

Abstract
Extracellular accumulation of amyloid beta protein (Aβ) plays a central role in Alzheimer’s disease (AD). Some metals, such as copper, lead, and aluminum can affect the Aβ accumulation in the brain. However, the effect of mercury on Aβ accumulation in the brain is not clear. Thus, this study was proposed to estimate whether mercury concentration affects Aβ accumulation in PC12 cells. We treated 10, 100, and 1000 nM HgCl2 (Hg) or CH3HgCl2 (MeHg) for 48 hr in PC12 cells. After treatment, Aβ40 in culture medium increased in a dose- and time-dependent manner. Hg and MeHg increased amyloid precursor protein (APP), which is related to Aβ production. Neprilysin (NEP) levels in PC12 cells were decreased by Hg and MeHg treatment. These results suggested that Hg induced Aβ accumulation through APP overproduction and reduction of NEP.
Keywords: Alzheimer’s disease, Amyloid beta, Mercury, Amyloid precursor protein, β-Secretase, Neprilysin
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia and a prevalent neurodegenerative disorder in the current society (1). Currently, there are ~35.6 million people living with dementia worldwide in 2010 (2). AD is agerelated, and its clinical features include cognitive decline and behavioral disorders (3). Despite the extensive research, the pathophysiologic mechanism of AD is not clear; however, two types of accumulation in the brain feature it: extracellular senile plaques and intracellular neurofibrillary tangles (4). The major components of senile plaques are amyloid beta (Aβ) (1,5,6). Most of all, Aβ accumulation and senile plaques played an important role in the development of the AD (7-9). Aβ are derived by cleavage of amyloid precursor protein (APP). There are two sequential cleavages, mediated by β-secretase (aspartyl protease; β-APP-site cleaving enzyme, BACE) and γ-secretase (10,11). In addition, Aβ are degraded by several proteases, such as neprilysin (NEP), insulin-degrading enzyme (IDE), and endothelin-coverting enzyme (ECE) (12,13). Using mouse models, it was found that NEP is the major Aβ-degrading enzyme in the brain (14,15). NEP expression was decreased in AD patients, and APP expression was increased in earlyonset familial AD patients (EOFAD) (13,16).
Recently, a number of metal ions have been suggested to be risk factors associated with the pathogenesis of AD. Copper, iron, and aluminum concentrations were increased in senile plaques and neurofibrillary tangles of AD patients (17,18). Some studies reported copper and iron levels in serum and CSF increased in AD (19,20). Aluminum, copper, iron, and lead induced the Aβ production using in vitro and in vivo studies (21-25). Mercury is a well-known neurotoxic metal that is ubiquitous in the environment. In general population, the main source of human exposure to mercury is fish consumption. It has been reported that fish consumption is positively related to the blood levels of mercury (26,27). Methylmercury can easily cross the bloodbrain barrier, and targets and kills neurons in specific areas of the nervous system including the visual cortex, cerebellum, and dorsal root of ganglia (28). The potential role of mercury toxicity in AD has been studied from diverse approaches. First, in vitro exposure to mercury could cause neurodegeneration (29). In addition, mercury is able to induce oxidative stress and cell cytotoxicity (30,31). Some autopsy studies reported mercury concentrations in brain of AD patients (32,33). However, the results of mercury levels in blood, urine, and CSF were controversial (20,34-37). Even if Hg exposure is related to AD, whether Hg can affect Aβ accumulation in the brain is unclear.
The present study was conducted to determine whether mercury affect Aβ accumulation mediated by imbalance between Aβ synthesis and degradation. Aβ40 levels in culture medium were analysed, and protein and mRNA levels of APP, BACE1, and NEP in PC12 cells were assessed.
MATERIALS AND METHODS
Cell culture. Rat pheochromcytoma cells (PC12 cells) were obtained from Korean Cell Line Bank (KCLB). The cells were cultured in RPMI 1640 media (Welgene, Daegu, Korea) supplemented with 5% fetal bovine serum, 10% heat-inactivated horse serum, and 100 U/ml penicillin & streptomycin, and maintained at 37℃ with 5% CO2.
