

Abstract
Simple supported lipid bilayers do not accurately reflect the complex heterogeneity of cellular membranes; however, surface modification makes it possible to tune membrane properties to better mimic biological systems. Here, 3-[2-(2-aminoethylamino)ethylamino]propyl-trimethoxysilane (DETAS), a silica modifier, facilitated formation of supported lipid bilayers on silica nanoparticles. Evidence for a stable supported bilayer came from the successful entrapment of a soluble fluorophore within an interstitial water layer. A fluorescence-quenching assay that utilized a pore-forming peptide was used to demonstrate the existence of two separate lipid leaflets. In this assay, fluorescence was quenched by dithionite in roughly equal proportions prior to and after addition of melittin. When a hydrophobic modifier, octadecyltriethoxysilane, was co-deposited on the nanoparticles with DETAS, there was a decrease in the amount of supported bilayer on the nanoparticles and an increase in the quantity of hybrid membrane. This allowed for a controlled mixture of two distinct types of membranes on a single substrate, one separated by a water cushion and the other anchored directly on the surface, thereby providing a new mimic of cellular membranes.
Keywords: Silica Nanoparticles, Phosphatidylcholine, Calcein, Melittin, Thermogravimetric Analysis, Fluorescence Quenching
INTRODUCTION
In living systems, complex heterogeneous lipid bilayers serve to compartmentalize cellular functions and are critical to mediating signaling, transport, and other biological functions by providing defined interfaces for recognition and interaction.1 Supported lipid bilayers are a well-established mimic of biological membranes and consist of two opposing leaflets of lipids supported at the aqueous interface of a hydrophilic solid.2-4 These mimics are generally formed by liposome rupture upon contact with a planar or colloidal surface, with lipids then spreading to enclose an interstitial water layer.5-7 Interest in supported lipid bilayers stems primarily from their usefulness as simplified model systems for characterizing the structure, assembly, dynamics, and functions of biological membranes.8-10 Supported membranes are also important in the design of biocompatible surfaces, biosensors, and as analytical platforms for assaying protein-membrane interactions.9,11,12
Although supported lipid bilayers are powerful as simplified models, large discrepancies in lipid diffusion rates between supported bilayers and true cellular membranes have been reported.13 Fluorescence tracking measurements have revealed compartmentalization of lipids that lowers the diffusion rate, and it has been proposed that this compartmentalization is caused by attachement of the membrane to the cytoskeleton.14 Membrane-spanning proteins can form semipermeable boundaries that permit rapid diffusion within domains while slowing diffusion between domains.14,15 This picket fence structure alters lipid mobility because the membrane skeleton influences the movements of membrane molecules.14,16 New model systems are needed that better mimic this compartmentalized behavior of biological membranes.
Changes to the surface chemistry of solid supports can alter the lipid membrane interfaces, thereby modifying the physical properties of membranes. This provides a simple and effective means to control membrane properties including spatial molecular distributions, compositional heterogeneity, lateral tension, packing density, diffusion rates, curvature, and membrane morphology.17 For example, hydrophobic surface modifiers have been used on both planar18-20 and nanoparticle surfaces21-26 to create hybrid membrane structures using both silica and gold surfaces. Advances in patterning and surface modification methods27,28 have allowed for the fabrication of surfaces with chemical and topographical variation at micrometer to nanometer length scales. Mixtures of supported and hybrid membranes (Scheme 1) could function to mimic the picket fence structure of membranes and patterning has allowed the creation of surfaces with co-existing regions of supported lipid bilayers and hybrid membranes on the same substrate.12,18,20,29-33 While nanoparticle templates have been used for both supported25,34 and hybrid membranes21,23,25 individually, they have not been used to prepare nanoparticles containing both types of membranes. Creating such mimics on nanoparticle templates would allow for additional methods of studying the membranes and their interactions with proteins.
Scheme 1.
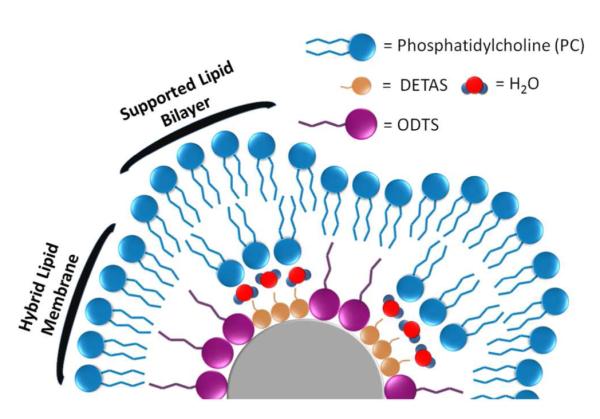
Schematic depiction of a heterogeneous distribution of DETAS and ODTS around a SiNP, resulting in a mixture of hybrid membrane and supported bilayer regions. Illustration is not drawn to scale.
We explored methods of preparing silica nanoparticle (SiNP) surfaces with a heterogeneous combination of hydrophilic and hydrophobic silane modifiers, DETAS and octadecyltriethoxysilane (ODTS). These two trialkoxysilanes were anchored on the SiNPs at different ratios and the SiNPs were then coated with L-α-phosphatidylcholine (PC). Both the silane-modified SiNPs and the PC coating were studied using thermogravimetric analysis (TGA) to quantify the amount of each silane on the surface and to determine how much PC was required to coat a single SiNP. Fluorophore release and quenching studies were employed to characterize the PC-coated SiNPs. Increasing the fraction of hydrophobic silane on the SiNP surface led to an increasing proportion of hybrid membrane.
EXPERIMENTAL METHODS
Materials
3-[2-(2-Aminoethylamino)ethylamino]propyl-trimethoxysilane (DETAS) was from Acros Organics (Pittsburgh, PA); anhydrous calcium chloride, octadecyltriethoxysilane (ODTS), 3-aminopropyltriethoxysilane (APTES), and sodium dithionite were from Alfa Aesar (Ward Hill, MA). A 1 M solution of 2-amino-2-hydroxymethyl-propane-1,3-diol (TRIS), pH 8.0, was from Ambion (Grand Island, NY); Soy PC, and 1-palmitoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphocholine (NBD-PC) were from Avanti Polar Lipids (Alabaster, AL). Ammonium hydroxide (28-30%), N-2-hydroxyethylpiperazine-N’-2-ethane sulphonic acid (HEPES), pH 6.5, and ethanol (190 proof) were from VWR Scientific (Radnor, PA); glacial acetic acid and sodium chloride were from Mallinckrodt Chemicals (Phillipsburg, NJ); chloroform was from VWR Scientific (Radnor, PA) and was filtered through basic alumina (Dynamic Adsorbents, Nocross, Georgia) prior to use. Tetraethyl orthosilicate (TEOS) was from Sigma Aldrich (St. Louis, MO) and Sephadex G-100 Superfine was from Pharmacis Fine Chemicals (Uppsala, Sweden). 18 megaohm Ultrapure water was from a Milli-Q Integral Water Purification System by EMD Millipore (Billerica, MA).
