

Abstract
Th17 cells are a distinct subset of T cells that have been found to produce interleukin 17 (IL-17), and differ in function from the other T cell subsets including Th1, Th2, and regulatory T cells. Th17 cells have emerged as a central culprit in overzealous inflammatory immune responses associated with many autoimmune disorders. In this method we purify T lymphocytes from the spleen and lymph nodes of C57BL/6 mice, and stimulate purified CD4+ T cells under control and Th17-inducing environments. The Th17-inducing environment includes stimulation in the presence of anti-CD3 and anti-CD28 antibodies, IL-6, and TGF-β. After incubation for at least 72 hours and for up to five days at 37 °C, cells are subsequently analyzed for the capability to produce IL-17 through flow cytometry, qPCR, and ELISAs. Th17 differentiated CD4+CD25- T cells can be utilized to further elucidate the role that Th17 cells play in the onset and progression of autoimmunity and host defense. Moreover, Th17 differentiation of CD4+CD25- lymphocytes from distinct murine knockout/disease models can contribute to our understanding of cell fate plasticity.
Keywords: Immunology, Issue 79, Cellular Biology, Molecular Biology, Medicine, Infection, Th17 cells, IL-17, Th17 differentiation, T cells, autoimmunity, cell, isolation, culture
Introduction
CD4+ T lymphocytes (T cells) play a critical role in immune system-mediated defense against infectious microorganisms. Conversely, T cells are also intimately associated with the onset and progression of autoimmune diseases such as type 1 diabetes, systemic lupus erythematosus, and rheumatoid arthritis. CD4+ T lymphocytes become activated through a combination of T cell receptor (TCR) interactions with cognate antigen/major histocompatibility complex II (MHCII) molecules, and CD28 receptor interactions with B7.1/B7.2 ligands15. In addition to the provision of TCR stimulation and CD28 co-stimulation, antigen-presenting cells also provide a cytokine environment, which determines the differentiation state of the T lymphocyte, thereby directing the T lymphocyte's response to the given antigen. Distinct pathogen/antigen-presenting cell interactions create distinct cytokine environments, which skew T lymphocytes down distinct pathways focused on the elimination of the initiating pathogen. Unfortunately, T lymphocyte effector pathways, originally designed to eradicate invading pathogens, can be erroneously directed against self-tissues15. Therefore, better understanding of each distinct CD4+ T cell subset's differentiation state is critical for our understanding of how to modulate the balance between elimination of pathogens and tolerance to self.
In addition to the Th1, Th2, and inducible regulatory T cell differentiation pathways, naïve T lymphocytes can also be driven by cytokines down the Th17 pathway. Whereas Th1 cells combat intracellular pathogens, Th2 cells eliminate extracellular pathogens, and regulatory T cells (Tregs) minimize inflammatory responses1, 16; Th17 cells play an important role in the elimination of extracellular bacteria and fungi. Th17 cells are generally denoted by expression of the lineage-specific transcription factor RORγT and production of IL-17A, which promotes the activation of macrophages and neutrophils1, 7.
Th17 cells have been implicated in several autoimmune disorders, and their associated rodent models. For example, it has been demonstrated that IL-23 (which is required to sustain the Th17 phenotype), but not IL-12, was the central culprit in experimental autoimmune encephalitis (EAE), the rodent disease model for MS. It has subsequently been shown that reductions in IL-17 production are correlated to EAE prevention2, 6, 17. Moreover, Th17 cells have been associated with other autoimmune disorders including arthritis and systemic lupus erythematosus (SLE)10, 16. IL-23 deficient p19-/- mice were shown to have very low numbers of Th17 cells, and are resistant to developing not only EAE, but also collagen-induced arthritis, a model for rheumatoid arthritis10, 18. In addition, mice treated with neutralizing IL-17A antibodies after the onset of collagen-induced arthritis were also found to have resolution of joint damage18. It should be noted that the role of Th17 cells in the progression of autoimmune disease remains to be characterized as recent research has also shown a protective role of Th17 cells in Type 1 diabetes9, 11 and intestinal inflammation14. These studies confirm the importance of Th17 differentiation in autoimmunity.