Mercury exposure. PC12 cells were exposed to mercury as follows: 0, 10, 100, 1000 nM of HgCl2 (Hg) or CH3HgCl2 (MeHg) for 48 hr at 37℃.
Measurement of Aβ40. For the present studies, cells were plated at a density of 1 × 106 cells/ml on poly-L-lysine coated 35 mm plate. Cells were allowed to attach and grown to 40-50% confluence. After treatment of Hg or MeHg into the cells, the culture media were collected at 0, 6, 12, 24, and 48 hr to measure the Aβ40. The levels of Aβ40 in the culture media were measured with a Human/Rat beta- Amyloid ELISA kit (Wako Pure Chemical Industries, Osaka, Japan). Briefly, 100 μl of 5-fold diluted media were placed in each well of a 96-well plate coated with monoclonal antibody specific for BNT77 and was incubated overnight at 4℃. On the following day, the solution was discarded and the plate washed 5 times with wash solution. 100 μl of HRP-conjugated antibody (BA27) were added into the wells and incubated at 4℃ for 2 hr. The antibody solution was removed from the wells and washed 5 times with wash solution. 100 μl of TMB solution were added and incubated in the dark for 30 min at room temperature. The reaction was terminated by adding 100 μl of stop solution, and the colorimetric absorbance was read at 450 nm. The levels of Aβ40 in the media were calculated using the standard curve.
Quantification of mRNA expression by real-time RTPCR. Total RNA was isolated from PC12 cells with the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Firststrand cDNA was synthesized from 1.0 μg of total RNA with RT-&GO Mastermix (MP Biomedicals, Solon, USA) and random nanomer (Takara, Tokyo, Japan) according to the manufacturer’s instruction. Real-time RT-PCR was performed with Mx3005P QPCR systems (Agilent Technologies, Forster city, USA) to quantify the mRNA levels of APP, BACE1, and β-actin. Each PCR reaction contained 2 μl cDNA, 10 μl of SYBR Premix Ex TaqTM (TaKaRa, Tokyo, Japan), and 125-500 nM of the forward and reverse primers (Table 1). After an initial denaturation at 95℃ for 30 sec, the amplification program consisted of 40 cycles of denaturation at 95℃ for 5 sec, annealing at 55℃ for 30 sec, and extension at 72℃ for 30 sec. Semi-nested RT-PCR method was used to quantify the mRNA expression of NEP. The relative differences in mRNA expression in the Hg- or MeHg-treated cells were calculated and expressed as a relative increase, setting the control at 100%.
Table 1.
Primers used for real time RT-PCR analysis
| mRNA | Forward primer | Reverse primer |
|---|---|---|
|
| ||
| APP | AACATGTGCGCATGGTGGA | CACGGCAGGGACGTTGTAGA |
| BACE1 | TGGTGGACACGGGCAGTAGTA | TCGGAGGTCTCGGTATGTACTGG |
| NEP | CCCAGTGTATGGTGTACCAG | TGGCCGGTAGGTTCCACACC |
| β-actin | GGAGATTACTGCCCTGGCTCCTA | GACTCATCGTACTCCTGCTTGCTG |
Western blot analysis. PC12 cells (1 × 107 cell/ml) were seeded on poly-L-lysine coated 100 mm plates. After 48 hr of treatment, cells were lysed with 200 μl Protein Extraction Solution (Elpis Biotech, Taejeon, Korea) containing 0.1% protease inhibitor (Sigma-Aldrich Co., St. Louis, USA). The lysates were then centrifuged at 12,000 g for 10 min at 4℃, and the supernatants were mixed with sample buffer and boiled for 10 min. Protein concentrations were determined by Quick Start Bradford Protein Assay Kit (Bio-rad, USA). Proteins were isolated by 10% SDS-polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride (PVDF) membranes (GE Healthcare, Pittsburgh, USA). Non-specific binding was blocked by incubation with 5% skim milk in TBS at room temperature for 1 hr. Membranes were incubated with primary antibody (APP: rabbit polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, USA), 1:200; NEP: mouse monoclonal IgG (Santa Cruz Biotechnology, Santa Cruz, USA), 1:50; Actin: goat polyclonal IgG (Santa Cruz Biotechnology, Santa Cruz, USA), 1:250) for 2 hr. Subsequently, the membranes were incubated with appropriate secondary antibodies (antirabbit IgG (Invitrogen, Carlsbad, USA), 1:2500; anti-mouse IgG (Koma Biotech Inc., Seoul, Korea), 1:2500; anti-goat IgG (Invitrogen, Carlsbad, USA), 1:2500) for 1 hr. The blots were developed using PowerOpti-ECL Western blotting detection reagent (Animal Genetics Inc., Hwasung, Korea) and LAS-1000 plus (Fujifilm, Tokyo, Japan). The blots were analyzed quantitatively using UN-SCAN-IT (Silk Scientific Inc., Orem, USA).