Synthesis of Silica Nanoparticles
Spherical SiNPs were synthesized using the Stöber method.35 Briefly, ethanol (40 mL) and ammonium hydroxide (1.0 mL) were stirred for 7 min in a glass flask before TEOS (0.5 mL) was added. The resulting mixture was stirred overnight at 30-35 °C. The synthesized SiNPs were purified by centrifugation (9,400×g, 10 min) using an Eppendorf 5424 Microcentrifuge (Eppendorf International, Hamburg, Germany). The pellet was resuspended by sonication in ethanol. This purification was repeated three times to remove unreacted precursors and impurities. The diameter of the SiNPs was 107 ± 3 nm by dynamic light scattering (DLS).
Functionalization of SiNPs
Ten milliliters of a 5 weight percent SiNP solution in ethanol was added to a mixture of DETAS and ODTS, keeping the total silane concentration at approximately 0.1 volume percent. All input ratios are given as a mole percentage of DETAS where the remaining fraction is ODTS. The resulting solutions were diluted in ethanol to a total volume of 50 mL and the reaction was stirred while refluxing overnight. The resulting functionalized SiNPs were washed by centrifugation (9,400×g, 10 min) three times with 190 proof ethanol (35 mL) then once with MilliQ water (35 mL). The average diameter of the functionalized SiNPs was ~113 ± 2 nm by DLS.
Thermogravimetric Analysis
TGA was performed on a TA Instruments Q50 Thermogravimetric Analyzer (New Castle, DE). The platinum pan was loaded with approximately 6-10 mg of dry sample or 60-80 mg of wet sample and the temperature was increased to 800 °C at a ramp rate of 2 °C/min. Dry samples were prepared by overnight evaporation of solvent in an oven at 110 °C. PC-coated samples were analyzed after purification, without drying.
The weight loss for each sample that was attributable to DETAS pyrolysis was estimated using the weight loss ratio (WLR) of loss that occurred from 150 °C - 440 °C to the loss that occurred from 440 °C - 800 °C. For SiNP samples containing both silanes, the fraction of DETAS was calculated from these values by referencing samples known to contain exclusively DETAS or ODTS using:
| (1) |
where WLRSiNP is the weight loss ratio for a given functionalization, WLRODTS is the weight loss ratio for 100% ODTS SiNPs, and WLRDETAS is the weight loss ratio for 100% DETAS SiNPs.
Preparation of SiNP Supported Lipid Bilayers
Stock solutions of PC (4 mM) were prepared in chloroform. Lipid films were dried by evaporation of the solvent under a stream of nitrogen, followed by overnight vacuum at room temperature. The samples were kept at −20 °C prior to use. The lipid films were redispersed in either HEPES buffer (10 mM HEPES, 50 mM NaCl, 2 mM CaCl2) or TRIS buffer (10 mM TRIS, 50 mM NaCl, 2 mM CaCl2). The resulting cloudy solutions were sonicated in a cuphorn sonicator (Branson Sonifier 450, Branson Ultrasonics, Danbury, CT) for 15 min at 70% power output. Sonication was followed by extrusion using a Mini Extruder (Avanti Polar Lipids, Alabaster, AL) with a 200 nm pore size polycarbonate filter. Approximately 1 mL of sonicated lipid solution was passed through the membrane at least 15 times, resulting in a clear solution. The liposome diameter (192 ± 5 nm) was measured by DLS. Adsorption of lipids onto the SiNPs was accomplished by stirring an aqueous solution of purified SiNPs with an equal volume of sonicated, extruded PC liposomes (3 mM) for 2 hr at room temperature. The PC-coated SiNPs were centrifuged (9,400×g, 10 min) and the supernatant was discarded. Additional buffer was added and these washing steps were repeated 5 times. After the final centrifugation, the sample was redispersed in 1 mL of HEPES or TRIS buffer for fluorescence measurements or in water for TGA analysis.
The weight loss attributed to PC on the SiNP was determined by subtracting the weight loss for an uncoated SiNP from the PC-coated SiNP. Then the relative PC coverage on the SiNP was calculated from the TGA data with reference to the 100% DETAS sample using:
| (2) |
where WLDETAS is the weight percent change for PC pyrolysis on the 100% DETAS sample and WLSiNP is the weight percent change for PC pyrolysis on the given sample. Also, the predicted PC coverage for a given surface coverage of DETAS was calculated using:
| (3) |
where fDETAS is the calculated fraction of DETAS from equation 1.
NBD-PC Fluorescence Quenching
Solutions of PC in chloroform were mixed with 1 mol% NBD-PC, formed into liposomes, and coated onto SiNPs using the above methods. Samples were purified by 5 centrifugation washes (9,400×g, 10 min). The lipid distributions were investigated by measuring NBD fluorescence on an LS 55 luminescence spectrometer (Perkin Elmer, Waltham, MA) with excitation at 475 nm (2.5 nm slit width) and emission recorded at 534 nm (20 nm slit width). The samples were run by collecting baseline fluorescence for 10 min followed by addition of sodium dithionite in HEPES buffer (final concentration was 5 mM) and mixing. Five min after the addition of sodium dithionite, the fluorescence was collected for 15 min and averaged. Melittin was added (final concentration was 1.3 μM) and mixed, followed by fluorescence measurement for 15 min. The mean and standard deviation of fluorescence changes were calculated from 5 independently prepared and assayed samples.
The change in fluorescence for the outer leaflet (ΔF1) was calculated as:
| (4) |
and the change in fluorescence for the inner leaflet (ΔF2) was calculated as:
| (5) |
where FI is the initial fluorescence, FD is the fluorescence after dithionite is added, and FM is the fluorescence after melittin is added.
To calculate the percentage of supported bilayer on each SiNP using fluorescence quenching data, each sample was normalized to the 100% DETAS sample, which is assumed to be exclusively supported bilayer, using:
| (6) |
where ΔF2SiNP is the decrease in fluorescence for the inner leaflet of the SiNP sample and ΔF2DETAS is the decrease for the inner leaflet on the 100% DETAS sample.
Measuring Interstitial Volume
Calcein-loaded liposomes were prepared using dehydrated PC films that were resuspended and extruded in 60 mM calcein dye in TRIS buffer. Adsorption of the liposomes onto the SiNPs was accomplished by stirring purified SiNPs with the calcein-loaded liposomes for 2 hr at room temperature using an established method.36 Samples were then purified using size exclusion chromatography on a Sephadex G-100 column. The first, salmon-colored fraction was collected. The SiNPs in this fraction were pelleted by centrifugation (9,400×g, 10 min), washed, and redispersed in 1 mL of TRIS buffer. Samples were excited at 490 nm (2.5 nm slit width), with emission recorded at 513 nm (10 nm slit width). Initial fluorescence was measured for 10 min. Melittin was added to the sample (final concentration of 1.3 μM) with gentle mixing, and incubated for 5 min. Fluorescence was measured for 15 min. In other experiments, 3 μL of 10% Triton-X was added in place of melittin.