In vitro Th17 differentiation is a necessary method in T cell research because there are at least two perplexing questions that require further investigation: 1) How exactly does IL-6 regulate the balance between Treg and Th17 differentiation, and 2) what are the exact mechanisms behind IL-17-induced inflammatory disorders? Our method employs CD4+CD25- T cells from the spleens and lymph nodes of the C57BL/6 mouse. It is important to note that although it is possible to induce Th17 differentiation using an impure population, acquiring at least an 80% pure CD4+CD25- T cell population negates any worry of contamination and ensures more successful Th17 differentiation results. In order to achieve proper Th17 differentiation, CD4+CD25- T cells are incubated in the presence of anti-CD3 and anti-CD28, which provide activation signals, 1 and 2, respectively, and IL-6, and TGF-β. Although it has been reported that IL-23 alone can be used to achieve Th17 differentiation, it was later demonstrated that IL-23 is necessary for the stability of the Th17 cell population, but IL-6 and TGF-β are essential for Th17 differentiation3, 18, 19. Murine studies have shown that the IL-23 receptor is expressed on CD4+ T cells only after they have been stimulated with IL-6 and TGF-β13, 18. Also, Th17 cells will successfully develop in the presence of IL-23-blocking antibodies as long as IL-6 and TGF-β are present18, 19. As such, this Th17 differentiation protocol provides the appropriate conditions to successfully induce Th17 differentiation. Development of a better understanding of the mechanisms underlying Th17 differentiation and IL-17 production present the opportunity for the development of better therapeutics aimed at autoimmune disorders13.
Protocol
All animal use was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee.
1. Preparation of Mixes and Media
Sterile PBS pH: 7.3 (1 L) 0.23 g NaH2PO4 1.15 g Na2HPO4 9.0 g NaCl Take up volume with DI water. Sterilize with autoclave.
Cell Culture Media (100 ml) 89 ml RPMI 10 ml 10% FBS 1 ml Antibiotic Antimycotic (ABAM) 100 μg 50 mM 2-mercaptoethanol (3.5 μg stock 2-mercaptoethanol into 96.5 μg PBS)
FACS Buffer (Fluorescent Activating Cell Sorting) (To be used during flow cytometry) 2% FBS in sterile PBS
iTh17 Mix (Based on number of samples, determine the volume needed for all mixes and media) 1.5 μg/ml anti-CD28 20 ng/ml IL-6 5 ng/ml TGF-β
2. Cells Will Be Plated in Triplicate under the Following Conditions
Plate bound anti-CD3, anti-CD28 (this is the activation control mix).
iTH17: plate bound anti-CD3, anti-CD28, IL-6, TGF-β.
3. Plate-bound anti-CD3 (10 μg/ml)
It is recommended that preparation of plate-bound anti-CD3 plates is done at least 4 hr prior to the time cells will be added to the plates.
Add 30 μl anti-CD3 to wells (anti-CD3 is diluted in sterile PBS) of a new 96 well U bottom Microtest tissue culture plate, and tap the sides of the plate to ensure uniform coverage of the wells. Incubate at 37 °C for 4 hr, and then refrigerate until it is time to add the mixes and cells to the plate.
4. Mouse Dissection
This protocol is based on the use of C57BL/6 mice, 3-8 months of age, and purchased from The Jackson Laboratory (Bar Harbor, ME).
Sacrifice mouse using CO2 asphyxiation, and confirm death with subsequent cervical dislocation.
Sterilize mice dissection tools and incision area with 70% ethanol and begin dissection.
From the ventral view, grab the skin that is anterior to the urethral opening and begin cutting with scissors up the ventral midline until reaching the chin area. Take precautions not to tear or cut into the lining of the peritoneal wall.
Pull back on the skin and pin down to allow comfortable access to lymph nodes during removal.
- The lymph nodes and spleens collected will all be removed with forceps, and placed in a sterile Petri dish containing 5 ml of autoMACS Running Buffer purchased from Miltenyi Biotec (refer to Figures 2 and 3 for lymph node and spleen removal diagrams).
- Remove the axillary lymph nodes that are found near the axilla (armpit) behind the pectoral muscles of each mouse.
- Remove the brachial lymph nodes that are located in the connective tissues located near each axilla.
- Remove the superficial cervical lymph nodes found in the neck of each mouse.
- Remove the inguinal lymph nodes which are located in the hip region at the conjunction of the 3 blood vessels.
- To access the mesenteric lymph nodes, cut through the peritoneal lining up the ventral midline. Mesenteric lymph nodes are found in the connective tissue that holds the intestines together. They are generally found in a string of 4-8 nodes, and may appear as a "string of pearls". Make sure to pull out the entire string.
- The spleen is located in the abdominal area, behind the stomach and intestines. Remove it by pulling and detaching it from the pancreas.
Grind organs under a sterile hood using 2 frosted microscope slides. Place lymph nodes or spleen on the frosted side of one microscope slide, and rub with frosted side of the second slide until disintegrated. Repeat until all lymph nodes and spleen have been ground.
Note: It is recommended to start with the lymph nodes and finish with the spleen, as the blood will make it difficult to see the remaining lymph nodes in the autoMACs buffer.
To filter ground organs into a single cell suspension, begin by folding over a piece of 40 μm nylon material several times, and placing in the top of a 15 ml centrifuge tube. The nylon material should be roughly 3 x 3 in.
