

Abstract
While the Cysteine-Rich Secretory Proteins (CRISPs) have been broadly proposed as regulators of reproduction and immunity, physiological roles have yet to be established for individual members of this family. Past efforts to investigate their functions have been limited by the difficulty of purifying correctly folded CRISPs from bacterial expression systems, which yield low quantities of correctly folded protein containing the eight disulfide bonds that define the CRISP family. Here we report the expression and purification of native, glycosylated CRISP3 from human and mouse, expressed in HEK 293 cells and isolated using ion exchange and size exclusion chromatography. Functional authenticity was verified by substrate-affinity, native glycosylation characteristics and quaternary structure (monomer in solution). Validated protein was used in comparative structure/function studies to characterise sites and patterns of N-glycosylation in CRISP3, revealing interesting inter-species differences.
Expression of the Cysteine-Rich Secretory Proteins (CRISPs) occurs primarily in the male reproductive tissues and salivary glands; in mammals, expression is biased to the reproductive tract, while in many poisonous non-mammalian vertebrates such as snakes and lizards, CRISPs are secreted into venom where they exert toxicity through ion channel inhibition1,2,3,4,5. The mechanisms by which CRISPs function in the mammalian reproductive tract are less clear, however individual roles in the processes of decapacitation, acrosome reaction, sperm-oocyte fusion, flagellar motility and immunosuppression have recently been proposed6,7,8,9,10,11,12,13,14. The latter function has been attributed to equine CRISP3, which is present in high concentrations in seminal plasma and has been shown to inhibit sperm-neutrophil interaction in horses14. Whether this function of equine CRISP3 extends to its mouse and human orthologs remains to be determined.
Structurally, CRISPs constitute one of eleven mammalian subfamilies of the CAP (CRISP, Antigen 5 and Pathogenesis-Related 1 proteins) superfamily, to which they belong by virtue of their N-terminal CAP domain2. They are characterised by the presence of a C-terminal CRISP domain, a collective term encompassing an ion channel regulator (ICR) domain and the ‘hinge' which links the ICR and CAP domains (Figure 1). As its name suggests, the CRISP domain comprises an unusually cysteine-rich sequence in which ten cysteine residues form five disulfide bonds; the CAP domain of the protein encodes a further six cysteine residues which are also exclusively disulfide bonded2,15. These eight disulfide bonds confer to CRISPs their high degree of structural complexity. Unsurprisingly therefore, recombinant expression of functional CRISPs has proven difficult, with a number of previous attempts resulting in the production of misfolded, insoluble protein aggregates by bacterial systems; this material must then be subjected to an involved and often inefficient refolding process12,13,16. This, along with the problem of often inadequate post-translational modification of mammalian proteins by bacteria, creates a need for an improved approach to CRISP expression and purification.
Figure 1. CRISP domain organisation and tertiary structure.
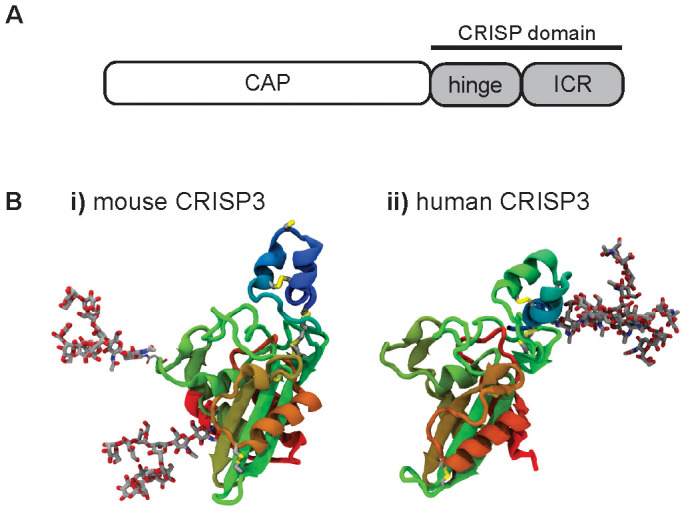
(A) CRISP domain organisation and (B) homology models of mouse and human CRISP3 based on the crystal structure of Natrin. The CRISP domain is shown in blue/aqua; disulfide bonds by white/yellow linkages. N-glycosylation is represented by high mannose (mouse CRISP3) and complex (human CRISP3) glycans (ball and stick models).
Here we used a mammalian protein expression system to generate useful quantities of soluble, correctly folded and post-translationally modified human CRISP3, as validated by interaction with the known serum binding partner alpha-1-B glycoprotein17,18. We also show that this validated CRISP3 preparation can provide informative results in structure/function analysis. In particular, differences in glycosylation between human and mouse CRISP3 were characterised.
Results
Expression of soluble human CRISP3
Stable, inducible HEK 293 cell lines expressing human or mouse CRISP3 were generated. Following 96–120 hours of induction, human His-tagged CRISP3 constituted approximately 1.8% of total protein in the cell media (1.5 μg/ml by comparison with a CRISP3 standard). Protein stability and lack of toxicity was indicated by a proportional increase in CRISP3 concentration with induction time (Figure 2A). Deglycosylase treatment with Peptide -N-Glycosidase F (PNGase F) confirmed that like native CRISP3, the recombinant protein was secreted in unglycosylated and N-glycosylated forms (Figure 2B)19.