The fluorescence measured after release was used to calculate the total interstitial volume:
| (7) |
where [calcein]cuvette is the concentration of calcein obtained from a standard curve, Vcuvette is the total sample volume (800 μL), and [calcein]interestitial is the concentration of calcein loaded on the SiNP (60 mM).
The interstitial volume per particle was then calculated as:
| (8) |
where the SiNPindividual mass was calculated from the average SiNP diameter of 113 ± 2 nm and a density of 2.65 g/mL37 and SiNPtotal mass was the mass of SiNPs in 800 μL of solution (obtained from a TGA experiment).
The thickness of the water layer was then calculated. First, the SiNP volume was combined with the interstitial volume and the diameter of a sphere needs to be encompass this combined volume was found using:
| (9) |
where Vtotal is the combination of VSiNP and the volume of an SiNP (calculated from the DLS diameter). Then, the thickness of the interstitial water layer was calculated from:
| (10) |
where dSiNP is the average diameter of the SiNPs.
Membrane Stability Assay
PC-coated, calcein-loaded SiNPs were mixed with HEPES buffer to a total volume of 800 μL. Triton-X was added to a concentration ranging from 0 to 0.4 mM and the fluorescence of each was measured for 10 min. Fluorescence background was subtracted and then normalized to 100 for the highest Triton-X concentration. A non-linear regression was used to calculate the concentration of Triton-X that resulted in 50% fluorescence intensity increase (F50) of the respective sample using GraphPad Prism. All R2 values were greater than 0.96.
Dynamic Light Scattering
DLS measurements were performed on a Zeta Sizer Nano S90 (Malvern Instruments, Westborough, MA). Data was recorded for samples with no visible precipitation. Average size was obtained from the intensity distribution.
Statistical Aanalysis
Statistical significance was evaluated using Student’s t-test and a p-value of <0.05 was considered as significant.
RESULTS
Thermal Analysis of Functionalized SiNPs
Samples with different functionalization ratios were synthesized by adding different input ratios of DETAS and ODTS (Scheme 2) at a total of 0.1 volume percent of silane. The relative amount of each silane on the SiNPs was determined using thermal analysis. Thermal analysis was performed on bare SiNP and SiNPs with 9 different functionalization ratios (Fig 1A and B) and data was normalized to 100% at 150 °C. To better observe differences in weight loss across the series and to identify a cut-off temperature for distinguishing DETAS from ODTS, a differential thermal analysis (DTA) was performed by calculating the rate of change in weight loss at each temperature (Fig 1C and D). The functionalized SiNPs showed changes in weight loss that varied with the input ratios of silanes used in the synthesis. Samples with 100% DETAS functionalization have three characteristic DTA peaks at approximately 225 °C, 335 °C, and 460 °C.38 As the input fraction of ODTS in the SiNPs increased, the two lower temperature peaks decreased in intensity, while the peak at 460 °C increased (Fig 1C and D).
Scheme 2.
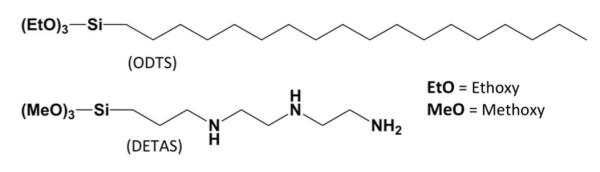
Silanes used for functionalization of SiNPs
Figure 1.

Thermal analysis of SiNPs displayed as (A) the weight percent lost and (B) the differential thermal analysis. Samples were functionalized with: 100% DETAS (dark red), 95% DETAS (red), 88% DETAS (orange), 81% DETAS (lime-green), 73% DETAS (dark green), 65% DETAS (light blue), 39% DETAS (blue), 17% DETAS (lavender), 100% ODTS (violet), and bare (black). Data was normalized to 100% at 150 °C for each sample.
To investigate the relationship between the ratio of silanes observed on the surface of the SiNPs by TGA and the input ratio of silanes used in the synthesis, the surface density of silanes was calculated from the TGA data. This was accomplished by calculating SiNP volume from average diameter, then converting the volume to an individual nanoparticle mass using the density of SiO2 as 2.65 g/mL.37 A surface concentration of 4 silanols per square nanometer was then used to estimate the total number of silanols available on the SiNP.37 Finally, the number of silanes per surface silanol were calculated (Table 1) using the total silane mass lost, the weighted average of the combustible portion of the molecular weights for each functionalization ratio, and an average SiNP diameter of 113 nm. Bare SiNPs had a 1.97% weight loss over the temperature range measured, indicating only minor condensation of the particles.35 The 100% DETAS SiNPs had the highest overall weight loss of 11.1% (Table 1). As a higher fractio of ODTS was used, there was a decreasing amount of weight loss.
Table 1.
Characterization of SiNPs by DLS and TGA. All data reported as mean ± SD, n=3.
| input DETAS (volume %) |
input DETAS (mol%) |
wt% loss ± SD < 440 °C |
wt% loss ± SD > 440 °C |
total wt% loss ± SD |
calculated f DETASa |
silanes per surface silanolb |
|---|---|---|---|---|---|---|
| 100 | 100 | 7.5 ± 0.1 | 3.6 ± 0.1 | 11.1 ± 0.3 | 1.00 | 2.58 |
| 90 | 95 | 7.1 ± 0.2 | 3.8 ± 0.2 | 10.9 ± 0.4 | 0.88 | 2.34 |
| 80 | 88 | 6.7 ± 0.1 | 4.3 ± 0.2 | 11.0 ± 0.3 | 0.67 | 2.15 |
| 70 | 81 | 5.7 ± 0.2 | 4.2 ± 0.2 | 9.9 ± 0.3 | 0.55 | 1.77 |
| 60 | 73 | 5.7 ± 0.1 | 4.2 ± 0.2 | 9.9 ± 0.3 | 0.54 | 1.75 |
| 50 | 65 | 4.9 ± 0.2 | 4.3 ± 0.1 | 9.2 ± 0.3 | 0.39 | 1.50 |
| 25 | 39 | 4.3 ± 0.1 | 4.4 ± 0.1 | 8.7 ± 0.2 | 0.30 | 1.35 |
| 10 | 17 | 3.6 ± 0.1 | 4.4 ± 0.1 | 8.0 ± 0.3 | 0.20 | 1.17 |
| 0c | 0 | 2.4 ± 0.1 | 4.6 ± 0.2 | 7.0 ± 0.3 | 0 | 0.97 |
Calculated from TGA data using eq (1) with a cut-off of 440 °C.