Use a 5 ml syringe and 21 G needle to aspirate the 5 ml of autoMACs Running buffer and ground organs. Dispense slowly into the folded nylon material. Take care to avoid puncturing the material with the needle.
Note: Once single cell suspension is achieved, the solution should appear as a pale, consistent solution with no visible pieces of solid tissue. If solids remain, re-aspirate and dispense through a new folded piece of nylon placed in a new 15 ml centrifuge tube.
Always keep cells on ice when not in use.
5. Cell Separation
For optimal results, obtain at least an 80% pure CD4+CD25- cell population through autoMACS Pro Cell Separator using the MACS CD4+CD25+ regulatory T cell isolation kit, mouse. The protocol for the negative retention of the CD4+CD25- cell population is obtained through Miltenyi Biotec.
6. Th17 Differentiation
Collect the CD4+CD25- cell population after separation with the autoMACS Pro Cell Separator.
Count cells diluted in a 1:1000 ratio with trypan blue using a hemocytometer.
Once concentration has been determined from cell counts, dilute cell suspension to 1 x 106 cells/ml in cell culture media.
Take out 96 well plates with plate bound anti-CD3 after 4 hr.
- Wash anti-CD3-coated wells with 200 μl of PBS. Repeat 2 times.
- To wash add 200 μl of sterile PBS to anti-CD3-coated wells and remove PBS into a waste container.
Add 100 μl of the iTh17 mix or activation control mix (see point #2) to the wells in triplicate.
Add 100 μl of cells to each well in which either the Th17 mix or the activation control mix has been placed.
Incubate cells for at least 72 hr or up to 5 days (Th17 differentiation can be obtained after 72 hr or after 5 days.)
Th17 differentiation can be assessed by intracellular staining and flow cytometric analysis, ELISA, or qPCR
7. Cell Activation (Only Necessary for Intracellular Staining)
- Remove 96 well plates from incubators at the end of either 72 hr or 5 days
- Recall that Th17 differentiation can be achieved after either 72 hr or 5 days.
For each condition (e.g. activation control or iTh17), transfer cells that are in triplicate into one well of a 24 well cell culture plate. The cells for one condition have now been pooled into one well of the 24 well culture plate, as opposed to being in triplicate in the 96 well U bottom culture plate.
The total volume of each well in the 24 well cell culture plate is now 600 μl. Raise the volume of each well to 1 ml with the cell culture media.
Add PMA (phorbol myristate acetate) (50 ng/ml), ionomycin (1 μM), and BFA (Brefeldin-A) (10 μg/ml) to each well in the 24 well cell culture plate at the listed concentrations.
Incubate at 37 °C for 4 hr.
8. Intracellular Staining
- Stain cells with the desired extracellular and intracellular markers for flow cytometric analysis. To detect the presence of IL-17, intracellular staining is done with anti-IL-17A antibodies. Recommended extracellular surface markers include CD4, CD8, and CD25.
- Use the Intracellular Cytokine Staining Starter Kit-Mouse from BD Bioscience for IL-17 Intracellular staining.
- For each sample, pellet cells, remove supernatant, and resuspend cell pellet in 200 μl FACS buffer (2% FBS in PBS).
- Transfer resuspended cells to 96 cell flow cytometry plate.
- Spin cells down for 5 min at 1,200 rpm and discard supernatant.
- Add 200 μl PBS FACS buffer, centrifuge for 5 min at 1,200 rpm, and discard supernatant.
- Resuspend cells in 100 μl of FACS buffer and apply 100 μl of extracellular antibody (Ab) mixture (extracellular Ab mixture is made in FACS buffer). Incubate for 15 min at RT, covered with foil.
- Repeat step 8.1.4.
- Repeat step 8.1.5 2x.
- Resuspend cells in 100 μl of BD Cytofix/Cytoperm Buffer. Incubate for 20 min at RT, covered with foil.
- Add 100 μl of 1x BD Perm/Wash buffer, centrifuge for 5 min at 1,200 rpm, and discard supernatant. Repeat.
- Add 50 μl of intracellular Ab mixture. (Intracellular Ab mixture is made in 1x BD Perm/Wash) Incubate for 15 min at RT, covered with foil.
- Repeat step 8.1.10.
- Resuspend cells in 200 μl of BD Staining Buffer.
- Place resuspended cells into flow cytometry tubes containing 200 μl BD Staining Buffer (final volume is 400 μl).
- Store at 4 °C until samples are ready to be read.
9. Flow Cytometric Analysis
Gate live cell population.
From the live cell population, gate on CD4+CD8- population.
- From the CD4+CD8- population, gate on IL-17A+ population.
- Based on our previous experimental results, 100% of the IL-17A+ population will be CD25+.
