosphorylation of ABL2 on Y439 induced by high ROS levels promoted ABL2 ubiquitination and protein degradation95. In contrast, phosphorylation on Y261 enhanced ABL2 protein stability in response to low ROS levels95.
ABL kinases can mediate anti-proliferative and apoptotic responses induced by ROS and DNA damage86, 97, 98. Whether ABL1 functions to promote or inhibit cell growth may depend on the status of p53 (TP53) or the related p73 (TP73)99-103. ABL1 was shown to enhance cell proliferation of fibroblasts lacking p53 under non-stress conditions99, 104. However, in response to cellular stresses, including DNA damage, ABL1 induced cell growth arrest and/or apoptosis mediated by p53 or p73104. Interestingly, ABL1 can differentially modulate p53 activity and cellular responses through alteration of Mdm2, a regulator and transcriptional target of p53. Mechanistically, ABL1 protects p53 from the inhibitory effect of Mdm2, by promoting Mdm2 tyrosine phosphorylation, which restricts its interactions with p53, allowing for the accumulation of p53 and the promotion of apoptosis under stress conditions104. Recently, ABL1 was shown to be required for cell survival and activation of a p38 MAP kinase-p53-Mdm2 pathway in response to ligand-activated Met kinase in hepatocellular carcinoma cells15. Thus, ABL kinases can differentially disrupt the p53-Mdm2 regulatory loop to suppress or promote cell growth and survival depending on the upstream signals leading to ABL activation and the cellular context.
In addition to oxidative stress, growth factor deprivation was reported to activate ABL1 in mammary epithelial cells by inducing binding to the Mig6 (ERRFI1) adaptor and triggering p73-dependent apoptosis89. Silencing ABL1 in primary mammary epithelial cells (pMECs) in 3D cultures impaired lumen formation, which may be due, in part, to decreased cell death89. However, in comparison to wild-type pMECs, the ABL1-depleted pMECs failed to reconstitute the mammary gland after transplantation into the cleared mouse fat pads, suggesting impaired cell proliferation in the absence of ABL189. It is likely that activation of ABL kinases by oxidative stress and/or reduced nutrient availability prevalent in many solid tumors, may modulate tumor cell survival, invasion, and metastasis90, 91.
ABL family kinases regulate morphogenesis
ABL in cell polarity
Disruption of cell polarity is an early event in the evolution of epithelial tumors105. A role for ABL kinases in the regulation of epithelial apical-basal polarity was first described in Drosophila106-108. Drosophila Abl (D-Abl) also regulates planar cell polarity during axis elongation by modulating the tyrosine phosphorylation and localization dynamics of β-catenin109. Recently, D-Abl was shown to be required for planar cell polarity in the Drosophila eye and wing by regulation of Frizzled/Dishevelled signaling110. Furthermore, expression of activated ABL2, and to a lesser extent ABL1, induces a striking inversion of apical-basal polarity in epithelial cells grown in a 3D cell culture system in the presence of collagen111 (Fig. 3A). Inversion of apical-basal polarity by active ABL2 is mediated by at least two pathways, which include the disruption of β1-integrin localization and signaling downstream of Rap1, and impaired Rac1-mediated laminin assembly111 (Fig. 3B). Thus, ABL kinases may contribute to loss of epithelial polarity at early stages of tumor development.
Figure 3. Active ABL kinases regulate epithelial cell polarity through β1-integrin.

A. Epithelial cells expressing control vector or constitutively active forms of the ABL kinases were grown in collagen gels and stained for the apical polarity marker gp135, the adherens junction marker E-cadherin and the nuclear DAPI stain. Active ABL2, and to a lesser extent ABL1, promote polarity inversion. All pictures were taken at 40× magnification. Scale bar: 25 μm. B. Model for the inversion of epithelial cell polarity by active ABL kinase. Activation of ABL downstream of RTKs, chemokine receptors, oxidative stress and other signals leads to disruption of β1-integrin signaling and localization by regulation of the Rap1 GTPase via ABL2-mediated phosphorylation of CRK and disruption of the CRK-C3G (RAPGEF1) complex. Active ABL2 also impairs Rac1-mediated laminin assembly in epithelial cysts111.
Spindle orientation is important for morphogenesis, asymmetric cell division and stem cell self-renewal, processes that are deregulated during tumorigenesis112-115. Notably, a recent report showed that ABL1 regulates spindle orientation in epithelial cells in culture and the mouse epidermis116. Loss of ABL1 induces spindle misorientation through regulation of the LGN (LeuGly-Asn repeat-enriched protein; GPSM2) and nuclear mitotic apparatus (NuMA) polarity proteins. Whether the ABL1-induced changes in spindle orientation contributes to tumor progression remains to be determined. Interestingly, β1-integrin regulates apical-basal epithelial cell polarity and controls spindle orientation in embryonic mouse skin, and the localization of LGN and NuMA is disrupted in the epidermis of β1-integrin knockout mice117. Because expression of activated ABL2 disrupts β1-integrin localization and signaling in epithelial cells111, an intriguing possibility is that activated ABL kinases might disrupt spindle orientation indirectly through β1-integrin as well as through directly targeting the LGN and NuMA polarity proteins.
ABL in Epithelial-to-Mesenchymal Transition
The progression of solid tumors involves dramatic changes in cell morphology, adhesion, motility and polarity that are dependent on cytoskeletal rearrangements. Epithelial tumors can activate dormant developmental pathways that promote conversion from organized epithelial tissue into isolated cells with mesenchymal morphology through the Epithelial-to-Mesenchymal Transition (EMT) program118, 119. EMT is characterized by loss of cell polarity, disassembly of cell-cell junctions, acquisition of mesenchymal cell morphology and gene expression profiles, together with enhanced cell motility and invasion. Notably, these cellular processes are linked to signaling networks regulated by ABL family kinases. ABL1 was required for PDGF-induced EMT in colon cancer cells; this involved ABL1 phosphorylation of the p68 helicase (DDX5), which promoted β-catenin nuclear translocation9. Consequently, depletion and pharmacological inhibition of ABL kinases in colon and cervical cancer cells promoted epithelial phenotypes in cells that had been induced to undergo EMT9, 120. Recently, ABL1 was shown to be required for claudin-1-induced EMT in human liver cells and hepatocellular carcinoma cell lines by regulating the activation of RAS-RAF-ERK pathway121. The role of ABL1 in EMT may be context-dependent, however, as pharmacological inhibition or silencing of ABL kinases in normal epithelial cells results in dissolution of cell-cell junctions without inducing expression of genes required for the EMT program32, 111. Interestingly, alternative splicing promoted by EMT in breast cancer cells results in the formation of a spliced ABL2 isoform predominantly expressed in mesenchymal cells122. Roles for this ABL2 isoform specifically and the ABL kinases in general during EMT in solid tumors are poorly defined and require further study. The EMT program is linked to acquisition of therapeutic resistance in solid tumors118, and ABL signaling has recently been shown to be up-regulated in chemoresistant breast tumors123. Thus, an intriguing possibility to be evaluated is whether ABL kinases are important for induction and/or maintenance of EMT pathways which contribute to acquisition of therapeutic resistance in diverse solid tumors.