Calculated using average particle diameter of 113 nm.
Sample was functionalized with 100% ODTS.
There is a variation in the weight loss across the series of SiNPs (Fig 1A) with those containing a higher fraction of DETAS showing a greater loss than the ones with more ODTS. This trend is roughly linear with the input moles of silane. The silane density, expressed as silanes per surface silanol group showed a similar trend, with higher percent DETAS SiNPs having a higher silane coverage, suggesting that DETAS is more prone to deposition of polymerized silane coatings than is ODTS. Alternatively, DETAS may pack more tightly on the surface.
TGA of PC-Coated, Functionalized SiNPs
Thermal analysis of the PC-coated samples was used to investigate how much PC adsorbs to the differently functionalized SiNPs. Thermal analysis of PC-coated 17% DETAS SiNPs (Fig 2, blue) is typical of the PC-coated SiNPs examined; it has two significant weight loss regions at 240 °C - 270 °C and 440 °C - 520 °C. The peak in the 240 °C - 270 °C region is primarily attributed to PC as it is unique to the PC-coated samples and is consistent with previous studies of PC pyrolysis.39 The weight loss from a corresponding uncoated SiNP was subtracted from the coated sample to determine the amount of PC on the SiNP (Table 2). Attempts at lipid coating SiNPs with 100% ODTS functionalization resulted in rapid aggregation in aqueous solutions. Thus, for all experiments involving lipids, 83% ODTS was the highest mole percent used. The significance of the weight percent change from PC loss was compared to the 100% DETAS sample (Table 2).
Figure 2.
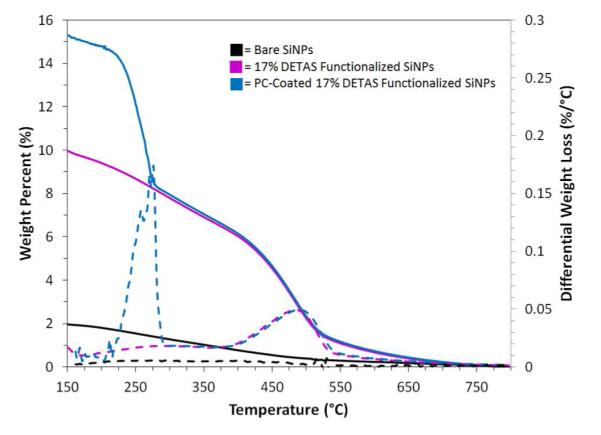
TGA (solid) and DTA (dashed) curves of bare SiNP (black), 17% DETAS functionalized SiNPs (pink), and PC-coated 17% DETAS functionalized SiNPs (blue). Data was set to 0 weight percent at 800 °C.
Table 2.
Thermal analysis of PC-coated functionalized SiNPs with varying input DETAS % (n = 5). Data was compared to 100% DETAS to determine p-value.
In the absence of normalization, each sample has slightly more PC than would be expected based on calculating each leaflet as a sphere of PC using the DLS data to determine the sphere size. That is, the experimental weight loss values (e.g. for 100% DETAS the weight loss was 10.9%) are somewhat higher than the expected weight percent assuming PC to be a homogeneous spherical shell. This could reflect instrumental error in either the TGA or DLS measurements. In particular, small errors in the diameter measured will have a large influence on the amount of PC calculated as required to coat the SiNPs. Since an alternate explanation is that excess liposomes were bound to the SiNPs, the ability of 5 wash steps to remove free liposomes2 was confirmed by a spiking experiment (Figure S3). Another explanation for this increase could be that an additional fraction of interdigitated PC is present on the SiNPs that is exclusively in the outer leaflet, as previously reported for PC-coated SiNPs.40,41
The amount of PC adsorbed on the functionalized SiNPs decreased with increasing coverage of hydrophobic ODTS and this can be rationalized as a mixture of supported and hybrid membrane on the surface (Scheme 1). The relative % PC coverage was calculated by normalization to the 100% DETAS SiNPs, thus providing an estimate of how much less PC was required due to the presence of ODTS on the SiNP. This decrease in amount of PC required is consistent with a model in which the amount of ODTS present determines the amount of hybrid membrane present. ODTS regions would be coated with a hybrid membrane that has only one leaflet of PC while DETAS regions would have a two-leaflet PC bilayer. For comparison, a prediction was made as to the amount of PC needed based on the assumption that DETAS regions would be coated with 2 PC leaflets and the ODTS regions with 1 PC leaflet using eq (3).
NBD-PC Fluorescence Quenching
To measure the lipid distribution between the inner and outer leaflets of the membrane at each functionalization ratio, the functionalized SiNPs were coated using liposomes doped with NBD-PC. Sodium dithionite, which quenches the NBD fluorescence by chemically reducing the dye,39 was added to the PC-coated SiNPs. The fluorescence of each sample was measured and compared to its initial fluorescence. Melittin or Triton-X was added to the sample to expose the inner leaflet lipids to the dithionite quencher. Melittin was used to create membrane pores,42-45 while Triton-X was used to solubilize the PC from the SiNPs. A representative plot of fluorescence is shown for a sample of PC-coated 100% DETAS functionalized SiNP (Fig 3). Fluorescence intensity drops were completed within 5 min of dithionite or melittin addition. Changes in fluorescence were calculated using eqs (4) and (5) (Table 3). Controls were performed with both Triton-X plus dithionite as well as dithionite only; shown in the supporting information (Figure S1).
Figure 3.
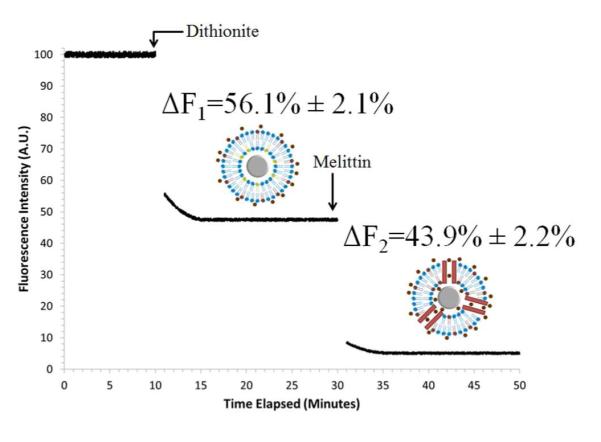
Dithionite quenching of PC/NBD-PC coated SiNPs, functionalized with 100% DETAS. Outer leaflet fluorescence was quenched by the addition of sodium dithionite (5 mM), followed by melittin (1.3 μM). The outer leaflet (ΔF1) and inner leaflet (ΔF2) fluorescence quenching was calculated using eqs (4) and (5). Data is presented as mean ± SD, n = 5. Plot is representative of multiple experiments.
Table 3.