*Total absolute cell counts were obtained after pooling the sample triplicates **Absolute number of CD4+CD25+IL-17A+ cells was determined by multiplying the total number of cells by the live gate percentage and the percentage of total cells bearing the lineage-specific markers, CD4, CD25, and IL-17A, as determined by flow cytometry.
10. qPCR and ELISA
Place cells not used for flow cytometric analysis in a 1.5 ml Eppendorf tube and centrifuge at 13,000 rpm for 5-10 min. The cells found in the cell pellet after centrifugation will be used for qPCR analysis. qPCR analysis was performed using PTC-200 Peltier Thermal cycler (Biorad).
Collect supernatant from the centrifuged tubes for ELISAs. IL-17A ELISAs were performed using antibody pair TC11-18H10 (capture, catalog number 555068) and TC11-8H4 biotin (detection, catalog number 555067) purchased from BD Biosciences. IL-17A ELISA standards were obtained from eBioscience (catalog number 14-8171-80).
- After removing the supernatant, resuspend the remaining cells to be used for qPCR in 175 μl RNA lysis buffer (use immediately for RNA extraction, cDNA synthesis, and qPCR, or store at -80 °C for later use).
- Refer to Table II for primer sequences for IL-17A and actin (house-keeping gene)
Representative Results
This Th17 differentiation protocol begins with the removal of the spleen and the axillary, brachial, mesenteric, cervical, and inguinal lymph nodes. A representation of the locations of each can be found in Figures 2 and 3. Figures 1 and 5 both provide a visual representation of the methods described in this protocol.
This Th17 protocol focuses on differentiation from the CD4+CD25- T lymphocyte population. It is important to note how efficiently the autoMACS Pro Cell separation enriches our desired CD4+CD25- population from the pooled total lymph node and spleen (Figure 4). The unfractionated, pooled lymph node and splenocytes, and the autoMACS-separated fractions were stained with antiCD4 and antiCD25 antibodies followed by flow cytometric analysis (Figure 4). Figure 4A shows a representative profile of the percentage of CD4+CD25+ and CD4+CD25- lymphocytes present in the unfractionated, pooled lymph nodes and spleen by flow cytometry. The isolation procedure also provides an enriched CD4+CD25+ Treg population from C57BL/6 mice which can be used in additional experiments (Figure 4B). Figure 4C shows our desired cell enrichment with a population that is 84% CD4+CD25- T cells, with the vast majority of CD4+CD25+ Tregs removed. We consistently have a small non-lymphoid cell population that is present in the enriched CD4+CD25-, but does it not interfere with the differentiation process (Figure 4C). Our enriched CD4+CD25- T cell population helps to assure a more successful Th17 differentiation.
Figure 5 shows a schematic of our Th17 differentiation procedure. Our enriched CD4+CD25- population is either incubated under Th17 differentiating conditions (anti-CD3, anti-CD28, IL-6, and TGF-β) or control (anti-CD3, anti-CD28). It can be seen in Figure 6 that whereas incubation of CD4+CD25- T lymphocytes with anti-CD3, anti-CD28 for 5 days yielded CD25+ T lymphocytes, incubation under Th17 inducing conditions yielded a subset of IL17 producing CD4+CD25+ lymphocytes. Please refer to Table 1 for examples of CD4+CD25+IL-17A+ absolute cell number yields after 5 day incubation under iTh17 conditions.
Th17 differentiation status can further be assessed through quantitative PCR and ELISA. Figure 7 presents data from enriched CD4+CD25- T lymphocytes incubated under control or Th17-inducing conditions. CD4+CD25- T lymphocytes incubated under Th17 conditions up-regulate IL17A, which can be ascertained through qPCR (Figure 7A) and ELISA (Figure 7B). The Th17-specific transcription factor RORγT is also upregulated by CD4+CD25- T cells incubated under Th17-inducing conditions, and can be ascertained through qPCR (Figure 7A).