Statistical analysis. All results are represented as mean ± SE. Statistical analyses were performed with one-way ANOVA following multiple-comparison tests using Duncan’s method. The level of statistical significance was set at p < 0.01 or p < 0.05. All statistical analyses were performed using the PASW statistics package for Windows (version 18.0).
RESULTS
The effect of Hg and MeHg on Aβ accumulation. In order to confirm whether Hg or MeHg affect Aβ concentration, the levels of Aβ in the medium were measured by ELISA method. Exposure of PC12 cells to various concentrations (10-1000 nM) of Hg or MeHg for 48 hr increased the levels of Aβ in a dose-dependent manner (Fig. 1A). These increases were significant at 100 nM in Hg treatment (p < 0.01) and 10 nM in MeHg treatment (p < 0.05). At 1000 nM there were 517% and 483% increase in Aβ compared to controls, respectively. The effect of time-course of Hg and MeHg on Aβ accumulation was evaluated (Fig. 1B). From 12 hr after administration, 100 nM Hg initiated an increase in Aβ level. 100 nM MeHg prompted an accumulation of Aβ from 6 hr after treatment.
Fig. 1. The effects of Hg or MeHg on secreted Aβ40. (A) PC12 cells were incubated with different concentrations (10, 100, and 1000 nM) of Hg or MeHg for 48 hr, and Aβ40 levels were measured in culture media by ELISA. (B) PC12 cells were treated with 100 nM of Hg or MeHg for 0, 6, 12, 24, and 72 hr, and Aβ40 levels were evaluated. Quantitative data are stated as mean ± SEM (n = 4); *p<0.05 vs. control, **p < 0.01 vs. control.

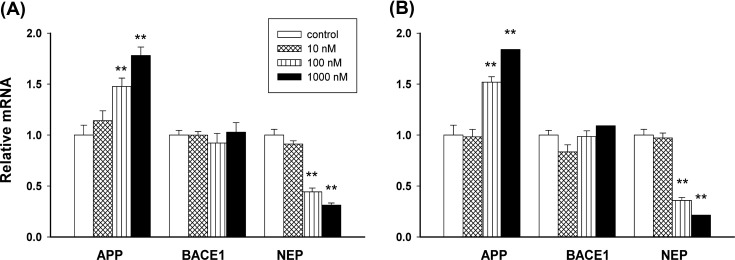
The effect on mRNA levels of APP, BACE1, and NEP. Quantitative real-time RT-PCR was performed to evaluate the effect of Hg and MeHg on intracellular APP, BACE1, and NEP expression (Fig. 2). mRNA expression of APP in PC12 cells that were treated with Hg or MeHg increased in a dose-dependent manner; these increase were significant at 100 nM in both treatment (p < 0.01, Fig. 2A and Fig. 2B). Fig. 2 shows that Hg or MeHg treatment had no significant effect on BACE1 expression, while increasing concentra-tions of Hg or MeHg reduced mRNA expression of NEP in PC12 cells (p < 0.01).