Inner and outer leaflet lipid distribution and fraction of supported bilayer calculated from NBD quenching data. Mean ± SE, n = 5.
| input DETAS (mol%) |
% quenched (outer leaflet)a |
% quenched (inner leaflet)b |
calculated % SLBc |
|---|---|---|---|
| 100 | 56.1 ± 2.1 | 43.9 ± 2.2 | 100 |
| 95 | 58.4 ± 1.9 | 41.6 ± 1.7 | 95 |
| 88 | 59.8 ± 2.0 | 40.2 ± 1.7 | 92 |
| 81 | 61.9 ± 2.0 | 38.1 ± 2.1 | 87 |
| 73 | 64.4 ± 2.4 | 35.6 ± 2.2 | 81 |
| 65 | 66.1 ± 2.6 | 33.9 ± 2.6 | 77 |
| 39 | 73.4 ± 2.2 | 26.6 ± 2.4 | 61 |
| 17 | 79.7 ± 2.1 | 20.3 ± 2.2 | 46 |
| Bare | 53.0 ± 2.1 | 47.0 ± 1.9 | N/A |
The same technique was employed for each functionalized SiNP (Table 3). For the 100% DETAS SiNPs, 56% quenching occurred in the first step (outer leaflet), with 44% in the second step (inner leaflet), consistent with a symmetric supported bilayer structure. As ODTS functionalization increased, the percentage of lipids present on the inner layer decreased. At the highest ODTS fraction, the lipids are distributed with 80% in the outer leaflet and 20% in the inner leaflet. This is consistent with a mixture of supported and hybrid membrane regions on the functionalized SiNPs (Scheme 1).
A second method of estimating the fraction of supported and hybrid membrane on the SiNPs was based on the quenching data. The fraction of supported bilayer was calculated using eq (6) (Table 3) and the higher the input ODTS the less suported bilayer there was on the SiNPs. The values obtained from this calculation are comparable to those obtained from the TGA data (Table 2).
Interstitial Volume Decreases with Increasing Fraction of Hydrophobic Coating
To investigate the relationship between interstitial volume and input functionalization ratios, SiNPs were incubated with PC liposomes that had been resuspended in a self-quenching calcein solution (60 mM) (Scheme 3). After purification, the PC-coated SiNPs were porated or lysed to determine the amount of calcein solution entrapped in the interstitial layer. Melittin has been shown to form a 2.5 – 3.0 nm diameter pore,45 large enough to allow passage of calcein, which has an estimated diameter of 1.2 nm.46 Release of the entrapped calcein resulted in a roughly 40-fold dilution of the dye into the cuvette, thereby decreasing the concentration to a level where it fluoresces.
Scheme 3.
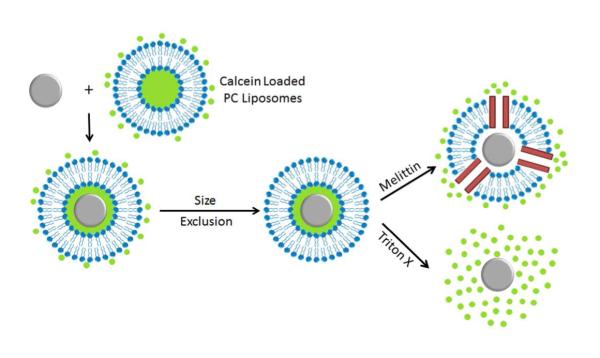
Assay for interstitial volume using melittin or Triton-X to release entrapped, self-quenching calcein.
The calcein concentration in the cuvette was obtained from the measured fluorescence intensity after release and was used to determine the interstitial volume. This was converted to an interstitial volume per SiNP using eq (7), (8), and (9) and a thickness of the interstitial water layer using eq (10) (Table 4). The more ODTS on the surface of the SiNPs, the smaller the interstitial volume was. PC-coated, 100% DETAS functionalized SiNPs showed a difference in water layer thickness that was not significantly different from the PC-coated bare SiNP control. Functionalization with the PC-coated, 17% DETAS SiNPs showed a decrease in water layer thickness by approximately 86% relative to the PC-coated bare SiNPs. A trend of decreasing fluorescence can be observed with decreasing water layer thickness as the DETAS percentage decreases (Table 4, Fig 4).
Table 4.
Calculated interstitial water layer volume and thickness.
Figure 4.
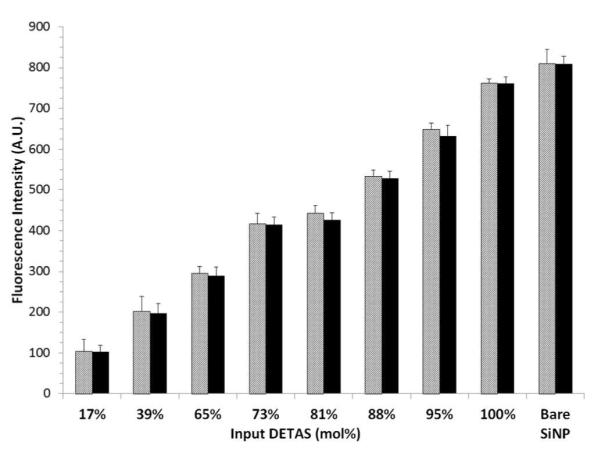
Interstitial volume of PC-coated SiNPs measured by fluorescence intensity. Fluorescence was measured after purification of the SiNPs and mixing with melittin (light bars) or Triton-X (dark bars). Mean ± SD, n = 5.
Bilayer Stability Assay
To examine the stability of the PC-coated functionalized SiNPs, calcein-loaded SiNPs were exposed to increasing concentrations of melittin or Triton-X (Scheme 3). After measuring the fluorescence after release, F50 values were calculated as the concentration of Triton-X that resulted in a 50% increase in fluorescence intensity (Fig 5). The stability of 100% DETAS functionalized SiNPs was significantly lower than the 17% DETAS functionalized SiNPs, with F50 values of 0.092 mM and 0.133 mM, respectively, and this change was statistically different as evaluated by a Student’s t-test.
Figure 5.
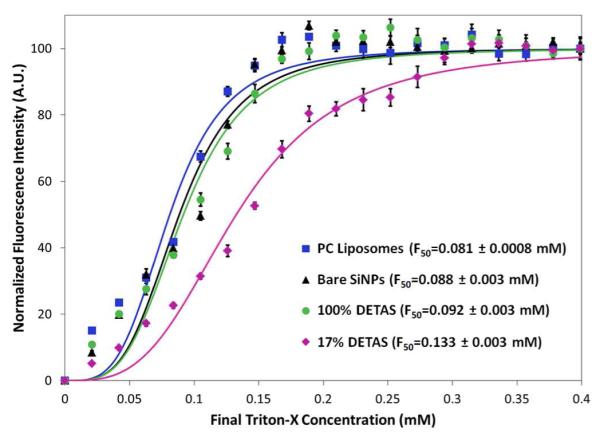
Fluorescence measurements to determine PC bilayer stability to Triton-X. Data was fitted with non-linear regression and the F50 values were calculated as the concentration of Triton-X that resulted in 50% drop in fluorescence intensity. Maximum absolute fluorescence was subtracted to 0 A.U. at 0 mM and normalized to 100 A.U. at 0.4 mM. Mean ± SD, n = 5.