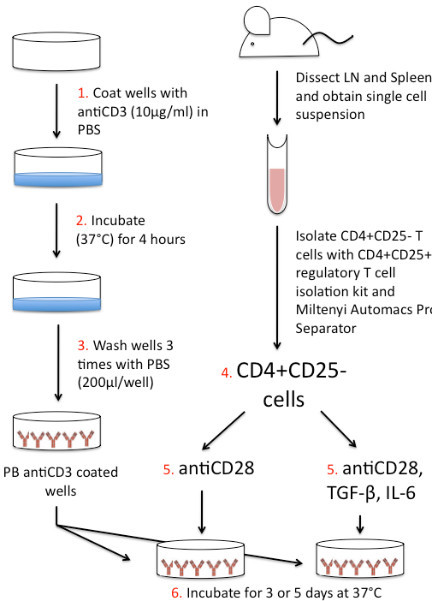 Figure 1. Th17 Differentiation schematic. 1. Coat wells of U-bottomed 96 well plate with anti-CD3 (10 μg/ml) diluted in PBS. 2. Place plate in incubator for 4 hr at 37 °C. While plate is incubating, dissect lymph nodes (LN) and spleen from mouse. Grind organs and filter to obtain a single cell suspension. Isolate CD4+CD25- T cells using the CD4+CD25+ Regulatory T cell Isolation Kit and Miltenyi autoMACS Pro Separator. CD4+CD25- cells should be diluted to 1 x 106 cells/ml. 3. Once 4 hr incubation of the 96 well plate is complete, wash wells with PBS (200 μl/well) 3x. 4. Add 100 μl CD4+CD25- T cells to plate-bound (PB) anti-CD3 coated wells. 5. Add 100 μl anti-CD28 or 100 μl iTh17 cocktail (antiCD28, TGF-β, and IL-6). 6. Incubate plate for 3 or 5 days at 37 °C. Following incubation assess differentiation by intracellular staining, qPCR, and/or ELISA.
Figure 1. Th17 Differentiation schematic. 1. Coat wells of U-bottomed 96 well plate with anti-CD3 (10 μg/ml) diluted in PBS. 2. Place plate in incubator for 4 hr at 37 °C. While plate is incubating, dissect lymph nodes (LN) and spleen from mouse. Grind organs and filter to obtain a single cell suspension. Isolate CD4+CD25- T cells using the CD4+CD25+ Regulatory T cell Isolation Kit and Miltenyi autoMACS Pro Separator. CD4+CD25- cells should be diluted to 1 x 106 cells/ml. 3. Once 4 hr incubation of the 96 well plate is complete, wash wells with PBS (200 μl/well) 3x. 4. Add 100 μl CD4+CD25- T cells to plate-bound (PB) anti-CD3 coated wells. 5. Add 100 μl anti-CD28 or 100 μl iTh17 cocktail (antiCD28, TGF-β, and IL-6). 6. Incubate plate for 3 or 5 days at 37 °C. Following incubation assess differentiation by intracellular staining, qPCR, and/or ELISA.
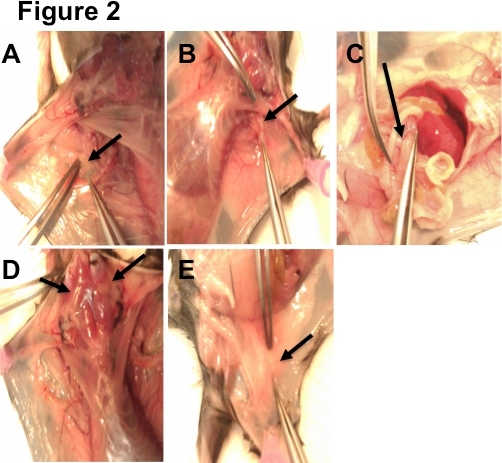 Figure 2. LN Dissection Guide. A) 1 brachial lymph node can be found in the connective tissue located near each axilla (armpit) of the mouse. B) 1 axillary lymph node can be found in each axilla, behind the pectoral muscles. C) Mesenteric lymph nodes are located in the connective tissue of the intestines. They resemble a string of pearls. D) 4 superficial cervical lymph nodes are found in the neck of the mouse. E) 2 inguinal lymph nodes (1 on each side) can be located in the hip region at the location where 3 blood vessels meet.
Figure 2. LN Dissection Guide. A) 1 brachial lymph node can be found in the connective tissue located near each axilla (armpit) of the mouse. B) 1 axillary lymph node can be found in each axilla, behind the pectoral muscles. C) Mesenteric lymph nodes are located in the connective tissue of the intestines. They resemble a string of pearls. D) 4 superficial cervical lymph nodes are found in the neck of the mouse. E) 2 inguinal lymph nodes (1 on each side) can be located in the hip region at the location where 3 blood vessels meet.
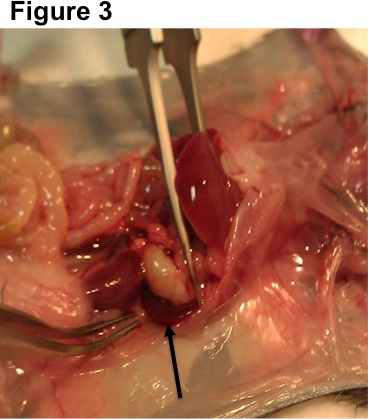 Figure 3. Spleen dissection guide. Mouse spleens are found on the left side of the body behind the stomach and intestines. Simply remove by pulling and separating with the forceps.
Figure 3. Spleen dissection guide. Mouse spleens are found on the left side of the body behind the stomach and intestines. Simply remove by pulling and separating with the forceps.