Figure 2. HEK 293 cells efficiently secrete unglycosylated and N-glycosylated recombinant human CRISP3.
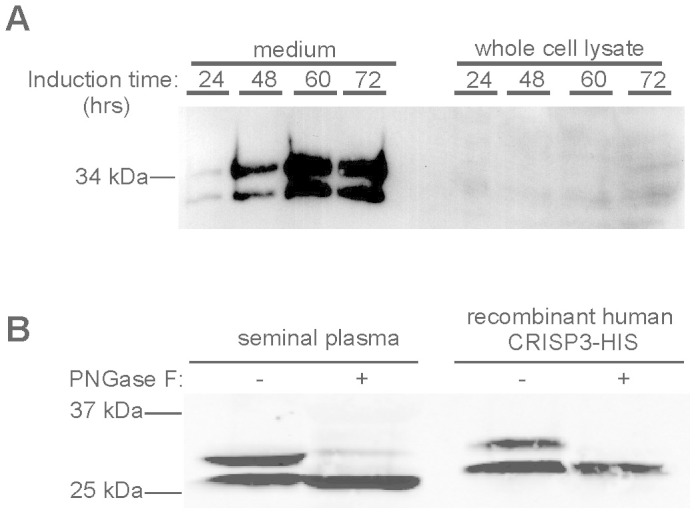
(A) Western blot with human CRISP3 antibody showing CRISP3 induction. (B) PNGase F treatment selectively affects the larger CRISP3 variant in both native (human seminal plasma, first two lanes) and recombinant (‘rCRISP3-His', last two lanes) protein. Full length gels are available under supplementary data.
Purification and validation of human CRISP3
We found that the nickel/cobalt coated resins used for His-tag purification lacked specificity with conditioned HEK cell medium, such that many secreted proteins bound to the column despite optimisation attempts. As an alternative enrichment method, we employed carboxymethyl (CM) cellulose cation exchange to achieve a fifteen-fold purification and 75% recovery. Subsequent size-exclusion chromatography achieved a further 2.5-fold enrichment with near-quantitative recovery and 67% purity (Figure 3B and Table 1). Immunoreactivity, SDS-PAGE analysis and glycosylation profile data were supported by mass spectrometry analysis of tryptic peptides from the predominant 26-kDa bands, which confirmed their identity as mouse and human CRISP3 (three high-quality sequence tags for each, not shown).
Figure 3. CRISP3 is enriched 15-fold following CM cellulose exchange.

(A) Silver-staining at various purification stages. Lane 1: starting material (250 μl conditioned media), lane 2: CM cellulose flow through, lanes 3–10: CM cellulose exchange gradient elutions (CRISP3 elutes in lanes 7–8, representing 110 and 240 mM NaCl respectively). (B) CRISP3 gel filtration to over 60% purity. (C) Mouse CRISP3 purified by the same method (two bands, 26 and 33 kDa). The 48 kDa contaminant is alpha-enolase. (D) Gel filtration absorbance trace with human CRISP3 peak indicated by the arrow. All gels were run under the same conditions.
Table 1. CRISP3 purification from HEK 293 conditioned media.
| Step | Volume (ml) | Protein (mg)* | hCRISP3 (μg)† | Purification (fold) | Recovery (%) |
|---|---|---|---|---|---|
| 1. Media | 500 | 40 | 720 | 1 | 100 |
| 2. CM cellulose | 10 | 2.2 | 540 | 15 | 75 |
| 3. S75 | 10 | 0.75 | 500 | 37 | 70 |
*Determined by Bradford assay.
†Determined by western blot comparison with commercial HIS-tagged CRISP3 standard and densitometry of silver-stained SDS PAGE (steps 1 and 2–3 respectively).
A single major contaminating protein in both human and mouse preparations was identified by mass spectrometry as alpha-enolase, a 48 kDa glycolytic enzyme, with a Mascot score of 159 (6 peptides, 19% sequence coverage). Secreted alpha enolase binds CM cellulose20,21,22,23,24.
Recombinant human CRISP3 binds A1BG
In the absence of a standard CRISP3 activity assay, we tested the ability of purified human CRISP3 to perform its only reported biological function, that of binding the serum protein alpha-1-B glycoprotein (A1BG), itself a protein of uncertain function17,18. Silver staining of CRISP3-binding human and bovine serum proteins revealed bands corresponding to A1BG in CRISP3-treated samples but not in untreated controls (Figure 4A). Mass spectrometry analysis confirmed their identity as human and bovine A1BG with Mascot scores of 343 (10 peptides, 26% sequence coverage) and 170 (4 peptides, 12% sequence coverage) respectively. Reduction and alkylation of recombinant CRISP3 practically abolished the CRISP3-A1BG interaction, indicating that the recombinant protein contains tertiary structure elements required for A1BG recognition (Figure 4B).