DISCUSSION
The surface chemistry required for preparing supported bilayers and hybrid lipid membranes differs, making it a challenge to identify conditions that are compatible with both types of membrane. Supported bilayers can be formed directly on hydrophilic surfaces, such as the silanol coating of unmodified silica.2,3,40,41,47,48 In contrast, hybrid membranes or tethered membranes49 are formed on hydrophobic surfaces. For example, alkanethiol monolayers have been used as hybrid membrane supports on planar gold19 and on gold nanoparticles.21-23 Similarly, silica nanoparticles have been made hydrophobic by a condensation reaction with octadecanol, providing a support for hybrid lipid membranes.24,25 In other work, 13-(chlorodimethylsilylmethyl)heptacosane was used as a hydrophobic support for a hybrid membrane.26 In each case, a complete coating of the hydrophobic group was used, resulting in exclusively hybrid membrane formation after exposure to lipids. Here, we utilized a mixture of hydrophilic (DETAS) and hydrophobic (ODTS) silanes to obtain heterogeneous surface coatings that template regions of both supported bilayer and hybrid membrane on the same nanoparticle. From this study of surface modifiers, we identified the chemical properties that are essential to preparing a mixture of supported and hybrid membranes. Initially, we used the more common silane modifier, (3-Aminopropyl)triethoxysilane (APTES). However, unlike DETAS, we found that the lipid layers formed on APTES-coated SiNPs were unstable and permeable to dithionite (Figure S2). Previous lipid mobility measurements on APTES-modified mesoporous silica surfaces showed that APTES reduces lipid diffusion.50 We suggest that APTES may hydrogen-bond directly to the lipids, reducing mobility. Given the importance of a hydration layer between the lipids and the substrate, we reasoned that DETAS would provide more hydration due to it having more hydrogen-bonding groups and therefore would result in better supported bilayers. One possible role of the water layer could be to extend the hydrogen bonding network that connects the lipid to the surface, and APTES may provide too direct of a coupling while DETAS creates a larger water layer. The interstitial water layer measured is higher than typical for supported bilayers and suggests that polymerized silanes on the surface result in increased porosity or void spaces. Alternatively, the SiNPs may have a rough surface that creates water-filled regions between the SiNP surface and the lipid. Another possible explanation would be multi-layering of lipids with entrapped layers of water, although large regions of multi-layering are ruled out by TGA analysis. ODTS was selected as a hydrophobic modifier rather than 13-(chlorodimethylsilylmethyl)heptacosane or octadecanol which are much more and much less reactive than DETAS, respectively. We reasoned that the methoxy and ethoxy groups on DETAS and ODTS would react at similar rates and under mild conditions allowing for a simultaneous deposition and that the relative input ratios of silane would determine the amount of hydrophobic and hydrophilic surface coverage.
TGA was used to confirm coating of the nanoparticles and to estimate the relative amount of each silane on the SiNP surface similar to previous methods.41 The weight percent loss range for SiNPs used in functionalization was equivalent to 0.97 to 2.58 silanes per silanol (Table 1) and is consistent with polymerized silanes on the surface, suggesting that no more free silanol groups are available on the surface. Although the two silanes have unique signatures in the DTA, the weight loss for each molecule occurs across a broad and overlapping temperature range. After measuring the weight loss that occurs above and below a cut-off of 440 °C, it was possible to estimate the fraction of each silane on the surface by referencing the 100% DETAS and 100% ODTS samples using eq (1). From this analysis, it can be seen that each sample is within 10% of the input ratio of silanes used (Table 1). Using different temperature cut-offs in this analysis has minimal effect on this conclusion (not shown). Given the very low water solubility and high turbidity of the 100% ODTS functionalized SiNPs, we can rule out complete partitioning of each silane to different populations of SiNPs in the same reaction pot; if all the ODTS in the reaction added to one subset of the SiNPs, they would be insoluble. In all but one of the SiNP functionalizations, ODTS is present in a greater amount than the mole fraction of ODTS added. On average, there is 12% more ODTS on the SiNPs than would be predicted by the input mol percent. Only in the 17% mole DETAS sample is DETAS over-represented compared to its input mol percent. Phase separation of the two silanes during the deposition process could explain these deviations from the input ratios. Binding of ODTS to the SiNP surface above a threshold may increase the likelihood of additional ODTS binding to that SiNP. We anticipate that some degree of phase separation is likely, as depicted in Scheme 1. Heterogeneous patches of ODTS and DETAS are also reasonable based on the tendency of phase separation to occur during self-assembly of hydrogen-bonding pre-cursors28 and is consistent with our other evidence that supported bilayers and hybrid membranes co-exist on the SiNPs.
Thermal analysis of the PC-coated SiNPs supports a correlation between the fraction of hydrophobic modification and the fraction of the surface covered by a hybrid membrane. Our method for PC coating the functionalized SiNPs is similar to methods reported for coating bare or hydrophobic SiNPs.2,26,41 After coating, each sample was isolated from free liposomes and the quantity of PC on each SiNP was measured by TGA. The amount of PC contained on the SiNPs decreased with an increasing amount of ODTS modification, consistent with the idea that ODTS is coated by a hybrid membrane (with 1 leaflet of PC) and that DETAS is coated with a supported bilayer (with 2 leaflets of PC) (Scheme 1). By assuming that the 100% DETAS modified SiNPs contain exclusively supported bilayers, it was possible to estimate using eq (2) the percentage of supported bilayer on each SiNP using the TGA data (Table 2). This analysis follows the same trend as the fraction of DETAS (Table 1) and is consistent with a mixture of supported and hybrid membranes on the SiNPs that is determined by the amount of ODTS available.
For all of the SiNP samples, the fraction of supported bilayer is somewhat higher than the fraction of DETAS observed by TGA (Tables 1 and 3), suggesting that the supported bilayer regions have more than a bilayer of PC associated with them. Regions of multilayered PC or attached liposomes would increase the amount of PC needed to cover the particles. If some regions of non-phase separated ODTS and DETAS exist on the surface and they are coated with bilayer, this similarly would account for the discrepancy. Another explanation could arise from inclusion an additional fraction of interdigitated PC that is exclusively in the outer leaflet. Such structures have been proposed for PC coated bare SiNPs.40,41,47 If the degree of interdigitation is determined largely by the membrane curvature, the excess PC in the outer leaflet will be similar throughout this series.