Fig. 2. The mRNA levels of APP, BACE1, and NEP after Hg or MeHg treatment. PC12 cells were cultured with medium only or with various concentrations (10, 100, and 1000 nM) of Hg (A) or MeHg (B) for 48 hr. Data are expressed as mean ± SEM (n = 4). *p < 0.05 vs. control, **p < 0.01 vs. control.
The effect of Hg and MeHg on protein expression of APP and NEP. As, Hg and MeHg affect mRAN expression of APP and NEP, the protein level of APP and NEP according to exposure of Hg and MeHg were examined (Fig. 3). PC12 cells exposed to increasing doses of Hg or MeHg (10-1000 nM) showed a dose-dependent increase in APP protein expression. At 100 nM of Hg or MeHg, there were significantly increased in APP levels in both treatments (p < 0.01). In addition, a decline in NEP protein expression following Hg or MeHg exposure was observed, with the most significant decrease at 100 and 1000 nM concentrations (p < 0.01).
Fig. 3. The protein expression levels of APP and NEP. PC12 cells were cultured with culture medium only or media with various concentrations (10, 100, and 1000 nM) of Hg (A, B) or MeHg (A, C) for 48 hr. (A) Cell lysates were immunoblotted for APP or NEP, and Actin was used as a loading control. Data are expressed as mean ± SEM (n = 4), **p < 0.01 vs. control.

DISCUSSION
The present study was performed to demonstrate whether mercury induces Aβ accumulation in the brain and, if it does, what the mechanism of the accumulation is. PC12 cells were treated with Hg or MeHg at 10-1000 nM, equivalent to around 2-200 μg Hg/L. These concentrations are not too high, because brain mercury levels in the general population are around 20 μg/L and can reach 174 μg/L in Greenlanders (33,38). MeHg is easily absorbed by the gastrointestinal tract and dispersed by blood throughout the body, including the brain. However, the main mercury species in the brain is inorganic mercury because demethylation of MeHg occurs in the brain (32,39). Thus, two different species of mercury - mercuric chloride (inorganic mercury) and methylmercury chloride (organic mercury) - were used for treatment.
In the results, Hg and MeHg increased Aβ40 levels in the medium in a dose- and time-dependent manner. Especially, Aβ40 increased significantly at 100 nM (20 μg/L) Hg or MeHg, which is similar to human brain levels in general population. These days, mercury is considered as one of the potential exogenous factors responsible for AD (40,41). Olivieri et al. (31) reported that mercury (50 μg/L) increased Aβ and tau phosphorylation in SHSY5Y neuroblastoma cells. However, they did not mention the mechanism of Aβ accumulation.
Aβ accumulation depends on the balance between Aβ production and degradation in the brain (42). The mRNA expression levels of APP and BACE1, which are related to Aβ production, were measured. Both Hg and MeHg increased mRNA expression of APP at 100 nM (Fig. 2). mRNA expression level of BACE1 did not change after exposure of Hg and MeHg. The findings matched those in the protein expression level of APP (Fig. 3). Pb, Mn, and Cu could induce Aβ accumulation through overproduction of APP (21,43-46). Nevertheless, the present study is the first report that Hg also increased Aβ deposit mediated by overproduction of APP without interaction with BACE1.
NEP is an important Aβ-degrading enzyme in the mammalian central nervous system. It has been reported that the expression or activity of NEP was reduced in the rat brain by Pb and Cu (12,13,21,44). However, little is known about the impact of Hg on NEP expression. In the present results, moderate dose (100 nM) of Hg and MeHg decreased NEP expression in the mRNA and protein levels dramatically.
In conclusion, these results show that Hg and MeHg exposure may be a risk factor of AD due to Aβ accumulation in the brain, and this Aβ accumulation is mediated by overproduction of APP and reduction of NEP.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (No. 2011-0012890).