Figure 4. Recombinant human CRISP3 binds human and bovine A1BG.

(A) Gel analysis of proteins purified on recombinant CRISP3-coated cyanogen bromide resin showed CRISP3 interaction with A1BG from both human (lane 3, ~85 kDa band) and bovine (lane 3, ~65 kDa band) serum, as confirmed by mass spectrometry. Elution control samples were loaded as follows; lane 1: uncoated beads incubated with serum, lane 2: CRISP3-coated beads incubated with PBS, lane 3: CRISP3-coated beads incubated with serum. In the case of human serum, all eluted proteins in lane 3 other than A1BG bound non-specifically to the beads (compare with lane 1). (B) Denatured CRISP3 (lane 3) was unable to bind A1BG. Full length gels are available under supplementary data; gels were run under the same conditions.
Identification of mouse and human CRISP3 N-glycosylation sites
Purification of mouse CRISP3 (Figure 3C) revealed that its putative glycosylated form possessed lower electrophoretic mobility than glycosylated human CRISP3 (Figure 5A). To identify the site(s) of N-glycosylation in mouse CRISP3, site-directed mutagenesis was performed at three NXS/T motifs; N99, N113 and N156, where amino acid number reflects post-cleavage position. These were mutated individually and in combination to produce the mutants N99Q, N113Q, N156Q, N99Q + N113Q and N99Q + N113Q + N156Q. Western blot analysis showed that N99Q and N113Q (but not N156Q) mutations affected protein size, indicating glycosylation at both sites in the wild type protein (Figure 5B, C). Interestingly, we observed that two mutants (N99Q + N113Q and N99Q + N113Q + N156Q) incapable of undergoing glycosylation were of relatively low abundance in the cell media (Figure 5C).
Figure 5. Recombinant mouse CRISP3 is glycosylated at N99 and N113.

Western blotting with His tag antibody showed glycosylated human and mouse CRISP3-His to be 29 and 33 kDa respectively (A). (B) Peptide pre-absorption of mouse CRISP3 antibody. (C) Mouse CRISP3 N99Q and N113Q mutant proteins were smaller than wild type (WT) mouse CRISP3 (PNGase F untreated samples); N156Q was unaffected. Two mutants unable to undergo glycosylation (N99Q + N113Q and N99Q + N113Q + N156Q) were of relatively low abundance. ‘NT' represents non-transfected HEK cell media. Full length gels are available under supplementary data; gels were run under the same conditions.
N-glycosylation of human CRISP3 at N220 only was confirmed, with no observable glycosylation of hCRISP3-N220Q (Figure 6B, last lane), and no impairment in protein secretion.
Figure 6. Mouse CRISP3 is incompletely digested by Endo H.
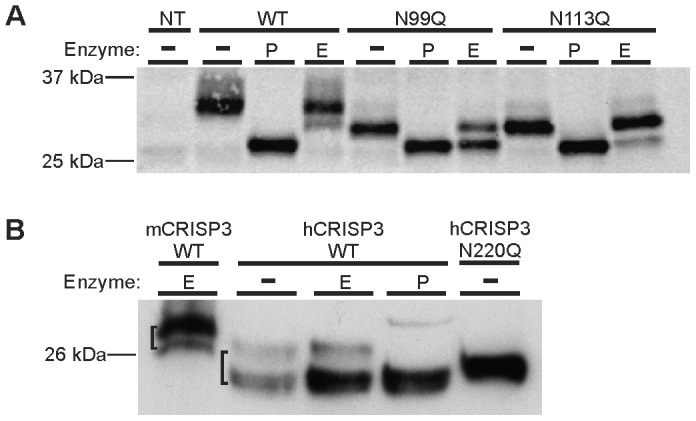
(A) Mouse CRISP3 wild type, N99Q and N113Q are partially digested by Endo H (E). Complete digestion with PNGase F (P) is included for size comparison. (B) A size comparison of mouse and human CRISP3 (mCRISP3 and hCRISP3, respectively) glycans (square brackets). Full length gels are available under supplementary data; gels were run under the same conditions.
Partial characterisation of mouse CRISP3 N-glycosylation types
To investigate the nature of mouse CRISP3 glycosylation, the N99Q and N113Q mutants were exposed to the deglycosylase Endoglycosidase H (Endo H). Endo H leaves complex oligosaccharides intact while cleaving high mannose and some hybrid N-linked oligosaccharides. Under standard reaction conditions, partial digestion of the mouse wild type, N99Q and N113Q proteins was observed following Endo H treatment (Figure 6A); none of a range of time and temperature conditions employed improved sensitivity to the enzyme. Human CRISP3 was resistant to Endo H, as expected19. Western blot comparison of mouse and human glycans revealed a size difference consistent with complex and high mannose modifications respectively (Figure 6B, lanes 1 and 2).
Finally, in a comparison of the relative mobility of salivary gland CRISP3 with three post-translationally modified variants of the recombinant protein, only the unglycosylated form of mouse CRISP3 could be observed in whole salivary gland (Figure 7).
Figure 7. CRISP3 from whole mouse salivary gland is unglycosylated.