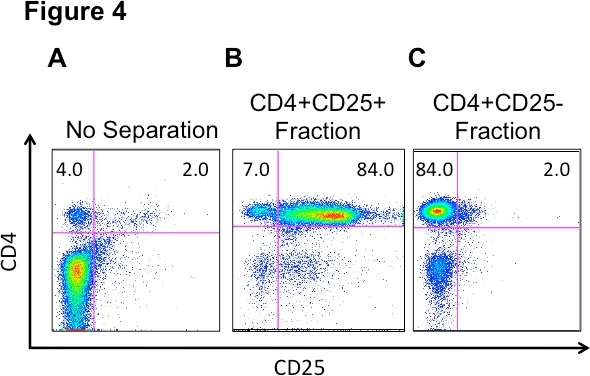 Figure 4. Cell fractions from pooled LN and spleen cells before and after autoMACS Pro separation. A) LN and spleen cells were stained, ex vivo, to establish baseline cell populations prior to separation. Following separation, CD4+CD25+ (B) and CD4+CD25- (C) fractions were stained to assess purity of CD4+CD25+ and CD4+CD25- T lymphocyte populations, respectively. All samples were stained with CD4+ and CD25+ antibodies and analyzed by flow cytometry.
Figure 4. Cell fractions from pooled LN and spleen cells before and after autoMACS Pro separation. A) LN and spleen cells were stained, ex vivo, to establish baseline cell populations prior to separation. Following separation, CD4+CD25+ (B) and CD4+CD25- (C) fractions were stained to assess purity of CD4+CD25+ and CD4+CD25- T lymphocyte populations, respectively. All samples were stained with CD4+ and CD25+ antibodies and analyzed by flow cytometry.
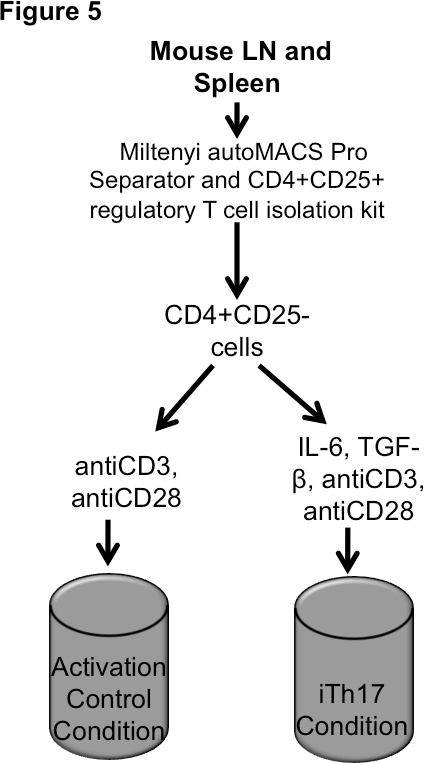 Figure 5. Th17 Differentiation. Incubate cells for up to 5 days. Harvest cells for intracellular staining and qPCR; harvest supernatants for ELISA.
Figure 5. Th17 Differentiation. Incubate cells for up to 5 days. Harvest cells for intracellular staining and qPCR; harvest supernatants for ELISA.
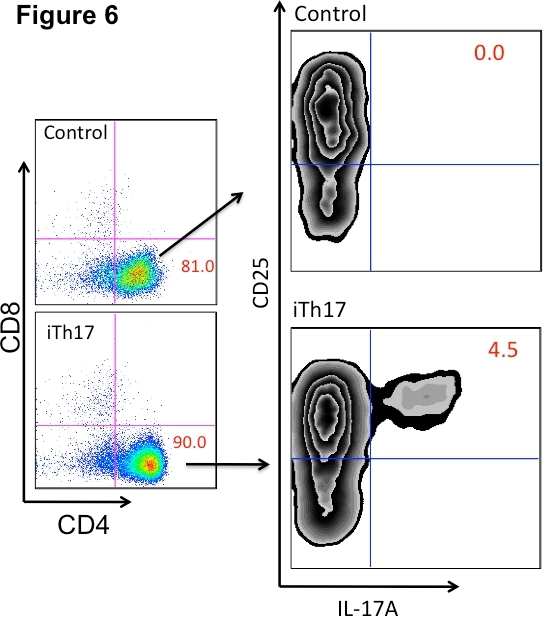 Figure 6. Th17 differentiation assessed by flow cytometry. CD4+CD25- T lymphocytes were incubated in the presence of plate-bound anti-CD3, anti-CD28, IL-6, and TGF-β (iTh17) or plate-bound anti-CD3 and anti-CD28 alone (control) for 5 days. Cells were then activated with PMA and Ionomycin for 4 hr at 37 °C. Following activation, cells were stained with anti-CD4, anti-CD8, anti-CD25, and anti-IL-17A antibodies for flow cytometric analysis to assess Th17 differentiation. iTh17 = Th17 inducing conditions.