References
- 1.Goedert M., Spillantini M.G. A century of Alzheimer’s disease. Science. (2006);314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer’s Disease International. World Alzheimer Report 2010. The global economic impact of dementia. World Alzheimer Report; (2010). pp. 1–52. [Google Scholar]
- 3.Van Den Heuvel C., Thornton E., Vink R. Traumatic brain injury and Alzheimer’s disease: a review. Prog. Brain Res. (2007);161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong R.A. The pathogenesis of Alzheimer’s disease:a reevaluation of the amyloid cascade hypothesis. Int. J.Alzheimers Dis. (2011);7:630985. doi: 10.4061/2011/630865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattson M.P. Pathways towards and away from Alzheimer’s disease. Nature. (2004);430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selkoe D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. (2001);3:75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- 7.Kounnas M.Z., Danks A.M., Cheng S., Tyree C., Ackerman E., Zhang X., Ahn K., Nguyen P, Comer D, Mao L., Yu C., Pleynet D., Digregorio P.J., Velicelebi G., Stauderman K.A., Comer W.T., Mobley W.C., Li Y.M., Sisodia S.S., Tanzi R.E., Wagner S.L. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. (2010);67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. (2003);300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 9.Hyman B.T. The neuropathological diagnosis of Alzheimer’s disease: clinical-pathological studies. Neurobiol. Aging. (1997);18:S27–32. doi: 10.1016/S0197-4580(97)00066-3. [DOI] [PubMed] [Google Scholar]
- 10.Carter M.D., Simms G.A., Weaver D.F. The development of new therapeutics for Alzheimer’s disease. Clin. Pharmacol. Ther. (2010);88:475–486. doi: 10.1038/clpt.2010.165. [DOI] [PubMed] [Google Scholar]
- 11.Nagga K., Gottfries J., Blennow K., Marcusson J. Cerebrospinal fluid phospho-tau, total tau and β-amyloid (1-42) in the differentiation between Alzheimer’s disease and vascular dementia. Dementia Geriatr. Cognit. Disord. (2002);14:183–190. doi: 10.1159/000066023. [DOI] [PubMed] [Google Scholar]
- 12.Guan H., Liu Y., Daily A., Police S., Kim M.H., Oddo S., LaFerla F.M, Pauly J.R., Murphy M.P., Hersh L.B. Peripherally expressed neprilysin reduces brain amyloid burden: A novel approach for treating Alzheimer’s disease. J. Neurosci. Res. (2009);87:1462–1473. doi: 10.1002/jnr.21944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miners J.S., Van Helmond Z., Chalmers K., Wilcock G., Love S., Kehoe P.G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. (2006);65:1012–1021. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- 14.Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N.P., Gerard C., Hama E., Lee H.J., Saido T.C. Metabolic regulation of brain Abeta by neprilysin. Science. (2001);292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 15.Iwata N., Tsubuki S., Takaki Y., Watanabe K., Sekiguchi M., Hosoki E., Kawashima-Morishima M., Lee H.J., Hama E., Sekine-Aizawa Y., Saido T.C. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. (2000);6:143–150. doi: 10.1038/77399. [DOI] [PubMed] [Google Scholar]
- 16.Carl W.C., Christian J.P. Alzheimer disease. Raven Press; New York: (1994). pp. 305–326. [Google Scholar]
- 17.Armendariz A.D., Gonzalez M., Loguinov A.V., Vulpe C.D. Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App. Physiol. Genomics. (2004);20:45–54. doi: 10.1152/physiolgenomics.00196.2003. [DOI] [PubMed] [Google Scholar]