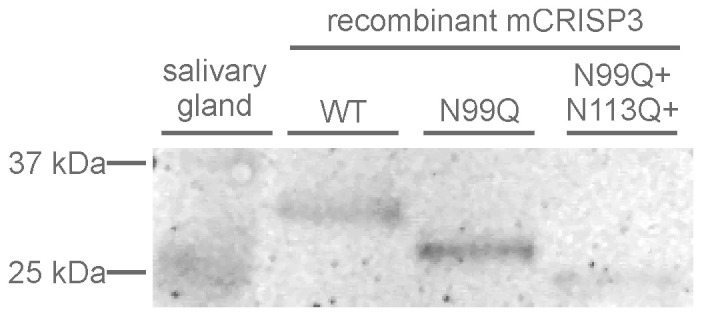
Western blot comparison of wild type salivary gland CRISP3 with recombinant mouse CRISP3 glycosylated at two sites (wild type, lane 1), one site (N99Q, lane 2) and unglycosylated (N99Q + N113Q + N156Q, lane 3). Full length gel is available under supplementary data.
Discussion
The structural complexity conferred by the high cysteine content of CRISPs has repeatedly caused purification attempts from bacterial cells to result in the production of insoluble inclusion bodies even under conditions intended to favour disulfide bond formation12,13. Alternative purification methods from neutrophil extracts have employed cation exchange and affinity chromatography, but these approaches are limited by tissue availability18,19. Here we demonstrate that recombinant human CRISP3 expression and two-step purification from HEK 293 cells yields milligram quantities of soluble protein suitable for structure/functional analyses. Indications of functional authenticity included efficient secretion, post-translational modification and confirmed interaction with the known binding partner A1BG, which was abolished with denaturation of CRISP3. The recombinant protein reproduced the native CRISP3 glycosylation profile, with site-directed mutagenesis confirming N220 as the exclusive site of N-glycosylation. A published report of Endo H resistance suggesting a hybrid or complex carbohydrate modification was confirmed19.
We present the first report of mouse CRISP3 immunodetection by western blotting using a mouse CRISP3-specific antibody of our own production, as well as the first evidence of N-glycosylation at two sites (N99 and N113). While incomplete digestion by Endo H of both glycosylation sites may reflect either imperfect enzyme access to cleavage sites or a mixture of glycosylation types, both N99 and N113 likely represent sites of high mannose or hybrid glycosylation to some degree. A relationship between glycosylation state and protein function (affecting protein stability or intracellular retention) was suggested by the relatively low abundance of unglycosylated mouse CRISP3. We speculate that glycosylation of mouse, but not human CRISP3, is required for escape from the ER quality control system25.
A gel comparison of salivary gland CRISP3 to three post-translationally modified variants of the recombinant protein indicated that in whole salivary gland at least, mouse CRISP3 is wholly or predominantly unglycosylated. Clarification of glycosylation patterns in other tissues is required; as we have demonstrated a link between glycosylation and secretion efficiency, it will be of particular interest to compare the glycosylation profile of CRISP3 from whole tissues with that obtained from endocrine secretions. Finally, we note that while human CRISP3 is glycosylated in the CRISP domain only (N220), both mouse glycosylation sites N99 and N113 are found in the N-terminal CAP domain (Figure 1B); this difference in glycosylation pattern may indicate distinct functions. This glycosylation site distribution appears to reflect a broader species-specific pattern in which human CRISPs 1 and 3 are N-glycosylated in the ICR domain with no N-glycosylation motifs in the CAP domain, while all four mouse CRISPs have potential N-glycosylation sites in their CAP but not CRISP domains. The major form of seminal plasma human CRISP1 contains a 4 kDa glycan, consistent with complex modification26. N-glycosylation motif sequences are absent in human CRISP2 and equine CRISP3, to which a neutrophil-suppressive function has been attributed14.
Sequence differences between pseudechetoxin and pseudecin, two cyclic nucleotide-gated ion channel blocking venom CRISPs with a 15–30-fold difference in affinity, suggest that most of the critical functional residues responsible for contact with target ion channels are concentrated on the concave surface of the proteins spanning the CAP-CRISP domain interface27,28. That this cleft formed by the bridge between CRISP and CAP domains is involved in substrate binding is supported by evidence that the ICR domain of CRISPs alone provides inadequate or suboptimal ion-channel inhibition, so that the CAP domain is believed to provide additional specificity to the ion channel interaction12,28. Indeed, sequence homology studies have identified a collection of residues within snake venom CRISPs which form a continuous line along the protein backbone and have been subject to high frequency evolutionary variation29. Sequence variability in these regions is therefore likely to confer selectivity and affinity for binding partners, accounting for the diverse range of CRISP ion channel targets. A comparison of CRISP and CAP proteins from multiple species similarly identified three hypervariable regions in the CAP domain which were postulated to confer substrate specificity30. The mouse CRISP3 glycosylation site N113 lies within one of these hypervariable regions, corresponding to G109-V117, in the CAP domain. Due to the high degree of rotational freedom of the CAP and ICR domains and the lack of an ICR domain in many CAP proteins, a separate, non-ion channel binding function has often been postulated for the CAP domain. If the G109-V117 sequence confers specificity to either an ion-channel interaction or distinct CAP domain function, then carbohydrate addition within this region may have significant functional implications. 25 and 12 amino acids separate the critical 5-amino acid variable ion-channel binding sequence in the ICR domain from the human CRISP3 and CRISP1 N-glycosylation sites, respectively. Despite the addition of a large, complex sugar moiety in close proximity to the CRISP3 binding cleft, Udby et al. have previously shown that interaction with at least one binding partner, A1BG, occurs independently of CRISP3 glycosylation status18.