Quenching assays using NBD-PC allowed for quantification of the relative fraction of PC present in the outer and inner leaflets, providing further confirmation that supported and hybrid membrane regions co-exist on each SiNP (Table 3). In contrast to using APTES as a surface modifier (Figure S2), the 100% DETAS coated SiNPs form a stable and completely ion-impermeable bilayer when coated with PC (Fig 3). The NBD-PC fluorescence intensity drops by 56% after the initial introduction of dithionite and drops an additional 44% after the membrane is porated by introduction of melittin (Fig 3) or Triton-X (Figure S1). This difference can largely be explained by an 8 nm difference in diameter for the inner and outer leaflets expected for a lipid bilayer on a sphere. For example, a SiNP with a 116 nm diameter and an 9 nm water layer is expected to have an inner leaflet diameter of 134 nm with 81,000 lipids and an outer leaflet diameter of 142.2 nm with 91,000 lipids. The lipid would be distributed with 53% in the outer leaflet and 47% in the inner leaflet. Thus, our NBD-PC measurements are consistent with previous studies of NBD-PC doped lipid bilayers that indicate no preferential distribution of NBD-PC to inner or outer bilayer leaflets.51
The fact that the second intensity drop after melittin addition is rapid and comparable to that induced by Triton-X suggests a substantial water layer under the lipids through which the dithionite migrates after melittin addition. The relative amount of the first NBD-PC fluorescence drop also scales with the fraction of ODTS on the SiNP surface (Table 3). As more ODTS is added, more hybrid membrane is formed. This is consistent with the ODTS modified regions containing a hybrid membrane in which there is no inner leaflet. Therefore all the NBD-PC is in the outer leaflet and is accessible to dithionite prior to any disruption of the membrane.
The method of calculating the fraction of supported bilayer from TGA data provides similar results to the method using NBD-PC quenching. The TGA method attributes a slightly smaller fraction to being supported bilayer than does the NBD-PC quenching method (~7% less on average). In neither case do we account for the expected difference in size of the inner leaflet and outer leaflet, although this difference is small (~3%). In the case of NBD-PC quenching, the data do not suggest that there is a complete interdigitated bilayer of PC which would have resulted in a much larger outer leaflet fluorescence drop. Using NBD-PC to test for interdigitation is only valid if the NBD-PC partitioning is comparable to and therefore representative of the PC partitioning. The similarity of the TGA and quenching analyses suggests that the NBD-PC does not accumulate within either leaflet more than would be expected for a PC liposome of comparable size.
By entrapping a self-quenching concentration of calcein dye inside the interstitial water layer, it was possible to correlate surface functionalization with changes in interstitial volume. The volume measured for the 100% DETAS SiNPs is consistent with a 89 Å water layer (Table 4). As the hybrid fraction increases with increasing ODTS, the interstitial water layer thickness decreases as expected. Both melittin and Triton-X release a comparable amount of calcein in each sample, supporting the idea that DETAS regions are coated with a single supported bilayer. This data also provides confirmation that the purification of PC-coated SiNPs is successful at removing free liposomes. This data provides additional support for a model with separate regions of supported and hybrid membrane (Scheme 1) in which polymerized silanes entrap more water. One motivation for designing hybrid or tethered bilayer systems is to increase membrane stability. Here, we show improved stability for the PC-coated SiNPs that have a higher fraction of hybrid membrane. The F50 values for SiNPs containing 100% DETAS (0.092 ± 0.003 mM) and 17% DETAS (0.133 ± 0.003 mM) reveal that the hybrid membranes are significantly more stable to surfactant than the supported bilayer (Fig 5). Similarly, we have reported high stability of hybrid membranes on gold nanoparticles (using a gold etchant assay) including stability to surfactants.21 For these heterogeneous SiNPs, any access of Triton-X through the supported bilayer regions may disrupt adjacent hybrid membrane regions, and therefore this may not represent the highest possible stability for a hybrid membrane on SiNPs.
CONCLUSIONS
Although the surface modification necessary for supported and hybrid membranes are dissimilar, methods are reported here whereby patches of hydrophilic and hydrophobic silanes can be attached to SiNPs. These surfaces allow for templating of both supported lipid bilayers and hybrid lipid membranes on the same nanoparticle. While planar substrates facilitate microscopic investigation of the surface heterogeneity, there are advantages to nanoparticle substrates. Here TGA was employed to quantify the nature of the silane coating and the PC coating. Similarly, fluorescence assays were used to probe symmetry of the leaflets and volume of the interstitial layer. Ultimately, these model systems will be useful in developing an understanding of how surface anchoring alters membrane properties. In particular, this work is a substantial first step toward creating models of picket fence membrane structures which could improve understanding of protein-membrane interactions. As the nanoparticle size is readily tunable, it will also be possible to access different membrane curvatures, providing a mimic of biological membranes that exist at very high curvature.52
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge Dr. Min S. Wang and Reid E. Messersmith’s assistance with data analysis and Heather L. Hodges for assistance with initial melittin studies. Support from NSF CBET-1033161 (SMR), NIH 2R15GM088960-02 (SMR) and a GREET award from the American Chemical Society is acknowledged.