- 18.Lovell M.A., Robertson J.D., Teesdale W.J., Campbell J.L., Markesbery W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. (1998);158:47–52. doi: 10.1016/S0022-510X(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 19.Squitti R., Lupoi D., Pasqualetti P., Dal Forno G., Vernieri F., Chiovenda P., Rossi L., Cortesi M., Cassetta E., Rossini P.M. Elevation of serum copper levels in Alzheimer’s disease. Neurology. (2002);59:1153–1161. doi: 10.1212/WNL.59.8.1153. [DOI] [PubMed] [Google Scholar]
- 20.Basun H., Forssell L.G., Wetterberg L., Winblad B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Parkinson’s Dis. Dementia Sect. (1991);3:231–258. [PubMed] [Google Scholar]
- 21.Kim D.K., Song J.W., Park J.D., Choi B.S. Copper induces the accumulation of amyloid-bet in the brain. Mol. Cell. Toxicol. (2013);9:57–66. doi: 10.1007/s13273-013-0009-0. [DOI] [Google Scholar]
- 22.Singh I., Sagare A.P., Coma M., Perlmutter D., Gelein R., Bell R.D., Deane R.J., Zhong E., Parisi M., Ciszewski J., Kasper R.T., Deane R. Low levels of copper disrupt brain amyloid-â homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. U. S. A. (2013);110:14771–14776. doi: 10.1073/pnas.1302212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walton J.R., Wang M.X. APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J. Inorg. Biochem. (2009);103:1548–1554. doi: 10.1016/j.jinorgbio.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 24.Zawia N.H., Lahiri D.K., Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radical Biol. Med. (2009);46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng S.Y., Trombetta L.D. The induction of amyloid precursor protein and alpha-synuclein in rat hippocampal astrocytes by diethyldithiocarbamate and copper with or without glutathione. Toxicol. Lett. (2004);146:139–149. doi: 10.1016/j.toxlet.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Myrtd G.J., Davidson P.W., Cox C., Shamlaye C.F., Palumbo D., Cernichiari E., Sloane-Reeves J., Wilding G.E., Kost J., Huang L.S., Clarkson T.W. Prenatal methylmercury exposure from ocean fish consumption in the Seychelles child development study. Lancet. (2003);361:1686–1692. doi: 10.1016/S0140-6736(03)13371-5. [DOI] [PubMed] [Google Scholar]
- 27.Grandjean P., Weihe P., White R.F., Debes F., Araki S., Yokoyama K., Murata K., Sorensen N., Dahi R., Jorgensen P.J. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol. Teratol. (1997);19:417–428. doi: 10.1016/S0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- 28.Clarkson T.W. Mercury: major issues in environmental health. Environ. Health Perspect. (1993);100:31–38. doi: 10.1289/ehp.9310031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leong C.C., Syed N.I., Lorscheider F.L. Retrograde degeneration of neurite membrane structural integrity of nerve growth cones following in vitro exposure to mercury. Neuroreport. (2001);12:733–737. doi: 10.1097/00001756-200103260-00024. [DOI] [PubMed] [Google Scholar]
- 30.Olivieri G., Novakovic M., Savaskan E., Meier F., Baysang G., Brockhaus M., M?ller-Spahn F. The effects of beta-estradiol on SHSY5Y neuroblastoma cells during heavy metal induced oxidative stress, neurotoxicity and beta-amyloid secretion. Neuroscience. (2002);113:849–855. doi: 10.1016/S0306-4522(02)00211-7. [DOI] [PubMed] [Google Scholar]
- 31.Olivieri G., Brack C., Müller-Spahn F., Stähelin H.B., Herrmann M., Renard P., Brockhaus M., Hock C. Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J. Neurochem. (2000);74:231–236. doi: 10.1046/j.1471-4159.2000.0740231.x. [DOI] [PubMed] [Google Scholar]