In conclusion, this study describes a new method of producing CRISP3 of sufficient abundance and purity for structure/function analysis, as demonstrated by elucidation of the sites of glycosylation of mouse CRISP3, about which very little is currently known.
Methods
Cloning, transfection and cell culture
Human and mouse CRISP3 cDNA were amplified from total RNA extracts of the LNCaP prostate cancer cell line and wild type C57BL/6 mouse salivary gland respectively. cDNAs were cloned following addition of attB sites by PCR using the Gateway system (Life Technologies), and the pcDNA5/FRT/TO-CRISP3 product obtained. AttB primers used for human CRISP3 were: forward primer 5′ggggacaagtttgtacaaaaaagcaggcttcttcaccatgacattattcccagtgctgttg, reverse primer 5′ggggaccactttgtacaagaaagctgggtcttaataaatgctgtttgaacaattgc. For mouse CRISP3, the primers used were: forward primer 5′ ggggacaagtttgtacaaaaaagcaggcttcttcaccatggcattaatgcttgtg, reverse primer 5′ ggggaccactttgtacaagaaagctgggtcttaacattggcatgtagc. The QuickChange Lightning Site-Directed Mutagenesis kit (Stratagene) was used to introduce N-terminal 6xHis tags to human and mouse CRISP3 following the third post-cleavage amino acid. For human CRISP3, these primers were: forward primer 5′catcatcaccatcaccacgcaaatgaagataaggatcccgcttttactgctttg, reverse primer 5′ ttcatttgcgtggtgatggtgatgatgttcatttgctggaaaagatggaagcag. Mouse His tag primers were: forward primer: 5′ caagataaccatcatcaccatcaccaccaagataactctcaggagaacagtcttgag, reverse primer 5′ gttatcttggtggtgatggtgatgatggttatcttgaagaagggatggggg. Glycosylation site mutants contained the following amino acid changes: N99Q, N113Q, N156Q, N99Q + N113Q, N99Q + N113Q + N156Q (mouse CRISP3) and N220Q (human CRISP3).
Stable transfections were performed by co-transfection of CRISP3 and Flp-recombinase (pOG44) expression vectors into FlpIn T-Rex HEK 293 cells (Life Technologies) using FuGENE HD. Transfected cells were selected and maintained in DMEM containing 10% FBS and 100 μg/ml hygromycin.
Expression and purification of human and mouse CRISP3
Transfected HEK cells were grown in 175 cm2 flasks until confluent, when culture media was removed and cells washed twice with PBS. Protein expression was induced for 96–120 hours in FBS-free DMEM containing 1 μg/ml tetracycline. Media was cleared with a 0.2 micron filter and supplemented with 1 mM PMSF upon collection.
Conditioned media was concentrated 4-fold with 10 kDa Amicon Ultra-15 Centrifugal Filter Units (Millipore). Concentrate was diluted with 7 volumes of binding buffer (10 mM HEPES, pH 6.5) to reduce NaCl concentration from 160 mM to 20 mM, and the pH adjusted to 6.5 with 1 M HCl. CM cellulose (Sigma-Aldrich), previously washed according to instructions and resuspended in dH2O, was adjusted to pH 6.5 and added as a 50% slurry to the protein solution to a 1% final volume. This mixture was incubated at 4°C for 1 hour with gentle rotation. Unbound protein was removed through a sintered glass funnel under vacuum, the resin washed twice with 10 times the resin volume of binding buffer, and adjusted to a 50% slurry. This was packed into a 1 × 50 cm Econo-Column (BioRad) column and washed until the resin bed was compact. The following gradient elution program was carried out at a flow rate of 1 ml/minute; 5 minute wash, 30 minute gradient elution of 0–1 M NaCl in wash buffer, 5 minute wash with 1 M NaCl in wash buffer, 5 minute wash with wash buffer.
Gel filtration was performed on a Hi Load 16/60 Superdex 75 column (GE Healthcare Life Sciences). Samples were diluted in PBS and 1 ml fractions were collected at 1.2 ml per minute at 4°C. CRISP3-containing fractions were identified by immunoblotting and correlated with the absorbance trace.