REFERENCES
- (1).Alberts B. Molecular Biology of the Cell. 5th ed Garland Science; 2008. [Google Scholar]
- (2).Bayerl TM, Bloom M. Biophys. J. 1990:1–6. doi: 10.1016/S0006-3495(90)82382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Johnson SJ, Bayerl TM, McDermott DC, Adam GW, Rennie AR, Thomas RK, Sackmann E. Biophys. J. 1991;59:289–294. doi: 10.1016/S0006-3495(91)82222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Koenig B, Kruger S, Orts W, Majkrzak C, Berk N, Silverton J, Gawrisch K. Langmuir. 1996;12:1343–1350. [Google Scholar]
- (5).Cremer P, Boxer S. J. Phys. Chem. B. 1999;103:2554–2559. [Google Scholar]
- (6).Keller C, Kasemo B. Biophys. J. 1998;75:1397–1402. doi: 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Reviakine I, Brisson A. Langmuir. 2000;16:1806–1815. [Google Scholar]
- (8).Boxer S. Curr. Opin. Chem. Biol. 2000;4:704–709. doi: 10.1016/s1367-5931(00)00139-3. [DOI] [PubMed] [Google Scholar]
- (9).Castellana ET, Cremer PS. Surf. Sci. Rep. 2006;61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Vereb G, Szollosi J, Matko J, Nagy P, Farkas T, Vigh L, Matyus L, Waldmann T, Damjanovich S. Proc. Natl. Acad. Sci. USA. 2003;100:8053–8058. doi: 10.1073/pnas.1332550100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bayley H, Cremer P. Nature. 2001;413:226–230. doi: 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- (12).Parikh AN, Groves JT. MRS Bull. 2006;31:507–512. [Google Scholar]
- (13).Bussell SJ, Koch DL, Hammer DA. Biophys. J. 1995;68:1828–1835. doi: 10.1016/S0006-3495(95)80359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, Kusumi A. J. Cell. Biol. 2002;157:1071–1081. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nicolau D, Burrage K, Parton R, Hancock J. Mol. Cell. Biol. 2006;26:313–323. doi: 10.1128/MCB.26.1.313-323.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hancock J. Nat. Rev. Mol. Cell. Bio. 2006;7:456–462. doi: 10.1038/nrm1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Parikh AN. Biointerphases. 2008;3:FA22–FA32. doi: 10.1116/1.2889055. [DOI] [PubMed] [Google Scholar]
- (18).Howland M, Sapuri-Butti A, Dixit S, Dattelbaum A, Shreve A, Parikh A. J. Am. Chem. Soc. 2005;127:6752–6765. doi: 10.1021/ja043439q. [DOI] [PubMed] [Google Scholar]
- (19).Plant A. Langmuir. 1999;15:5128–5135. [Google Scholar]
- (20).Jenkins A, Bushby R, Evans S, Knoll W, Offenhausser A, Ogier S. Langmuir. 2002;18:3176–3180. [Google Scholar]
- (21).Sitaula S, Mackiewicz MR, Reed SM. Chem. Commun. (Camb) 2008:3013–3015. doi: 10.1039/b801525b. [DOI] [PubMed] [Google Scholar]
- (22).Kundu J, Levin CS, Halas NJ. Nanoscale. 2009;1:114–117. doi: 10.1039/b9nr00063a. [DOI] [PubMed] [Google Scholar]
- (23).Yang JA, Murphy CJ. Langmuir. 2012;28:5404–5416. doi: 10.1021/la300325p. [DOI] [PubMed] [Google Scholar]
- (24).Koole R, van Schooneveld MM, Hilhorst J, Castermans K, Cormode DP, Strijkers GJ, de Mello Donegá C, Vanmaekelbergh D, Griffioen AW, Nicolay K, Fayad ZA, Meijerink A, Mulder WJM. Bioconjug. Chem. 2008;19:2471–2479. doi: 10.1021/bc800368x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Linseisen FM, Hetzer M, Brumm T, Bayerl TM. Biophys. J. 1997;72:1659–1667. doi: 10.1016/S0006-3495(97)78811-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang L-S, Wu L-C, Lu S-Y, Chang L-L, Teng I-T, Yang C-M, Ho J-AA. ACS Nano. 2010;4:4371–4379. doi: 10.1021/nn901376h. [DOI] [PubMed] [Google Scholar]
- (27).Geissler M, Xia Y. Adv. Mater. 2004;16:1249–1269. [Google Scholar]
- (28).Smith R, Lewis P, Weiss P. Prog. Surf. Sci. 2004;75:1–68. [Google Scholar]
- (29).Duschl C, Liley M, Corradin G, Vogel H. Biophys. J. 1994;67:1229–1237. doi: 10.1016/S0006-3495(94)80592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Han X, Critchley K, Zhang L, Pradeep SND, Bushby RJ, Evans SD. Langmuir. 2007;23:1354–1358. doi: 10.1021/la062636v. [DOI] [PubMed] [Google Scholar]
- (31).Jenkins A, Boden N, Bushby R, Evans S, Knowles P, Miles R, Ogier S, Schonherr H, Vancso G. J. Am. Chem. Soc. 1999;121:5274–5280. [Google Scholar]
- (32).Lenz P, Ajo-Franklin C, Boxer S. Langmuir. 2004;20:11092–11099. doi: 10.1021/la048450i. [DOI] [PubMed] [Google Scholar]
- (33).Jung M, Vogel N, Köper I. Langmuir. 2011;27:7008–7015. doi: 10.1021/la200027e. [DOI] [PubMed] [Google Scholar]
- (34).Mackiewicz MR, Ayres BR, Reed SM. Nanotechnology. 2008;19:115607. doi: 10.1088/0957-4484/19/11/115607. [DOI] [PubMed] [Google Scholar]
- (35).Stöber W, Fink A, Bohn E. Colloid Surface. 1968:62–69. [Google Scholar]
- (36).Liu J, Jiang X, Ashley C, Brinker CJ. J. Am. Chem. Soc. 2009;131:7567–7569. doi: 10.1021/ja902039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bergna H, R W, Hubbard A. Colloidal Silica: Fundamentals and Applications. 1st ed CSC Press; Boca Raton: 2005. [Google Scholar]
- (38).Chan CCP, Choudhury NR, Majewski P. Colloids Surfaces A. 2011;377:20–27. [Google Scholar]
- (39).Dayani Y, Malmstadt N. Langmuir. 2012;28:8174–8182. doi: 10.1021/la301094h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ahmed S, Nikolov Z, Wunder SL. J. Phys. Chem. B. 2011;115:13181–13190. doi: 10.1021/jp205999p. [DOI] [PubMed] [Google Scholar]
- (41).Ahmed S, Wunder SL. Langmuir. 2009;25:3682–3691. doi: 10.1021/la803630m. [DOI] [PubMed] [Google Scholar]
- (42).Bhalla A, Chicka MC, Tucker WC, Chapman ER. Nat. Struct. Mol. Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- (43).Ningsih Z, Hossain MA, Wade JD, Clayton AHA, Gee ML. Langmuir. 2012;28:2217–2224. doi: 10.1021/la203770j. [DOI] [PubMed] [Google Scholar]
- (44).Zeineldin R, Piyasena ME, Sklar LA, Whitten D, Lopez GP. Langmuir. 2008;24:4125–4131. doi: 10.1021/la703575r. [DOI] [PubMed] [Google Scholar]
- (45).Ladokhin AS, Selsted ME, White SH. Biophys. J. 1997;72:1762–1766. doi: 10.1016/S0006-3495(97)78822-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Edwards DA, Prausnitz MR, Langer R, Weaver JC. J. Control. Release. 1995;34:211–221. [Google Scholar]
- (47).Savarala S, Ahmed S, Ilies MA, Wunder SL. Langmuir. 2010;26:12081–12088. doi: 10.1021/la101304v. [DOI] [PubMed] [Google Scholar]
- (48).Mornet S, Lambert O, Duguet E, Brisson A. Nano Letters. 2005;5:281–285. doi: 10.1021/nl048153y. [DOI] [PubMed] [Google Scholar]
- (49).Basit H, Van der Heyden A, Gondran C, Nysten B, Dumy P, Labbé P. Langmuir. 2011;27:14317–14328. doi: 10.1021/la202847r. [DOI] [PubMed] [Google Scholar]
- (50).Worsfold O, Voelcker NH, Nishiya T. Langmuir. 2006;22:7078–7083. doi: 10.1021/la060121y. [DOI] [PubMed] [Google Scholar]
- (51).Bhalla A, Chicka MC, Tucker WC, Chapman ER. Nat. Struct. Mol. Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- (52).Zimmerberg J, Kozlov MM. Nat. Rev. Mol. Cell. Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.