- 32.Björkman L., Lundekvam B.F., Laegreid T., Bertelsen B.I., Morild I., Lilleng P., Lind B., Palm B., Vahter M. Mercury in human brain, blood, muscle and toenails in relation to exposure: an autopsy study. Environ. Health. (2007);6:30–43. doi: 10.1186/1476-069X-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehmann W.D., Markesbery W.R., Alauddin M., Hossain T.I., Brubaker E.H. Brain trace elements in Alzheimer’s disease. Neurotoxicology. (1986);7:195–206. [PubMed] [Google Scholar]
- 34.Lee J.Y., Kim J.H., Choi D.W., Lee D.W., Park J.H., Yoon H.J., Pyo H.S., Kwon H.J., Park K.S. The association of heavy metal of blood and serum in the Alzheimer’s diseases. Toxicol. Res. (2012);28:93–98. doi: 10.5487/TR.2012.28.2.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerhardsson L., Lundh T., Minthon L., Londos E. Metal concentrations in plasma and cerebrospinal fluid in patients with Alzheimer’s disease. Dementia Geriatr. Cognit. Disord. (2008);25:508–515. doi: 10.1159/000129365. [DOI] [PubMed] [Google Scholar]
- 36.Letz R., Gerr F., Cragle D., Green R.C., Watkins J., Fidler A.T. Residual neurologic deficits 30 years after occupational exposure to elemental mercury. Neurotoxicology. (2000);21:459–474. [PubMed] [Google Scholar]
- 37.Fung Y.K., Meade A.G., Rack E.P., Blotcky A.J., Claassen J.P., Beatty M.W., Durham T. Determination of blood mercury concentrations in Alzheimer’s patients . J. Toxicol.Clin. Toxicol. (1995);33:243–247. doi: 10.3109/15563659509017991. [DOI] [PubMed] [Google Scholar]
- 38.Pedersen M.B., Hansen J.C., Mulvad G., Pedersen H.S., Gregersen M., Danscher G. Mercury accumulations in brains from populations exposed to high and low dietary levels of methyl mercury. Concentration, chemical form and distribution of mercury in brain samples from autopsies. Int. J. Circumpolar Health. (1999);58:96–107. [PubMed] [Google Scholar]
- 39.Friberg L., Mottet N.K. Accumulation of methylmercury and inorganic mercury in the brain. Biol. Trace Elem. Res. (1989);21:201–206. doi: 10.1007/BF02917253. [DOI] [PubMed] [Google Scholar]
- 40.Mutter J., Curth A., Naumann J., Deth R., Walach H. Does inorganic mercury play a role in Alzheimer’s disease? A systematic review and an integrated molecular mechanism. J. Alzheimers Dis. (2010);22:357–374. doi: 10.3233/JAD-2010-100705. [DOI] [PubMed] [Google Scholar]
- 41.Mutter J., Naumann J., Sadaghiani C., Schneider R., Walach H. Alzheimer disease: mercury as pathogenetic factor and apolipoprotein E as a moderator. Neuroendocrinol. Lett. (2004);25:331–339. [PubMed] [Google Scholar]
- 42.Farris W., Schütz S.G., Cirrito J.R., Shankar G.M., Sun X., George A., Leissring M.A., Walsh D.M., Qiu W.Q., Holtzman D.M., Selkoe D.J. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. (2007);171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y.Y., Chen T., Wan, Y., Xu S.Q. Lead exposure in pheochromocytoma cells induces persistent changes in amyloid precursor protein gene methylation patterns. Environ. Toxicol. (2012);27:495–502. doi: 10.1002/tox.20666. [DOI] [PubMed] [Google Scholar]
- 44.Huang H., Bihaqi S.W., Cui L., Zawia N.H. In vitro Pb exposure disturbs the balance between Aβ production and elimination: the role of AβPP and neprilysin. Neurotoxicology. (2011);32:300–306. doi: 10.1016/j.neuro.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basha M.R., Wei W., Bakheet S.A., Benitez N., Siddiqi H.K., Ge Y.W., Lahiri D.K., Zawia N.H. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J. Neurosci. (2005);25:823–829. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin R., Chen X., Li W., Han Y., Liu P., Pi R. Exposure to metal ions regulates mRNA levels of APP and BACE1 in PC12 cells: blockage by curcumin. Neurosci. Lett. (2008);440:344–347. doi: 10.1016/j.neulet.2008.05.070. [DOI] [PubMed] [Google Scholar]