Role of ABL kinases during invasion and metastasis
Tumor progression and metastasis require the induction of an invasive program that promotes dissemination into surrounding tissues by primary tumor cells, followed by intravasation, migration, extravasation, and formation of metastases at distant sites124. ABL kinases engage the actin polymerization machinery in response to growth factor and chemokine stimulation as well as adhesion cues, and promote formation of membrane protrusions, morphological changes, altered cell adhesion, migration and invasion of diverse cell types6, 37, 54, 70. ABL kinases are required for cell motility and invasion induced by IGF-1, EGF, serum and chemokines in breast, hepatocellular carcinoma, and melanoma cells8, 16, 33, 73, 84, 125. One or both ABL kinases are required for invasion in several cancer cell lines, but ABL2 is preferentially required for invasion of the MDA-MB-231 breast cancer cell line84. ABL kinases can regulate invasion by directly phosphorylating proteins required for invasive behavior, and also by regulating the expression of genes linked to invasion and cell motility (Fig. 4). ABL2 localizes to invadopodia, where it is required for both matrix degradation and invasion in breast cancer cells33, 73. Knockdown of ABL2 decreases matrix degradation and invasion by MDA-MB-231 breast cancer cells, and ABL2 regulates the maturation of invadopodia by linking activation of the EGFR and Src kinases to tyrosine phosphorylation of cortactin, which is required for Arp2/3-dependent actin polymerization73. In addition to cortactin, other actin regulatory proteins are targeted by ABL kinases at invadopodia (Fig. 4). Among these are the Abl interactor 1 (Abi1) adaptor protein, a component of the WAVE (WASF) protein complex, which is required for invadopodia formation, ECM degradation and induction of matrix metalloproteinase (MMP)-9 expression in MDA-MB-231 breast cancer cells126. ABL kinases also regulate the activity of MMPs during invadopodia maturation (Fig. 4). Active ABL2 interacts with and promotes phosphorylation of the membrane type 1-matrix metalloproteinase (MT1-MMP, also known as MMP14) and is required for its localization and function at invadopodia33. Both ABL1 and ABL2 kinases are required for melanoma cell invasion by regulating MMP expression through STAT3-dependent and independent pathways16 (Fig. 4). Other potential targets of ABL kinases at invadopodia include the N-WASP (WASL) actin regulatory protein, which is required for Arp2/3-mediated actin polymerization at invadopodia, and is phosphorylated by ABL kinases127-129. Recently, HEF1 (NEDD9) was identified as a direct substrate and binding partner of the ABL kinases in T cells54. The HEF1 protein also regulates cancer cell invasion, and ABL-mediated phosphorylation of HEF1 may be required for promoting invasive behavior as observed during chemokine-induced T cell migration130. Consistent with its ability to positively regulate invadopodia formation and function, knockdown of ABL2 decreased cancer cell invasion and intravasation following implantation of MDA-MB-231 cells in the mammary fat pad84.
Figure 4. ABL kinases regulate cancer cell invasion.
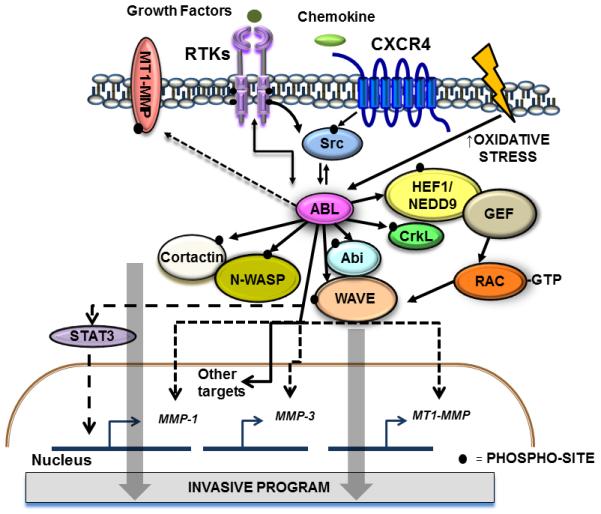
Activation of ABL kinases downstream of hyperactive RTKs, chemokine receptors and Src kinases, or in response to oxidative stress promotes cancer cell invasion by direct or indirect activation of actin regulatory proteins such as cortactin, N-WASP, Abl interactor 1 (Abi1), WAVE, CrkL, HEF1/NEDD9, and the Rac GTPase54, 73, 126-128, 130. ABL kinases regulate MT1-MMP localization and activity33. ABL2 interacts with MT1-MMP promoting its phosphorylation, and ECM degradation33. One or both ABL kinases are required for cancer cell invasion depending on the cellular context16, 73. ABL kinases also regulate cancer cell invasion by modulating expression of several MMPs and other genes involved in EGFR-mediated invasion16, 84. ABL1 regulates MMP1 expression through a STAT3-dependent pathway, and ABL2 regulates the expression of MMP1, MMP3, and MT1-MMP through yet to be identified transcription factors. Solid lines indicate direct links between ABL and target proteins; dotted lines indicate indirect links.