Figure 6. Th17 differentiation assessed by flow cytometry. CD4+CD25- T lymphocytes were incubated in the presence of plate-bound anti-CD3, anti-CD28, IL-6, and TGF-β (iTh17) or plate-bound anti-CD3 and anti-CD28 alone (control) for 5 days. Cells were then activated with PMA and Ionomycin for 4 hr at 37 °C. Following activation, cells were stained with anti-CD4, anti-CD8, anti-CD25, and anti-IL-17A antibodies for flow cytometric analysis to assess Th17 differentiation. iTh17 = Th17 inducing conditions.
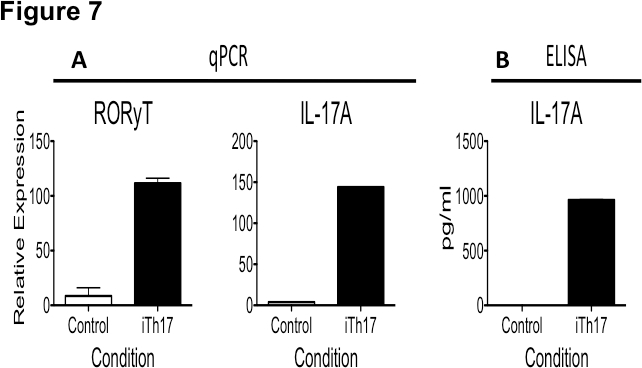 Figure 7. Th17 differentiation assessed by qPCR and ELISA. Lymphocytes from C57BL/6 mice were sorted and CD4+CD25- T cells were incubated in the presence of plate-bound anti-CD3 (10 μg/ml), anti-CD28 (1.5 μg/ml), IL-6 (20 ng/ml), and TGF-β (5 ng/ml) or plate-bound anti-CD3 and anti-CD28 alone (control) for 5 days. A) Cells were then harvested to assess RORγT and IL-17A message expression via qPCR. B) Supernatants were harvested to assess IL-17A protein expression by ELISA.
Figure 7. Th17 differentiation assessed by qPCR and ELISA. Lymphocytes from C57BL/6 mice were sorted and CD4+CD25- T cells were incubated in the presence of plate-bound anti-CD3 (10 μg/ml), anti-CD28 (1.5 μg/ml), IL-6 (20 ng/ml), and TGF-β (5 ng/ml) or plate-bound anti-CD3 and anti-CD28 alone (control) for 5 days. A) Cells were then harvested to assess RORγT and IL-17A message expression via qPCR. B) Supernatants were harvested to assess IL-17A protein expression by ELISA.
Discussion
Here we have described the protocol to achieve in vitro Th17 differentiation. The study of Th17 differentiation is important, as differentiation of T lymphocytes into the Th17 subset is critical for the effective elimination of human pathogens13. Conversely, IL17 production has been associated with autoimmune disease progression13. The method of Th17 differentiation is applicable to many research settings as it can be applied to numerous distinct murine models of immune regulation and autoimmunity. This method helps to answer questions regarding the ability of T lymphocytes, to differentiate into a Th17 cell, denoted in this protocol by the production of IL-17A and expression of the transcription factor RORγT, to modulate immune responses. Moreover, it has been shown by others that Th17 cells can also be identified by the production of IL17F, Il22, and GM-CSF 8, 12. This protocol is significant with respect to other known T lymphocyte differentiation protocols, such as Th1 or Th2 differentiation, because it can be used to complement those protocols in order to better understand how T lymphocyte effector functions relate to immune system operation.
There are several critical steps to consider when conducting these experiments. Th17 differentiation can be achieved through an impure CD4+CD25- population, but for the most effective and reliable results, a CD4+CD25- population of at least 80 % purity is desired. Our method utilizes the protocol provided by Miltenyi Biotec for their autoMACS Pro Cell Separator. The autoMACS CD4+CD25+ Regulatory T Cell Isolation Kit, Mouse is used for negative selection of the CD4+CD25- cell population. Negative selection allows for the use of naïve cells that have not been potentially altered by antibody interactions.
Using certain cell surface markers in flow cytometric analysis can confirm the purity of a population. The recommended surface markers are CD4, CD8, and CD25 as they can readily identify the desired CD4+CD25- population. B220 and CD11b may be used to test for B cell and macrophage contamination, respectively. Samples consistently diluted to 1 x 106 cells/ml is also recommended as we have found that this concentration yields optimal differentiation. In the past we have found that cell concentrations that were too high or too low often had perturbed cellular expansion and biased results (such as skewed IL-17A production).