Deglycosylase treatment
Human or mouse pcDNA5/FRT/TO-CRISP3 constructs were transiently transfected into HEK T-Rex cells as described. Protein expression was induced 24 hours after transfection and media collected 48 hours later. For both control and PNGase F/Endo H (New England Biolabs) treatments, 1 ml medium was concentrated to 20 μl with a 10 kDa Amicon Ultra-4 Centrifugal Filter Unit (Millipore) and samples were treated with deglycosylase buffers according to instructions; in control treatments, dH2O was substituted for enzyme. Samples were treated for 3 hours at 37°C before addition of Laemmli buffer.
Anti-mouse CRISP3 antibody production and validation
A mouse CRISP3-specific antibody was raised in goats against the peptide QYKDMSFWCKRLEYVC conjugated to diphtheria toxoid (Antibodies Australia, Clayton, VIC) and purified using a peptide affinity column as described previously31,32. 1 μg/ml was used for western blotting, with specificity validated by pre-absorption with 150-fold molar excess of immunising peptide (16 hour incubation at 4°C).
Western blotting
Conditioned medium was concentrated with an Amicon Centrifugal Filter Unit and boiled for 10 minutes in Laemmli buffer (2% β-mercaptoethanol). Proteins were separated on precast 4–15% acrylamide Criterion Tris/HCl SDS-PAGE gels (Bio-Rad) for 2.5 hours at 110 V. Transfers were perfomed at 4°C for 55 minutes at 100 V onto nitrocellulose membrane and blocked in 5% skim milk powder/TBS-T (0.05% Tween-20) for one hour at room temperature. Human CRISP3 antibody (R&D Systems, MAB23971) was used at 2.5 μg/ml in blocking buffer for one hour at room temperature. His-tag antibody (Cell Signalling Technology, 2366) was used at a 1:1000 dilution. Li-cor secondary antibodies were diluted 500-fold in blocking buffer and imaging performed on the Odyssey Infrared Imaging System (Li-cor Biosciences).
A1BG binding
A1BG binding was carried out using cyanogen bromide-activated-Sepharose 4B (Sigma Aldrich) essentially as described by Udby et al17. 50 μg purified human CRISP3 was used per treatment with 0.5 ml FBS or 0.1 ml human serum. Denatured CRISP3 was obtained by reduction with 20 mM DTT for 1 hour at 55°C followed by addition of iodoacetamide for 1 hour at room temperature in the dark. Buffer exchange to PBS was performed with a10 kDa Amicon Ultra-15 Centrifugal Filter Unit.
Mass spectrometry
Purified and silver stained human and mouse CRISP3 from SDS-PAGE were excised and subjected to in-gel trypsinolysis essentially as before33. Bands were reduced (2.5 mM DTT, 30 minutes, 60°C), alkylated (10 mM iodoacetamide, 30 minutes, room temperature in the dark), and digested (0.5 μg trypsin, 20 mM ammonium bicarbonate, 37°C for 14 hours). Tryptic peptides were analysed by LC-MS/MS using an HCT ULTRA ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) coupled to a nano HPLC (Ultimate 3000, Dionex Corporation, SunnyBrook, CA, USA), largely as done previously34. Identity searches were conducted using the MASCOT MS/MS Ions Search engine (www.matrixscience.com) against the NCBInr and Swiss-Prot databases with mammalian taxonomy selected. Search parameters were; fixed modifications, carbamidomethyl; variable modifications, oxidation (Met), up to one missed cleavage allowed.
Preparation of human tissues
The collection and use of tissues described in this study was approved by the Human Research and Ethics Committee of Southern Health (Monash Medical Centre, Monash Day Surgery and Monash University). Human seminal plasma was obtained with informed consent from Monash IVF (Clayton, Australia) and prepared for western blotting as described elsewhere35. Human blood was provided with informed consent by the Monash Medical Centre (Clayton Australia).
Preparation of mouse tissues
Animal procedures were performed in accordance with the Australian NHMRC Guidelines on Ethics in Animal Experimentation and approved by the Monash University Animal Experimentation Ethics Committees. Whole mouse salivary gland was obtained from 12 week old C57BL/6 mice, frozen in liquid nitrogen, powdered with a mortar and pestle and resuspended in tissue lysis buffer (1% SDS, 50 mM Tris, 150 mM NaCl, 1% Triton-X, pH8) containing protease inhibitors (Calbiochem, 539134). Samples were centrifuged at maximum speed (20 minutes, 4°C) and 100 μg of protein was prepared for western blotting.
Homology modelling
A mouse CRISP3 homology model based on the crystal structure template of Natrin, a snake venom CRISP from Taiwan Cobra (pdb 1XTA), was generated using the prime module from the Schrodinger molecular modelling software suite with missing residues added (Prime version 3.0 Schrödinger, Inc, New York, NY 2010). The model was further refined and visualized with VMD software36. A 50 nanosecond NAMD molecular dynamics simulation of the CRISP model was produced. Temperature was controlled using Langevin dynamics37. The representative glycan groups modelled were complex (tetraantennary with terminal sialic acid residues) and high mannose (mannose-9) for human and mouse CRISP3, respectively.
Author Contributions
M.V. performed the protein purification and glycosylation experiments. J.M. designed the protein purification strategy. P.M. and M.O. supervised the project. D.J. performed cDNA isolation, and both D.J. and R.D. provided technical assistance. M.V., J.M. and P.M. wrote the main manuscript text, and M.V. prepared all figures. All authors reviewed the manuscript.