Pharmacological inhibition of ABL kinases in cancer
To date, there are five approved ABL inhibitors for the treatment of BCR-ABL1+ CML (Box 1). The pharmacology and clinical data of these drugs has been recently reviewed131. While imatinib has proven remarkably successful for the treatment of CML, particularly those in chronic phase, up to one-third of patients will require alternative therapies132. The effectiveness of imatinib decreases during the acute and blast stages of CML. Development of imatinib resistance may result from the development of mutations in BCR-ABL1 that alter drug binding, including the T315I gatekeeper mutation. Other ATP-competitive inhibitors, such as nilotinib, dasatinib, and bosutinib are also ineffective against the T315I mutant form of BCR-ABL1131. Newer allosteric inhibitors have been tested in preclinical models of resistance; among these are the GNF-2 and GNF-5 allosteric inhibitors that target the myristate pocket of ABL kinases (Fig. 1C). Combining GNF-5 with imatinib or nilotinib prolonged survival of mice with imatinib-resistant BCR-ABL1 T315I-induced leukemia56. Another type of allosteric inhibitor, an engineered ABL1 SH2-binding fibronectin type III monobody, blocked the intramolecular interaction between the SH2 and kinase domains of BCR-ABL1, inhibited kinase activity in vitro and in vivo, impaired leukemogenesis in mice, and increased the sensitivity of imatinib-resistant BCR-ABL1 mutants to TKI therapies133. Thus, the combined use of allosteric and ATP competitive inhibitors may represent an effective strategy to overcome resistance to either type of compound alone.
Use of ABL kinase inhibitors for solid tumors: past and future
In contrast to the success of TKI therapies for the treatment of BCR-ABL1-induced leukemias, use of the ATP-competitive inhibitors imatinib, nilotinib and dasatinib has not achieved similar success for the treatment of solid tumors (reviewed in 134, 135). The variable clinical responses to these TKIs may reflect the heterogeneous nature of solid tumors, which often have acquired mutations in multiple tumor promoting pathways. In addition, recent work has shown that signaling networks can be rewired in response to treatment with single kinase inhibitors136, 137, which may underlie poor responses to treatment. Another mechanism that underlies the poor response to TKI therapy in some solid tumors and chemoresistant leukemias is the targeting of alternative pathways resulting in paradoxical activation of proliferative pathways. Treatment with imatinib, nilotinib, and dasatinib can activate BRAF/ CRAF (RAF1) complexes in cells expressing active RAS leading to enhanced activation of the MEK-ERK pathway in diverse cancer cell lines including melanoma, lung, colorectal and pancreatic carcinoma, as well as BCR-ABL1 TKI-resistant leukemic cells76. The paradoxical MEK-ERK activation by these TKIs is reminiscent of the increased MEK-ERK pathway activation induced by vemurafenib, a B-RAF inhibitor, in cancer cells expressing active RAS, which was reported to promote cell proliferation in soft agar and accelerate cutaneous squamous-cell tumors in mouse models and patients138. At the molecular level, whereas imatinib, nilotinib, and dasatinib promote the formation of B-RAF/C-RAF complexes in cells with activated RAS, these TKIs are weak RAF inhibitors in vitro76. Therefore, the indiscriminate use of imatinib, nilotinib, dasatinib and other ABL-targeted ATP-competitive TKIs is not recommended; rather, screening tumors for ABL amplification, enhanced expression and hyperactivation, as well as co-expression of other mutations associated with poor response to specific TKIs, is crucial for identifying those tumors that may benefit from therapies with selective ABL TKIs.
Notably, in contrast to the ABL-targeted ATP-competitive TKIs, the ABL allosteric inhibitor GNF-2 does not inhibit BRAF activity in vitro, fails to induce formation of BRAF/CRAF dimers, and thus, treatment with this compound does not elicit paradoxical activation of the MEK-ERK pathway76. Development of additional selective ABL TKIs and further mechanistic understanding of the signaling networks disrupted in response to distinct ABL TKIs, may better inform the selection of therapies that can be used in combination with ABL inhibitors. Thus, tumors with ABL2 genomic amplification and/or robust kinase activity (as detected by high levels of pCRKL) might be predicted to be good candidates for TKIs that are selective for ABL kinases (and do not target B-RAF or other kinases that may induce paradoxical activation of MEK-ERK and other tumor-promoting pathways). Additionally, developing an ABL signature (genomic, transcriptional, or phospho-proteomic) might be critical to identify subset of patients to rationally test the effects of these inhibitors.
One interesting possibility for the future of ABL TKIs in cancer therapy may lie in overcoming resistance to chemotherapies. Enhanced expression of ABL1 signaling networks was recently detected in endocrine-resistant breast cancer123. ABL1 and PDGFR were identified as the top adaptive pathways characteristic of estrogen receptor (ER)-positive breast cancers that acquired resistance to aromatase inhibitor (AI) therapy. Activation of the PDGFR and ABL1 pathways was linked to long-term estrogen deprivation in MCF7 breast cancer cells, and was associated with decreased anti-proliferative response to AI treatment in primary ER-positive breast carcinomas123, 139. Moreover, treatment of these cells with nilotinib (which inhibits both the PDGFR and ABL1), suppressed ER-mediated transcription by destabilizing the ER. This finding is consistent with reports that ABL1 regulates ERα-mediated transcription by inducing phosphorylation and stabilization of ERα140 and that silencing ABL1 in ER-positive breast cancer cells sensitized these cells to tamoxifen treatment141. Treatment of breast cancer cells with imatinib has been shown to sensitize these cells to selected drugs such as 5-fluorouracil, cisplatin and vinorelbine142, 143. Furthermore, imatinib treatment or ABL1 depletion was shown to restore sensitivity to the EGFR and ErbB2 inhibitor lapatinib in breast cancer cells that had acquired lapatinib-resistance144. Future studies are warranted to define how ABL1 inhibition sensitizes cancer cells to chemotherapies and whether such an approach may improve the effectiveness of chemotherapy in vivo.