It is also important to remember to keep the samples on ice at all times when not in use. Cell viability is an important factor in achieving positive Th17 differentiation. This can be confirmed through cell counting using trypan blue dilutions. Because this protocol employs manual tissue homogenization by grinding the lymph nodes and spleen with frosted microscope slides, this can be a time consuming method. One design modification is to utilize automatic tissue dissociators as an alternative option.
In the event that the desired Th17 differentiation is not achieved, there are several trouble shooting tips that should be considered. First, controls must also be carefully considered so that the true extent of Th17 differentiation can be properly measured. We typically determine the extent of Th17 differentiation by comparing these results to the CD4+ T lymphocytes receiving only anti-CD3 and anti-CD28 stimulation. Also remember that CD4+ T lymphocytes need to be activated in order differentiate. One should also visually inspect cultured cells under the microscope to confirm the presence of blasted/activated T cells. Activated T cells visually appear significantly larger than cells that have not been stimulated. Clonal expansion can also be observed through visible clusters of blasting cells which have been derived from a single T cell clone. Cell activation can also be verified through the use of flow cytometric analysis using anti-CD4 and anti-CD25 antibodies to identify the CD4+CD25+ activated cells4
It is also important to mention that this protocol has been optimized for use with C57BL/6 mice, however published data shows that Th17 differentiation may be achieved in other mouse strains including NOD and BALB/c mice5, 11, 20 . It should be noted that Th17 differentiation, including overall frequency and absolute number of CD4+ T lymphocytes undergoing differentiation, is dependent upon genetic factors (such as mouse strain)20, and environmental factors (such as colony location)5 and may require optimization.
This Th17 differentiation protocol can serve as an invaluable means to further answer questions regarding the function of cells in the Th17 subset. Our method of Th17 differentiation has several significant advantages. As previously mentioned the use of a minimum 80% pure CD4+CD25- cell population provides a more consistent and effective differentiation result. Ease of isolation through use of the autoMACS Pro cell separator is an additional advantage. Furthermore the use of the optimal cytokine and activation signals provides the T cells with the appropriate environment in which to differentiate. Future implications for this protocol include determining the mechanistic means by which Th17 cells and the production of IL-17A are involved in the onset and progression of autoimmunity.
Disclosures
No disclosures to declare.
Acknowledgments
This work is supported in part by the NIH/NCATS Clinical and Translational Science Awards to the University of Florida TL1 TR000066 and UL1 TR000064, a diversity supplement from Parent Grant R01AI056152 from the National Institute of Health, a BD Biosciences reagent grant, and the University of Florida.
References
- Chen Z, Lin F, et al. Foxp3 and RORγT: transcriptional regulation of Treg and Th17. International Immunopharmacology. 2011;11:536–542. doi: 10.1016/j.intimp.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:742–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- El-Behi M, Rostami A, et al. Current views on the roles of Th17 and Th17 cells in experimental autoimmune encephalitis. J. Neuroimmune Pharmacol. 2010;5:189–197. doi: 10.1007/s11481-009-9188-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Kroide S, et al. Properties of memory T lymphocytes isolated from the mixed leukocyte reaction. Proc Natl Acad Sci. 1985;82:7686–7690. doi: 10.1073/pnas.82.22.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L IvanovFrutosR, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;16:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager LD, Dabelic R, et al. The Kinase Inhibitory Region of SOCS-1 is Sufficient to Inhibit T helper-17 Function in Experimental Allergic Encephalomyelitis. J. Neuroimmunol. 2011;232:08–18. doi: 10.1016/j.jneuroim.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur. J. Immunol. 2010;40:1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, et al. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Kriegel MA, Seflk E, et al. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. PNAS. 2011;108:11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau K, Benitez P, et al. Inhibition of type 1 diabetes by a Lactobacillus johnsonii mediated Th17 bias. J. Immunol. 2011;186:3538–3546. doi: 10.4049/jimmunol.1001864. [DOI] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litmann DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140:845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- O'Connor W, Jr, Kamanaka M, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10:603–609. doi: 10.1038/ni.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picca CC, Larkin J, et al. Role of TCR specificity in CD4+CD25+ regulatory T cell selection. Immunol Rev. 2006;212:74–85. doi: 10.1111/j.0105-2896.2006.00416.x. [DOI] [PubMed] [Google Scholar]
- Shah K, Lee WW, et al. Disregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Research & Therapy. 2010;12:R53. doi: 10.1186/ar2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of Multiple Sclerosis. Annu. Rev. Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Stockringer B, Veldhoen M. Differentiation and function of Th17 T cells. Current Opinion in Immunology. 2007;19:281–286. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, et al. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Zou Y, Zhang H, et al. Strain-dependent production of interleukin-17/interferon-γ and matrix remodeling-associated genes in experimental Candida albicans keratitis. Mol Vis. 2012;18:1215–1225. [PMC free article] [PubMed] [Google Scholar]