Supplementary Material
Supplementary figures
Acknowledgments
We thank staff at the Monash Biomedical Proteomics Facility at Monash University, Australia, for support with proteomic analysis, and Monash IVF, Australia, for providing access to human seminal plasma samples. Homology modelling was carried out by Dr. Michael Kuiper, Victorian Life Science Computation Initiative (VLSCI). This work was funded by NHMRC grant 606563 to MO'B and PMc.
References
- Brown R. L., Haley T. L., West K. A. & Crabb J. W. Pseudechetoxin: a peptide blocker of cyclic nucleotide-gated ion channels. Proc Natl Acad Sci U S A 96, 754–759 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs G. M., Roelants K. & O'Bryan M. K. The CAP superfamily: cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins--roles in reproduction, cancer, and immune defense. Endocr. Rev. 29, 865–897, 10.1210/er.2008-0032 (2008). [DOI] [PubMed] [Google Scholar]
- Morrissette J. et al. Primary structure and properties of helothermine, a peptide toxin that blocks ryanodine receptors. Biophys. J. 68, 2280–2288, 10.1016/S0006-3495(95)80410-8 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y., Brown R. L. & Morita T. Purification and cloning of toxins from elapid venoms that target cyclic nucleotide-gated ion channels. Biochemistry 41, 11331–11337 (2002). [DOI] [PubMed] [Google Scholar]
- Zhou Q. et al. Structural and functional characterization of ryanodine receptor-natrin toxin interaction. Biophys. J. 95, 4289–4299, 10.1529/biophysj.108.137224 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts K. P., Wamstad J. A., Ensrud K. M. & Hamilton D. W. Inhibition of capacitation-associated tyrosine phosphorylation signaling in rat sperm by epididymal protein Crisp-1. Biol. Reprod. 69, 572–581, 10.1095/biolreprod.102.013771 (2003). [DOI] [PubMed] [Google Scholar]
- Nixon B. et al. The identification of mouse sperm-surface-associated proteins and characterization of their ability to act as decapacitation factors. Biol. Reprod. 74, 275–287, 10.1095/biolreprod.105.044644 (2006). [DOI] [PubMed] [Google Scholar]
- Da Ros V. G. et al. Impaired sperm fertilizing ability in mice lacking Cysteine-RIch Secretory Protein 1 (CRISP1). Dev. Biol. 320, 12–18, 10.1016/j.ydbio.2008.03.015 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busso D., Cohen D. J., Da Ros V., Fissore R. & Cuasnicu P. S. Studies on the participation of epididymal sperm protein DE/CRISP-1 in egg activation. Cell Mol Biol (Noisy-le-grand) 49, 407–412 (2003). [PubMed] [Google Scholar]
- Busso D., Goldweic N. M., Hayashi M., Kasahara M. & Cuasnicu P. S. Evidence for the involvement of testicular protein CRISP2 in mouse sperm-egg fusion. Biol. Reprod. 76, 701–708, 10.1095/biolreprod.106.056770 (2007). [DOI] [PubMed] [Google Scholar]
- Ellerman D. A. et al. Sperm protein “DE” mediates gamete fusion through an evolutionarily conserved site of the CRISP family. Dev. Biol. 297, 228–237, 10.1016/j.ydbio.2006.05.013 (2006). [DOI] [PubMed] [Google Scholar]
- Gibbs G. M. et al. The cysteine-rich secretory protein domain of Tpx-1 is related to ion channel toxins and regulates ryanodine receptor Ca2+ signaling. J. Biol. Chem. 281, 4156–4163, 10.1074/jbc.M506849200 (2006). [DOI] [PubMed] [Google Scholar]
- Gibbs G. M. et al. Cysteine-rich secretory protein 4 is an inhibitor of transient receptor potential M8 with a role in establishing sperm function. Proc Natl Acad Sci U S A 108, 7034–7039, 10.1073/pnas.1015935108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doty A. et al. Equine CRISP3 modulates interaction between spermatozoa and polymorphonuclear neutrophils. Biol. Reprod. 85, 157–164, 10.1095/biolreprod.110.084491 (2011). [DOI] [PubMed] [Google Scholar]
- Roberts K. P. et al. Structure and function of epididymal protein cysteine-rich secretory protein-1. Asian J Androl 9, 508–514, 10.1111/j.1745-7262.2007.00318.x (2007). [DOI] [PubMed] [Google Scholar]
- Udby L. et al. An ELISA for SGP28/CRISP-3, a cysteine-rich secretory protein in human neutrophils, plasma, and exocrine secretions. J. Immunol. Methods 263, 43–55 (2002). [DOI] [PubMed] [Google Scholar]
- Udby L., Johnsen A. H. & Borregaard N. Human CRISP-3 binds serum alpha(1)B-glycoprotein across species. Biochim. Biophys. Acta 1800, 481–485, 10.1016/j.bbagen.2010.01.011 (2010). [DOI] [PubMed] [Google Scholar]
- Udby L. et al. Cysteine-rich secretory protein 3 is a ligand of alpha1B-glycoprotein in human plasma. Biochemistry 43, 12877–12886, 10.1021/bi048823e (2004). [DOI] [PubMed] [Google Scholar]