Conclusions and perspectives
While the ABL1 kinase was first linked to the induction of human leukemia almost three decades ago2, a role for ABL family kinases in the progression of solid tumors has only recently been recognized. Over the last decade the use of imatinib, dasatinib and nilotinib to treat patients with BCR-ABL1-positive leukemias provided the best example for the successful use of TKI-targeted therapy. However, it is clear that the use of these drugs is inadequate for the treatment of solid tumors as monotherapies due to the complexity of mutations even in early-stage tumors, and the potential for inappropriate activation (rather than inhibition) of proliferative pathways by some TKIs with multiple protein targets. Ongoing preclinical and future clinical studies are expected to evaluate whether treatment of patients with tumors with ABL amplification, enhanced expression and hyper-activation could benefit from therapies with ABL-selective TKIs. The identification of new allosteric inhibitors with greater specificity against ABL family kinases may allow for evaluation of the effectiveness of targeting these kinases in the treatment of solid tumors with hyperactive ABL kinases, and whether anti-ABL therapies display greater effectiveness at specific tumor stages. Recent findings suggest that ABL1 and ABL2 may regulate overlapping as well as distinct cellular processes in various cell types, and may differentially contribute to tumor progression. Future studies will be required to evaluate unique roles for the ABL kinases not only in selected solid tumors, but also in cells in the tumor microenvironment. ABL kinases regulate diverse signaling pathways in fibroblasts, macrophages, and T cells34, 54, 70, 100, 145, 146. These cell types are known to contribute to tumor progression and metastasis in various cancers147-149, and a role for ABL kinases in tumor-promoting pathways in distinct cell types in the microenvironment should be an area of active enquiry. Exciting research over the next few years is expected to evaluate the contribution of ABL kinases to distinct tumor types and establish whether these kinases prove to be useful targets for the treatment of multiple solid tumor types.
Acknowledgements
The authors regret that due to space limitations, they could not directly cite the work of many investigators. We thank Ran Li for confocal images. The Pendergast laboratory is supported by grants from the National Cancer Institute, including NCI grants R01CA155160 and R01CA070940 to A.M.P. We also acknowledge the support by training grants from the Department of Defense Breast Cancer Research Program Fellowship W81XWH-10-1-0345 and the Pharmaceutical Research and Manufacturers of America to E.K.G., and ACS Spin-Odyssey Postdoctoral grant 11984-7-PF-10-228-01-CSM to P.S.S.P.
GLOSSARY
- 3D cultures
system of growing cells in a three-dimensional environment, matrix, or scaffold to more closely model physiological conditions
- Activation loop
conserved regulatory motif that extends into the active kinase domain and is phosphorylated in the active conformation of the kinase; the activation loop serves as a binding platform for the peptide substrate to be phosphorylated
- Adherens junctions
Intercellular adhesion structures that tightly seal the lateral spaces between cells in simple epithelia. They contain the intercellular adhesion molecule E-cadherin, and β- and α-catenin
- Apical-basal polarity
Epithelial cells are polarized, with an apical membrane that faces the external environment or a lumen and is opposite the basolateral membrane, which functions in cell–cell interactions and contacts the basement membrane
- Aromatase inhibitors (AIs)
Drugs that block aromatase, the enzyme that converts androgens to oestrogens; used to treat oestrogen receptor+ patients with breast cancer by decreasing circulating levels of oestrogenic compounds
- Arp2/3 complex
a seven subunit protein complex involved in regulation of the actin cytoskeleton; mediates nucleation of branched actin filaments
- ErbB receptors
family of structurally related receptor tyrosine kinases of which there are four members, ErbB1 (EGFR, Her1), ErB2 (Her2, Neu), ErbB3 (Her3), and ErbB4 (Her4)
- Focal adhesions
sites where integrins and proteoglycan-mediated adhesions connect to the actin cytoskeleton
- Invadopodia
actin-rich, protrusive structures in cancers cells that promote remodeling of the extracellular matrix during tumor invasion
- Monobody
genetically engineered proteins that recognize specific antigens
- Oxidative stress
An excess of reactive oxygen species (ROS), caused by an imbalance between the rate of reduction and oxidation of oxygen, leading to free radical generation and damage to cellular components such as DNA and lipids
- Phagocytic cups
actin-rich membrane invagination that closes to form phagosomes during phagocytosis
- Planar cell polarity
the coordinated polarization of cells in the plane of the tissue
- SH1 domain
SRC homology- 1 domain, refers to the tyrosine kinase domain, first identified in the SRC kinase
- SH2 domain
SRC homology-2 domain is a protein module that binds to tyrosine phosphorylated sites in a sequence-specific context
- SH3 domain
SRC homology-3 domain is a protein module that binds to proline-rich sequences
- WAVE protein complex
mediates activation of the Arp2/3 complex at the leading edge of migrating cells
Footnotes
Competing Interests Statement
The authors declare no competing financial interests.
Online links
DATABASES: http://cbioportal.org; TCGA database at www.oncomine.
FURTHER INFORMATION: Pendergast Lab homepage: PendergastLab.duhs.duke.edu
REFERENCES
- 1.Goff SP, Gilboa E, Witte ON, Baltimore D. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: studies with cloned viral DNA. Cell. 1980;22:777–85. doi: 10.1016/0092-8674(80)90554-1. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–4. doi: 10.1126/science.3460176. References 1 and 2 are historic manuscripts that describe the identification of normal and oncogenic forms of mouse and human ABL kinases.
- 3.Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–91. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- 4.Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- 5.Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 2010;3:re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J. Cell Sci. 2009;122:3441–54. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–74. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 8.Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–55. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell. 2006;127:139–55. doi: 10.1016/j.cell.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 10.Rikova K, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Lin J, et al. Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene. 2007;26:6989–96. doi: 10.1038/sj.onc.1210500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu CJ, et al. A predictive phosphorylation signature of lung cancer. PLoS One. 2009;4:e7994. doi: 10.1371/journal.pone.0007994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carretero J, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake JM, et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc. Natl. Acad. Sci. U.S.A. 2012;109:1643–8. doi: 10.1073/pnas.1120985109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furlan A, et al. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell Death and Differentiation. 2011;18:1608–16. doi: 10.1038/cdd.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganguly SS, et al. c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene. 2012;31:1804–16. doi: 10.1038/onc.2011.361. References 8 and 16 showed that ABL kinases are required for invasion of breast cancer cells and melanoma.
- 17.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412.
- 18.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sos ML, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J. Clin. Invest. 2009;119:1727–40. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simpson L, et al. Renal medullary carcinoma and ABL gene amplification. J. Urol. 2005;173:1883–8. doi: 10.1097/01.ju.0000158448.56888.09. [DOI] [PubMed] [Google Scholar]
- 21.Behbahani TE, et al. Tyrosine kinase expression profile in clear cell renal cell carcinoma. World J. Urol. 2012;30:559–65. doi: 10.1007/s00345-011-0767-z. [DOI] [PubMed] [Google Scholar]
- 22.Schwartzberg PL, et al. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–75. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 23.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–63. doi: 10.1016/0092-8674(91)90011-m. References 22 and 23 describe the consequences of genetic inactivation of the murine abl1 (c-abl) gene, which resulted in immune deficits and decreased viability.
- 24.Qiu Z, Cang Y, Goff SP. Abl family tyrosine kinases are essential for basement membrane integrity and cortical lamination in the cerebellum. J. Neurosci. 2010;30:14430–9. doi: 10.1523/JNEUROSCI.2861-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li B, et al. Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nature Genetics. 2000;24:304–8. doi: 10.1038/73542. [DOI] [PubMed] [Google Scholar]
- 26.Kua HY, et al. c-Abl promotes osteoblast expansion by differentially regulating canonical and non-canonical BMP pathways and p16INK4a expression. Nat. Cell Biol. 2012;14:727–37. doi: 10.1038/ncb2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koleske AJ, et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–72. doi: 10.1016/s0896-6273(00)80646-7. This hallmark paper revealed that genetic inactivation of murine abl1 and abl2 (arg) resulted in embryonic lethality, demonstrating that these two protein kinases have some critical redundant functions during development.
- 28.Gourley SL, Koleske AJ, Taylor JR. Loss of dendrite stabilization by the Abl-related gene (Arg) kinase regulates behavioral flexibility and sensitivity to cocaine. Proc. Natl. Acad. Sci. U.S.A. 2009;106:16859–64. doi: 10.1073/pnas.0902286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Etten RA, et al. The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J. Cell Biol. 1994;124:325–40. doi: 10.1083/jcb.124.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Miller AL, Mooseker MS, Koleske AJ. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14865–70. doi: 10.1073/pnas.251249298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller AL, Wang Y, Mooseker MS, Koleske AJ. The Abl-related gene (Arg) requires its F-actin-microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J. Cell Biol. 2004;165:407–19. doi: 10.1083/jcb.200308055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zandy NL, Playford M, Pendergast AM. Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc. Natl. Acad. Sci. U.S.A. 2007;104:17686–91. doi: 10.1073/pnas.0703077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith-Pearson PS, Greuber EK, Yogalingam G, Pendergast AM. Abl kinases are required for invadopodia formation and chemokine-induced invasion. J. Biol. Chem. 2010;285:40201–11. doi: 10.1074/jbc.M110.147330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greuber EK, Pendergast AM. Abl family kinases regulate FcgammaR-mediated phagocytosis in murine macrophages. J. Immunol. 2012;189:5382–92. doi: 10.4049/jimmunol.1200974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 36.Hantschel O, et al. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 2003;112:845–57. doi: 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 37.Plattner R, et al. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat. Cell Biol. 2003;5:309–19. doi: 10.1038/ncb949. References 36 and 37 showed that regulation of ABL kinases is mediated in part by lipids through distinct mechanisms. Ref. 36 showed that the intramolecular interaction of the myristoylated residue in the ABL N-terminus with a hydrophobic pocket in the kinase domain stabilizes the auto-inhibited conformation of the kinase. Ref. 37 showed that PtdIns(4,5)P2 inhibits ABL1 activity in vitro and in cells, and that decreasing PtdIns(4,5)P2 cellular levels by PLCγ1-mediated hydrolysis or dephosphorylation by an inositol-polyphosphate phosphatase results in dramatic increases in ABL kinase activity.
- 38.Cao X, Tanis KQ, Koleske AJ, Colicelli J. Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J. Biol. Chem. 2008;283:31401–7. doi: 10.1074/jbc.M804002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wen ST, Van Etten RA. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–67. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cong F, et al. Cytoskeletal protein PSTPIP1 directs the PEST-type protein tyrosine phosphatase to the c-Abl kinase to mediate Abl dephosphorylation. Mol. Cell. 2000;6:1413–23. doi: 10.1016/s1097-2765(00)00138-6. [DOI] [PubMed] [Google Scholar]
- 41.Tanis KQ, Veach D, Duewel HS, Bornmann WG, Koleske AJ. Two distinct phosphorylation pathways have additive effects on Abl family kinase activation. Mol. Cell Biol. 2003;23:3884–96. doi: 10.1128/MCB.23.11.3884-3896.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Echarri A, Pendergast AM. Activated c-Abl is degraded by the ubiquitin-dependent proteasome pathway. Curr. Biol. 2001;11:1759–65. doi: 10.1016/s0960-9822(01)00538-3. [DOI] [PubMed] [Google Scholar]
- 43.Wong S, Witte ON. The BCR-ABL story: bench to bedside and back. Annu. Rev. Immunol. 2004;22:247–306. doi: 10.1146/annurev.immunol.22.012703.104753. [DOI] [PubMed] [Google Scholar]
- 44.Kantarjian HM, et al. Significance of the P210 versus P190 molecular abnormalities in adults with Philadelphia chromosome-positive acute leukemia. Blood. 1991;78:2411–8. [PubMed] [Google Scholar]
- 45.Suryanarayan K, et al. Consistent involvement of the bcr gene by 9;22 breakpoints in pediatric acute leukemias. Blood. 1991;77:324–30. [PubMed] [Google Scholar]
- 46.Pane F, et al. Neutrophilic-chronic myeloid leukemia: a distinct disease with a specific molecular marker (BCR/ABL with C3/A2 junction) Blood. 1996;88:2410–4. [PubMed] [Google Scholar]
- 47.Wilson G, et al. BCR-ABL transcript with an e19a2 (c3a2) junction in classical chronic myeloid leukemia. Blood. 1997;89:3064. [PubMed] [Google Scholar]
- 48.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer. 2005;5:172–83. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 49.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer. 2012;12:513–26. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 50.McWhirter JR, Wang JY. Activation of tyrosinase kinase and microfilament-binding functions of c-abl by bcr sequences in bcr/abl fusion proteins. Mol. Cell Biol. 1991;11:1553–65. doi: 10.1128/mcb.11.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muller AJ, et al. BCR first exon sequences specifically activate the BCR/ABL tyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol. Cell Biol. 1991;11:1785–92. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin AS., Jr. A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998;12:968–81. doi: 10.1101/gad.12.7.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Titz B, et al. The proximal signaling network of the BCR-ABL1 oncogene shows a modular organization. Oncogene. 2010;29:5895–910. doi: 10.1038/onc.2010.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu JJ, Lavau CP, Pugacheva E, Soderblom EJ, Moseley MA, Pendergast AM. Abl Family Kinases Modulate T Cell-Mediated Inflammation and Chemokine-Induced Migration through the Adaptor HEF1 and the GTPase Rap1. Sci. Signal. 2012;5:ra51. doi: 10.1126/scisignal.2002632. This manuscript established a requirement for both ABL1 and ABL2 in chemokine-induced polarity and migration in T cells. A novel pathway was identified linking ABL kinases to HEF1/NEDD9-mediated regulation of Rap1 during chemokine-induced migration and polarization.
- 55.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 56.Zhang J, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–6. doi: 10.1038/nature08675. This paper describes allosteric inhibitors specific for the ABL kinases that target the myristate-binding pocket and impair downstream signaling as well as BCR-ABL-induced leukemia in mice.
- 57.Hagemeijer A, Graux C. ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes, Chromosomes and Cancer. 2010;49:299–308. doi: 10.1002/gcc.20743. [DOI] [PubMed] [Google Scholar]
- 58.De Braekeleer E, et al. ABL1 fusion genes in hematological malignancies: a review. European Journal of Haematology. 2011;86:361–71. doi: 10.1111/j.1600-0609.2011.01586.x. [DOI] [PubMed] [Google Scholar]
- 59.Ernst T, et al. Identification of FOXP1 and SNX2 as novel ABL1 fusion partners in acute lymphoblastic leukaemia. British Journal of Haematology. 2011;153:43–6. doi: 10.1111/j.1365-2141.2010.08457.x. [DOI] [PubMed] [Google Scholar]
- 60.De Braekeleer E, et al. ETV6 fusion genes in hematological malignancies: a review. Leuk. Res. 2012;36:945–61. doi: 10.1016/j.leukres.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 61.De Keersmaecker K, et al. Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Mol. Cell. 2008;31:134–42. doi: 10.1016/j.molcel.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 62.Koos B, et al. The tyrosine kinase c-Abl promotes proliferation and is expressed in atypical teratoid and malignant rhabdoid tumors. Cancer. 2010;116:5075–81. doi: 10.1002/cncr.25420. [DOI] [PubMed] [Google Scholar]
- 63.Chen G, et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 1999;274:12748–52. doi: 10.1074/jbc.274.18.12748. [DOI] [PubMed] [Google Scholar]
- 64.Crnogorac-Jurcevic T, et al. Expression profiling of microdissected pancreatic adenocarcinomas. Oncogene. 2002;21:4587–94. doi: 10.1038/sj.onc.1205570. [DOI] [PubMed] [Google Scholar]
- 65.Wu CW, et al. Arg tyrosine kinase expression in human gastric adenocarcinoma is associated with vessel invasion. Anticancer Research. 2003;23:205–10. [PubMed] [Google Scholar]
- 66.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruhe JE, et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines. Cancer Res. 2007;67:11368–76. doi: 10.1158/0008-5472.CAN-07-2703. [DOI] [PubMed] [Google Scholar]
- 68.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hammerman PS, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–11. doi: 10.1101/gad.13.18.2400. This manuscript demonstrated for the first time that endogenous ABL1 (cellular ABL) is activated by growth factors PDGF and EGF, and also by activated Src family kinases.
- 71.Sirvent A, Boureux A, Simon V, Leroy C, Roche S. The tyrosine kinase Abl is required for Src-transforming activity in mouse fibroblasts and human breast cancer cells. Oncogene. 2007;26:7313–23. doi: 10.1038/sj.onc.1210543. [DOI] [PubMed] [Google Scholar]
- 72.Srinivasan D, Sims JT, Plattner R. Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival. Oncogene. 2008;27:1095–105. doi: 10.1038/sj.onc.1210714. This manuscript showed that similar to growth factor-stimulated fibroblasts, maximal proliferation and survival of some breast cancer cells required the activity of ABL kinases.
- 73.Mader CC, et al. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–41. doi: 10.1158/0008-5472.CAN-10-1432. References 33 and 73 showed a role for ABL kinases in the formation and function of invadopodia. ABL2 localizes to invadopodia and regulates the function of invadopodia components including cortactin and MT1-MMP in breast cancer cells.
- 74.Iavarone C, et al. Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the RET proto-oncogene. J. Biol. Chem. 2006;281:10567–76. doi: 10.1074/jbc.M513397200. [DOI] [PubMed] [Google Scholar]
- 75.Hantschel O, Rix U, Superti-Furga G. Target spectrum of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib. Leuk. Lymphoma. 2008;49:615–9. doi: 10.1080/10428190801896103. [DOI] [PubMed] [Google Scholar]
- 76.Packer LM, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–27. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yuan BZ, Jefferson AM, Popescu NC, Reynolds SH. Aberrant gene expression in human non small cell lung carcinoma cells exposed to demethylating agent 5-aza-2′-deoxycytidine. Neoplasia. 2004;6:412–9. doi: 10.1593/neo.03490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sirvent A, Benistant C, Roche S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biologie Cellulaire. 2008;100:617–31. doi: 10.1042/BC20080020. [DOI] [PubMed] [Google Scholar]
- 79.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 80.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat. Rev. Cancer. 2010;10:415–24. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 81.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat. Rev. Cancer. 2013;13:246–57. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 82.Noren NK, Foos G, Hauser CA, Pasquale EB. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl-Crk pathway. Nat. Cell Biol. 2006;8:815–25. doi: 10.1038/ncb1438. [DOI] [PubMed] [Google Scholar]
- 83.Allington TM, Galliher-Beckley AJ, Schiemann WP. Activated Abl kinase inhibits oncogenic transforming growth factor-beta signaling and tumorigenesis in mammary tumors. FASEB J. 2009;23:4231–43. doi: 10.1096/fj.09-138412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gil-Henn H, et al. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene. 2012;31:1804–1816. doi: 10.1038/onc.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li X, et al. c-Abl and Arg tyrosine kinases regulate lysosomal degradation of the oncoprotein Galectin-3. Cell Death and Differentiation. 2010;17:1277–87. doi: 10.1038/cdd.2010.8. [DOI] [PubMed] [Google Scholar]
- 86.Shaul Y, Ben-Yehoyada M. Role of c-Abl in the DNA damage stress response. Cell Res. 2005;15:33–5. doi: 10.1038/sj.cr.7290261. [DOI] [PubMed] [Google Scholar]
- 87.Sun X, et al. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. J. Biol. Chem. 2000;275:17237–40. doi: 10.1074/jbc.C000099200. [DOI] [PubMed] [Google Scholar]
- 88.Cao C, et al. The ARG tyrosine kinase interacts with Siva-1 in the apoptotic response to oxidative stress. J. Biol. Chem. 2001;276:11465–8. doi: 10.1074/jbc.C100050200. [DOI] [PubMed] [Google Scholar]
- 89.Hopkins S, et al. Mig6 is a sensor of EGF receptor inactivation that directly activates c-Abl to induce apoptosis during epithelial homeostasis. Dev. Cell. 2012;23:547–59. doi: 10.1016/j.devcel.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grek CL, Tew KD. Redox metabolism and malignancy. Curr. Opin. Pharmacol. 2010;10:362–8. doi: 10.1016/j.coph.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer. 2008;8:425–37. doi: 10.1038/nrc2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 93.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004;44:239–67. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 94.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004;53:209–19. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 95.Cao C, et al. Ubiquitination and degradation of the Arg tyrosine kinase is regulated by oxidative stress. Oncogene. 2005;24:2433–40. doi: 10.1038/sj.onc.1208454. [DOI] [PubMed] [Google Scholar]
- 96.Cao C, Leng Y, Li C, Kufe D. Functional interaction between the c-Abl and Arg protein-tyrosine kinases in the oxidative stress response. J. Biol. Chem. 2003;278:12961–7. doi: 10.1074/jbc.M300058200. [DOI] [PubMed] [Google Scholar]
- 97.Kharbanda S, et al. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature. 1995;376:785–8. doi: 10.1038/376785a0. [DOI] [PubMed] [Google Scholar]
- 98.Yuan ZM, et al. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382:272–4. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- 99.Whang YE, et al. c-Abl is required for development and optimal cell proliferation in the context of p53 deficiency. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5486–91. doi: 10.1073/pnas.97.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Furstoss O, et al. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J. 2002;21:514–24. doi: 10.1093/emboj/21.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goldberg Z, et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J. 2002;21:3715–27. doi: 10.1093/emboj/cdf384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–13. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 103.Yuan ZM, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399:814–7. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- 104.Levav-Cohen Y, et al. C-Abl as a modulator of p53. Biochem. Biophys. Res. Commun. 2005;331:737–49. doi: 10.1016/j.bbrc.2005.03.152. [DOI] [PubMed] [Google Scholar]
- 105.Feigin ME, Muthuswamy SK. Polarity proteins regulate mammalian cell-cell junctions and cancer pathogenesis. Curr. Opin. Cell Biol. 2009;21:694–700. doi: 10.1016/j.ceb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baum B, Perrimon N. Spatial control of the actin cytoskeleton in Drosophila epithelial cells. Nat. Cell Biol. 2001;3:883–90. doi: 10.1038/ncb1001-883. [DOI] [PubMed] [Google Scholar]
- 107.Grevengoed EE, Loureiro JJ, Jesse TL, Peifer M. Abelson kinase regulates epithelial morphogenesis in Drosophila. J. Cell Biol. 2001;155:1185–98. doi: 10.1083/jcb.200105102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fox DT, Peifer M. Abelson kinase (Abl) and RhoGEF2 regulate actin organization during cell constriction in Drosophila. Development. 2007;134:567–78. doi: 10.1242/dev.02748. [DOI] [PubMed] [Google Scholar]
- 109.Tamada M, Farrell DL, Zallen JA. Abl regulates planar polarized junctional dynamics through beta-catenin tyrosine phosphorylation. Dev. Cell. 2012;22:309–19. doi: 10.1016/j.devcel.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Singh J, Aaronson SA, Mlodzik M. Drosophila Abelson kinase mediates cell invasion and proliferation through two distinct MAPK pathways. Oncogene. 2010;29:4033–45. doi: 10.1038/onc.2010.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li R, Pendergast AM. Arg kinase regulates epithelial cell polarity by targeting beta1-integrin and small GTPase pathways. Curr. Biol. 2011;21:1534–42. doi: 10.1016/j.cub.2011.08.023. This work describes a role for activated ABL kinases in the regulation of epithelial apical-basal polarity. Expression of active ABL2 induces striking inversion of epithelial apical-basal polarity by disrupting β1-integrin signaling and laminin assembly. Thus, activated ABL kinases may be involved in early steps of tumor initiation.
- 112.Quyn AJ, et al. Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell. 2010;6:175–81. doi: 10.1016/j.stem.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 113.Gonzalez C. Spindle orientation, asymmetric division and tumour suppression in Drosophila stem cells. Nat. Rev. Genet. 2007;8:462–72. doi: 10.1038/nrg2103. [DOI] [PubMed] [Google Scholar]
- 114.Fleming ES, Temchin M, Wu Q, Maggio-Price L, Tirnauer JS. Spindle misorientation in tumors from APC(min/+) mice. Molecular Carcinogenesis. 2009;48:592–8. doi: 10.1002/mc.20506. [DOI] [PubMed] [Google Scholar]
- 115.Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132:583–97. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 116.Matsumura S, et al. ABL1 regulates spindle orientation in adherent cells and mammalian skin. Nat. Commun. 2012;3:626. doi: 10.1038/ncomms1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275–80. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 119.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 120.Yasmeen A, Alachkar A, Dekhil H, Gambacorti-Passerini C, Al Moustafa AE. Locking Src/Abl Tyrosine Kinase Activities Regulate Cell Differentiation and Invasion of Human Cervical Cancer Cells Expressing E6/E7 Oncoproteins of High-Risk HPV. J. Oncol. 2010;2010 doi: 10.1155/2010/530130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Suh Y, et al. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene. 2012 doi: 10.1038/onc.2016.294. [DOI] [PubMed] [Google Scholar]
- 122.Shapiro IM, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weigel MT, et al. Enhanced expression of the PDGFR/Abl signaling pathway in aromatase inhibitor-resistant breast cancer. Annals of Oncology. 2012;24:126–33. doi: 10.1093/annonc/mds240. [DOI] [PubMed] [Google Scholar]
- 124.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 125.Yoon CH, et al. Claudin-1 acts through c-Abl-protein kinase Cdelta (PKCdelta) signaling and has a causal role in the acquisition of invasive capacity in human liver cells. J. Biol. Chem. 2010;285:226–33. doi: 10.1074/jbc.M109.054189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun X, et al. Abl interactor 1 regulates Src-Id1-matrix metalloproteinase 9 axis and is required for invadopodia formation, extracellular matrix degradation and tumor growth of human breast cancer cells. Carcinogenesis. 2009;30:2109–16. doi: 10.1093/carcin/bgp251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Burton EA, Oliver TN, Pendergast AM. Abl kinases regulate actin comet tail elongation via an N-WASP-dependent pathway. Mol. Cell Biol. 2005;25:8834–43. doi: 10.1128/MCB.25.20.8834-8843.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Oser M, et al. Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 2009;186:571–87. doi: 10.1083/jcb.200812176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Oser M, et al. Specific tyrosine phosphorylation sites on cortactin regulate Nck1-dependent actin polymerization in invadopodia. J. Cell Sci. 2010;123:3662–73. doi: 10.1242/jcs.068163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Singh M, Cowell L, Seo S, O’Neill G, Golemis E. Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochemistry and Biophysics. 2007;48:54–72. doi: 10.1007/s12013-007-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hantschel O, Grebien F, Superti-Furga G. The growing arsenal of ATP-competitive and allosteric inhibitors of BCR-ABL. Cancer Res. 2012;72:4890–5. doi: 10.1158/0008-5472.CAN-12-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Roychowdhury S, Talpaz M. Managing resistance in chronic myeloid leukemia. Blood Reviews. 2011;25:279–90. doi: 10.1016/j.blre.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 133.Grebien F, et al. Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell. 2011;147:306–19. doi: 10.1016/j.cell.2011.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ganguly SS, Plattner R. Activation of abl family kinases in solid tumors. Genes Cancer. 2012;3:414–25. doi: 10.1177/1947601912458586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. The Oncologist. 2011;16:566–78. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee MJ, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–94. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Duncan JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–21. doi: 10.1016/j.cell.2012.02.053. References 136 and 137 are two excellent papers that demonstrate the dynamic reprogramming of cancer cells in response to kinase inhibitors and show that understanding these responses will permit rational design of combination therapies to overcome resistance to chemotherapy.
- 138.Su F, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012;366:207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Weigel MT, et al. Preclinical and clinical studies of estrogen deprivation support the PDGF/Abl pathway as a novel therapeutic target for overcoming endocrine resistance in breast cancer. Breast Cancer Res. 2012;14:R78. doi: 10.1186/bcr3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.He X, et al. c-Abl regulates estrogen receptor alpha transcription activity through its stabilization by phosphorylation. Oncogene. 2010;29:2238–51. doi: 10.1038/onc.2009.513. [DOI] [PubMed] [Google Scholar]
- 141.Zhao H, et al. Enhanced resistance to tamoxifen by the c-ABL proto-oncogene in breast cancer. Neoplasia. 2010;12:214–23. doi: 10.1593/neo.91576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sims JT, et al. STI571 sensitizes breast cancer cells to 5-fluorouracil, cisplatin and camptothecin in a cell type-specific manner. Biochemical Pharmacology. 2009;78:249–60. doi: 10.1016/j.bcp.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weigel MT, et al. Combination of imatinib and vinorelbine enhances cell growth inhibition in breast cancer cells via PDGFR beta signalling. Cancer Letters. 2009;273:70–9. doi: 10.1016/j.canlet.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 144.Lo YH, Ho PC, Zhao H, Wang SC. Inhibition of c-ABL sensitizes breast cancer cells to the dual ErbB receptor tyrosine kinase inhibitor lapatinib (GW572016) Anticancer Research. 2011;31:789–95. [PubMed] [Google Scholar]
- 145.Wetzel DM, McMahon-Pratt D, Koleske AJ. The Abl and Arg kinases mediate distinct modes of phagocytosis and are required for maximal Leishmania infection. Mol. Cell Biol. 2012;32:3176–86. doi: 10.1128/MCB.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gu JJ, Zhang N, He YW, Koleske AJ, Pendergast AM. Defective T cell development and function in the absence of Abelson kinases. J. Immunol. 2007;179:7334–43. doi: 10.4049/jimmunol.179.11.7334. [DOI] [PubMed] [Google Scholar]
- 147.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 148.Pollard JW. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009;9:259–70. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer and Metastasis Reviews. 2010;29:309–16. doi: 10.1007/s10555-010-9223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Millot F, et al. Imatinib is effective in children with previously untreated chronic myelogenous leukemia in early chronic phase: results of the French national phase IV trial. Journal of Clinical Oncology. 2011;29:2827–32. doi: 10.1200/JCO.2010.32.7114. [DOI] [PubMed] [Google Scholar]
- 151.Schultz KR, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children’s oncology group study. Journal of Clinical Oncology. 2009;27:5175–81. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.O’Hare T, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bianchi C, et al. Eight full-length abelson related gene (Arg) isoforms are constitutively expressed in caki-1 cell line and cell distribution of two isoforms has been analyzed after transfection. J. Cell Biochem. 2008;105:1219–27. doi: 10.1002/jcb.21922. [DOI] [PubMed] [Google Scholar]
- 154.Adrian FJ, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2006;2:95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- 155.Choi Y, et al. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J. Biol. Chem. 2009;284:29005–14. doi: 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Abelson HT, Rabstein LS. Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Res. 1970;30:2213–22. [PubMed] [Google Scholar]
- 157.Green PL, Kaehler DA, Bennett LM, Risser R. Multiple steps are required for the induction of tumors by Abelson murine leukemia virus. J. Virol. 1989;63:1989–94. doi: 10.1128/jvi.63.5.1989-1994.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]