- Kjeldsen L., Cowland J. B., Johnsen A. H. & Borregaard N. SGP28, a novel matrix glycoprotein in specific granules of human neutrophils with similarity to a human testis-specific gene product and a rodent sperm-coating glycoprotein. FEBS Lett. 380, 246–250 (1996). [DOI] [PubMed] [Google Scholar]
- Chong U. R., Abdul-Rahman P. S., Abdul-Aziz A., Hashim O. H. & Junit S. M. Tamarindus indica extract alters release of alpha enolase, apolipoprotein A-I, transthyretin and Rab GDP dissociation inhibitor beta from HepG2 cells. PLoS One 7, e39476, 10.1371/journal.pone.0039476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De D. et al. Inactive enzymatic mutant proteins (phosphoglycerate mutase and enolase) as sugar binders for ribulose-1,5-bisphosphate regeneration reactors. Microb Cell Fact 4, 5, 10.1186/1475-2859-4-5 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima J. et al. Establishment of a human cell line secreting neuron-specific enolase from a primitive neuroectodermal tumor of the retroperitoneal cavity. Jpn J Cancer Res 86, 1172–1178 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell G. A., Dunbar B. & Fothergill-Gilmore L. A. The complete amino acid sequence of chicken skeletal-muscle enolase. Biochem. J. 236, 115–126 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui H., Imai H., Komatsuda A. & Miura A. B. Circulating antibodies against alpha-enolase in patients with primary membranous nephropathy (MN). Clin. Exp. Immunol. 118, 445–450 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L. & Helenius A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191, 10.1038/nrm1052 (2003). [DOI] [PubMed] [Google Scholar]
- Hayashi M. et al. Characterization of a human glycoprotein with a potential role in sperm-egg fusion: cDNA cloning, immunohistochemical localization, and chromosomal assignment of the gene (AEGL1). Genomics 32, 367–374, 10.1006/geno.1996.0131 (1996). [DOI] [PubMed] [Google Scholar]
- Shikamoto Y., Suto K., Yamazaki Y., Morita T. & Mizuno H. Crystal structure of a CRISP family Ca2+ -channel blocker derived from snake venom. J. Mol. Biol. 350, 735–743, 10.1016/j.jmb.2005.05.020 (2005). [DOI] [PubMed] [Google Scholar]
- Suzuki N. et al. Structures of pseudechetoxin and pseudecin, two snake-venom cysteine-rich secretory proteins that target cyclic nucleotide-gated ion channels: implications for movement of the C-terminal cysteine-rich domain. Acta Crystallogr D Biol Crystallogr 64, 1034–1042, 10.1107/S0907444908023512 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga Y. et al. Structural divergence of cysteine-rich secretory proteins in snake venoms. J Biochem 145, 365–375, 10.1093/jb/mvn174 (2009). [DOI] [PubMed] [Google Scholar]
- Guo M. et al. Crystal structure of the cysteine-rich secretory protein stecrisp reveals that the cysteine-rich domain has a K+ channel inhibitor-like fold. J. Biol. Chem. 280, 12405–12412, 10.1074/jbc.M413566200 (2005). [DOI] [PubMed] [Google Scholar]
- Jamsai D. et al. Characterization of gametogenetin 1 (GGN1) and its potential role in male fertility through the interaction with the ion channel regulator, cysteine-rich secretory protein 2 (CRISP2) in the sperm tail. Reproduction 135, 751–759, 10.1530/REP-07-0485 (2008). [DOI] [PubMed] [Google Scholar]
- Jamsai D. et al. A novel protein, sperm head and tail associated protein (SHTAP), interacts with cysteine-rich secretory protein 2 (CRISP2) during spermatogenesis in the mouse. Biol. Cell 102, 93–106, 10.1042/BC20090099 (2010). [DOI] [PubMed] [Google Scholar]
- Mangum J. E., Farlie P. G. & Hubbard M. J. Proteomic profiling of facial development in chick embryos. Proteomics 5, 2542–2550, 10.1002/pmic.200401207 (2005). [DOI] [PubMed] [Google Scholar]
- Mangum J. E., Veith P. D., Reynolds E. C. & Hubbard M. J. Towards second-generation proteome analysis of murine enamel-forming cells. Eur J Oral Sci 114 Suppl 1, 259–265; discussion 285–256, 382, 10.1111/j.1600-0722.2006.00310.x (2006). [DOI] [PubMed] [Google Scholar]
- Udby L. et al. Characterization and localization of cysteine-rich secretory protein 3 (CRISP-3) in the human male reproductive tract. J Androl 26, 333–342, 10.2164/jandrol.04132 (2005). [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A. & Schulten K. VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38 (1996). [DOI] [PubMed] [Google Scholar]
- Phillips J. C. et al. Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802, 10.1002/jcc.20289 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures