

Abstract
Congenital gonadotropin-releasing hormone (GnRH) deficiency is a treatable albeit rare form of reproductive failure that has revealed physiological mechanisms controlling human reproduction, but despite substantial progress in discovering pathogenic single-gene defects, most of the genetic basis of GnRH deficiency remains uncharted. Although unbiased genetic investigations of affected families have identified mutations in novel genes as causes of this disease, their application has been severely limited because of the negative effect of GnRH deficiency on fertility; moreover, relatively few of the many candidate genes nominated because of biological plausibility from in vitro or animal model experiments were subsequently validated in patients. With the advent of exciting technological platforms for sequencing, homozygosity mapping, and detection of structural variation at the whole-genome level, human investigations are again assuming the leading role for gene discovery. Using human GnRH deficiency as a paradigm, we discuss the emerging model of patient-focused clinical genetic research and its complementarities with basic approaches in the near future.
GNRH DEFICIENCY AS A PARADIGM OF TRANSLATIONAL MEDICINE
Puberty is arguably one of the most striking postnatal developmental processes in humans. It is accompanied by accelerated skeletal growth, acquisition of secondary sexual characteristics, onset of fertility, and psychosocial changes (1). At the neuroendocrine level, sexual maturation is initiated by the reawakening of the hypothalamic-pituitary-gonadal (HPG) axis—the combined, often cooperative actions of the hypothalamus, pituitary gland, and gonads—from its dormancy of mid-childhood that has been firmly in place after a window of activity—“mini-puberty”—in infancy (2). The pulsatile secretion of gonadotropin-releasing hormone (GnRH) by a specialized network of hypothalamic neurons serves as the biological alarm clock that initiates this process by calling on specific cells—the gonadotropes—in the anterior pituitary. In response, the gonadotropes release the gonadotropins, luteinizing hormone and follicle-stimulating hormone, which, in turn, stimulate gonadal production of sex steroids and gametogenesis (Fig. 1). Although puberty has intrigued clinicians and scientists alike for decades, the mechanisms that trigger its onset and modulate its progress and completion remain largely unknown. In its 125th year celebration issue published in 2005, Science magazine listed the problem of what triggers puberty as one of the “125 great unanswered scientific questions” (3). The human disease models of GnRH dysregulation in which the timing of puberty onset is altered are invaluable systems for deciphering this fundamental biological problem.
Fig. 1.

Genes mutated in IHH have critical roles in GnRH neuron ontogeny and physiology. See the text for details. KAL1, Kallmann syndrome 1 sequence, MIM: 308700; FGFR1, fibroblast growth factor receptor 1, MIM: 136350; FGF8, fibroblast growth factor 8, MIM: 600483; PROK2, prokineticin 2, MIM: 607002; PROKR2, prokineticin receptor 2, MIM: 607123; CHD7, chromodomain helicase DNA binding protein 7, MIM: 608765; NELF, nasal embryonic LHRH factor, MIM: 608137; DAX1, dosage-sensitive sex reversal, adrenal hypoplasia congenital (AHC) critical region on the X chromosome, gene 1, MIM: 300473; PC1, prohormone convertase 1, MIM: 162150; LEP, leptin, MIM: 164160; LEPR, leptin receptor, MIM: 601007; KISS1R, KISS1 receptor, MIM: 604161; TAC3, tachykinin 3, MIM: 162330; TACR3, tachykinin receptor 3, MIM: 162332; GNRH1, gonadotropin-releasing hormone 1, MIM: 152760; GNRHR, GnRH receptor, MIM: 138850.
CREDIT: C. BICKEL/SCIENCE TRANSLATIONAL MEDICINE
From a clinical perspective, abnormalities in the timing and progression of puberty take a substantial somatic and psychological toll on the affected child. Premature reawakening of the reproductive axis via pulsatile secretion of endogenous GnRH—“central precocious puberty”—can be readily arrested therapeutically by administration of long-acting GnRH analogs that inhibit the gonadotropes (4). On the other hand, delayed puberty presents a more difficult problem: in most cases, puberty is eventually initiated and completed—“constitutional delay of puberty, (CDP)”. In other cases, however, puberty never initiates—“absent puberty”. Moreover, pubertal development can initiate in a timely manner but then arrest after incomplete sexual maturation—“partial puberty”. When the activity of the HPG axis is suspended because of a deficiency in GnRH secretion or action while the other neuroendocrine axes are relatively normal, persistently absent or partial puberty is clinically diagnosed as Idiopathic Hypogonadotropic Hypogonadism (IHH). This condition is a rare (with an incidence of 1: 10,000 to 1: 86,000) and heritable form of GnRH deficiency (5). Investigations of its genetic basis have helped elucidate some of the genes and mechanisms involved in the initiation of puberty. These insights have been critical in opening new avenues for basic research into the neuroendocrine control of mammalian reproduction (6, 7). Clinically, however, a definitive differential diagnosis between CDP and IHH remains virtually impossible before age 16, a feature that does not help to alleviate the child’s developmental delay, its social and psychological consequences, and the family’s anxiety. The currently incomplete understanding of the genetics of IHH also makes it difficult to provide confident genetic counseling to GnRH-deficient patients and their relatives. Moreover, a better understanding of the central regulation of the HPG axis and the ability of the hypothalamic GnRH neurons to control this process could create new opportunities to discover effective and safe contraceptives for women and men. Lastly, abnormalities in the patterns of GnRH secretion underlie a group of common reproductive disorders that impair fertility in women, such as hypothalamic amenorrhea (8)—a suppression of reproductive function caused by energy deficits or psychological stress—and possibly polycystic ovarian disease (9), whose molecular pathophysiology and genetic underpinnings remain unknown and for which no curative treatments exist. These common forms of reduced fertility might well share genetic defects with the rare forms of congenital and more severe GnRH deficiency. Thus, research on GnRH deficiency is a good example of translational medicine providing new insights of broad biological and medical relevance from a human disease model.
CHARTING REPRODUCTIVE BIOLOGY THROUGH HUMAN GENETICS
The ability of the central nervous system (CNS) to control human reproduction is characterized by remarkable biological features. GnRH serves as “the pilot light of reproduction” in all mammals. In humans, a “mini-puberty” takes place during infancy when the HPG axis is activated by pulsatile GnRH secretion (2). Subsequently, GnRH secretion is silenced during childhood, only to be reinitiated when unknown metabolic signals cue the hypothalamus of the body’s readiness to enter puberty. Such precise CNS control of reproduction is accomplished by strikingly few (~1,200) and widely dispersed small neurons linked operationally into a single neuronal network. Remarkably, this diffuse and miniscule “parvocellular system” has a unique repertoire of sensing and signaling abilities which enable it to monitor shifting environmental conditions and adjust its neurosecretory activity accordingly. The GnRH neuronal network has likely acquired these functions through evolutionary pressures on mammalian reproduction. Over time, wide swings in nutrient availability during periods of feast and famine; dramatic changes in metabolic and caloric requirements during climate changes and population migrations; ever-changing light-dark cycles that cue seasonal breeding; and varying olfactory signals that indicate reproductive readiness or predatory threats—all have left their evolutionary markers on the neuronal control of reproduction. Remarkably, much of this complex central control of reproduction is mediated via the convergence of these regulatory mechanisms on the GnRH parvocellular system and/or its upstream hypothalamic neurons. In all mammalian systems, both GnRH and its cognate pituitary receptor, GnRHR, are encoded by single-copy genes. Thus, crucial biological mechanisms essential for reproduction are invested upon this single ligand-receptor pair, a circumstance that contrasts strikingly with most other essential biological systems. Typically, functions so critical to survival and evolution are equipped with fail-safe mechanisms in the form of biological redundancy: families of genes encoding receptors, signaling mediators, and transcription factors with overlapping functions. Thus, the curious lack of genetic redundancy in the crucial neuroendocrine regulator of sexual maturation and reproduction in mammals is surprising. It implies that substantial loss-of-function of GnRH or GnRHR as a result of mutations would cause infertility and thus immediately be selected against. On the other hand, this singularity of the GnRH system might facilitate the fine-tuning of its secretion by a complex genetic network of upstream regulators so that reproductive function can be tailored to and integrated with these varying hormonal, nutritional, psychological, and environmental conditions. Such a hierarchical organization might be particularly advantageous under certain adverse evolutionary circumstances. For example, shutting down reproduction by suppressing the HPG axis could result in a “natural contraception” period—in the form of seasonal breeding in some species or hypothalamic amenorrhea in women—that would preserve the body’s resources and postpone fertility for more favorable times.
As a result of these unusual properties, as well as the considerable species specificity of reproduction, our understanding of the complex developmental biology, physiology, and pathology of GnRH secretion is scant. Initially, knowledge on the subject was pieced together from in vitro studies of cellular systems and model organism approaches supplemented by very limited and difficult to perform investigations in humans. However, the human model of isolated GnRH deficiency has facilitated the discovery of numerous genes mutated in GnRH-deficient patients over the last two decades, and has thus directly charted the ontogeny of the GnRH neuronal network in the human (Fig. 1). Genes harboring defects that cause GnRH deficiency have been demonstrated to play critical roles in the ontogeny (i.e., fate specification, proliferation, developmental migration, homeostasis, secretory function, and apoptosis) of GnRH neurons when studied in species that allow reductionist experimental approaches not possible in humans (6, 7, 10).
In agreement with Murphy’s Law, such studies have shown that everything that can go wrong for GnRH biology sometimes does, resulting in the human model of isolated GnRH deficiency. For example, rodent studies revealed that GnRH neurons originate outside the CNS from precursor cells in the olfactory placode, an embryonic tissue that gives rise to the nose’s olfactory epithelium. Once their biological fate as GnRH neurons is specified by the expression of their hallmark neurohormone, they commence a long migration from the olfactory placode across the CNS via the axons of olfactory neurons and the olfactory bulbs and tracts. In patients with mutations in KAL1 (11, 12), this migratory path fails to form correctly, leading to arrested GnRH neuronal migration. The resulting combination of reduced or absent sense of smell with IHH is termed Kallmann syndrome (KS). Normally, migrating GnRH neurons ultimately divert from the olfactory tract pathway to assume their final residence within the hypothalamus. Once there, they extend axons into the median eminence (an area abutting the vasculature that transports hypothalamic hormones to the pituitary), interconnect into a coherent functional network, and coordinately produce secretory pulses of GnRH. To evoke physiological secretion of gonadotropins from the anterior pituitary gonadotropes, GnRH secretion from the hypothalamus must be pulsatile. This is achieved via pacemaker activity that may be intrinsic to the GnRH neurons and/or is powerfully modulated by afferent stimuli. In patients with mutations in KISS1R (13, 14) or TAC3/TACR3 (15, 16), the GnRH neurons complete their migration but do not receive sufficient stimulation from other cells in the hypothalamus to secrete GnRH in a normal manner. Mutations in these genes have been found in patients with normosmic IHH (nIHH), as is also the case with mutations in GNRHR (17), which have been associated with pituitary resistance to GnRH (18). Interestingly, some of the genes mutated in GnRH deficiency have additional roles outside the HPG axis, which accounts for the frequent presence of non-reproductive phenotypes in IHH patients. For example, unilateral renal agenesis (in which one kidney fails to develop and is absent at birth) can occur in association with KAL1 defects (19), whereas skeletal abnormalities and midline malformations can be present in patients with FGFR1 mutations (20, 21).
MENDING THE SCHISM BETWEEN MONOGENIC AND COMMON DISEASES
Traditionally, a wide conceptual, methodological, and “ideological” divide has separated the rare monogenic diseases like cystic fibrosis, sickle cell anemia, thalassemia, and isolated GnRH deficiency from the common multifactorial ones like diabetes, autism, schizophrenia, or inflammatory bowel disease. Whereas the former have been attributed to large effects of rare variants, i.e., single gene mutations with allelic frequencies of <1%, the latter have been thought to be caused by incremental contributions of multiple common genetic variants (polymorphisms) acting in synergy with environmental risk factors. Over the last five years, genome-wide association studies (GWASs) have uncovered common variants that are associated with increased risk for several complex disorders (22). Unexpectedly, however, such variants have accounted only for a minor fraction of the genetic predisposition to most common disorders (23). Therefore, it has now begun to be appreciated that rare variants not only underlie rare genetic diseases but may also make previously unsuspected genetic contributions to common disorders. In addition, the same genes that harbor common variants associated with differences in physiological parameters (such as plasma lipid concentrations) in the general population can also harbor mutations responsible for corresponding forms of rare genetic disease (such as familial dyslipidemias) (24). In fact, computer simulations have recently suggested that some of the associations credited to common variants may actually be “synthetic associations” involving nearby rare variants that occur, by chance, more frequently in association with one allele at a common variant site than with the other (25). Lastly, numerous genetic diseases originally thought to be monogenic, such as retinitis pigmentosa (26), nephronopthisis (27), Bardet-Biedl syndrome (28), and several others (29) have now been documented to be oligogenic, i.e., caused in each patient/family by contributions of rare variants in more than one gene. Because the architecture of oligogenic diseases shares genetic principles with both monogenic and polygenic/multifactorial disorders, their genetic characterization should help to model the role of rare variants in common diseases.
Like most other rare genetic diseases, GnRH deficiency was originally thought to be a strictly monogenic condition caused in each patient/family by a single mutation in one of a small number of genes. In addition to charting the regulation of GnRH neuron ontogeny and function, translational studies of IHH have challenged the monogenic view by demonstrating unexpected complexity in the genetic architecture of human GnRH deficiency (6, 7). With mutations in no fewer than 16 different genes described to date, the locus heterogeneity of IHH is impressive (Fig. 1 and Fig. 2). Considering that mutations in each of these genes occur in only a small fraction of patients with well-phenotyped GnRH deficiency (~2 to 10%) and that mutations remain to be discovered in nearly 60% of patients, the real number of genes accounting for GnRH deficiency is likely to be much higher. The allelic heterogeneity of the disease (the range of different mutations in each gene) is also striking, with few mutation hotspots in the IHH genes. In addition to this substantial genetic heterogeneity, GnRH deficiency is characterized by an equally rich phenotypic variability in its reproductive and non-reproductive manifestations both within and across families segregating identical mutations. Thus, rare variants that are known to be pathogenic often occur in family members who (i) are asymptomatic (incomplete penetrance); (ii) display only non-reproductive IHH-associated phenotypes; or (iii) suffer from milder forms of GnRH deficiency such as delayed puberty or hypothalamic amenorrhea. Importantly, some patients with GnRH deficiency harbor pathogenic rare variants in two different disease-associated genes, and the segregation pattern of the digenic mutations in their family pedigrees accounts, at least partially, for the observed phenotypic variability (30). Even though the extent of oligogenicity (the proportion of non-monogenic cases and number of interacting genes) in this condition remains to be fully characterized, it is already evident that human GnRH deficiency can no longer be considered a classic rare single-gene Mendelian disease.
Fig. 2.

Timeline of IHH gene discovery.
CREDIT: C. BICKEL/SCIENCE TRANSLATIONAL MEDICINE
Even more surprisingly, translational investigations of GnRH deficiency have shown that IHH, once thought to be exclusively both congenital and life-long, can actually develop in individuals who have had a normal puberty and demonstrated full reproductive function including fertility (adult-onset HH) (31). In addition, in as many as 10% of cases, the HPG axis can recover its functionality after treatment with exogenous gonadotropins or sex steroids, and such reversal can occur despite the presence of pathogenic mutations (32). Therefore, the clinical phenotypes of GnRH deficiency are not determined solely by genotype, but also by non-genetic influences that are likely hormonal and/or environmental. As a human disease model that integrates oligogenicity and non-genetic factors, GnRH deficiency thus offers an unusually rich opportunity to model the interplay of genetics and environment that also underlies common multifactorial diseases.
LESSONS FROM GENETIC RESEARCH IN ISOLATED GNRH DEFICIENCY: GRAND SLAMS AND STRIKEOUTS
Notwithstanding the substantial progress made through the genetic dissection of GnRH deficiency, the overall process of gene discovery has been rather piecemeal (Fig. 2). If discoveries continue at the same pace in the future, the thorough genetic elucidation of this condition could well take a few more decades. If so, this timetable implies that translation of these biological and genetic insights into clinical progress could take even longer. However, if the technological advances of the genomic era are properly harvested, mapping the genetic basis of GnRH deficiency (and other mono/oligogenic disorders) could be spectacularly expedited. Consequently, it is important to formulate a discovery strategy for the next decade that incorporates, or at least anticipates, some of the technological advances in genome science.
Naturally, devising such a strategic plan should begin with a review of the gene discovery experience to date, and with a reevaluation of the ways in which candidate genes for GnRH deficiency are nominated and validated. A brief review of successful (published) and unsuccessful (previously unreported, Table 1 and Supporting Online Material) efforts should help in extracting general principles about the comparative effectiveness of current gene discovery approaches. When retracing the discovery of each of the genes known to cause GnRH deficiency, it becomes evident that, similar to other mono/oligogenic diseases, two main strategies have been successfully employed in nominating candidate disease genes that were subsequently validated by the identification of mutations in patients (Fig. 2).
Table 1.
Screened candidate IHH genes in which no definitive pathogenic mutations were detected in their protein-coding sequences. The rationale for screening each gene and potential reasons for the absence of mutations are discussed in more detail in the Supporting Online Material.
| # | Symbol | Name | MIM # | Main nomination reason(s) | Potential caveat(s) | Patients screened | ||
|---|---|---|---|---|---|---|---|---|
| KS | nIHH | Total | ||||||
| 1 | CRTC1 | CREB regulated transcription coactivator 1 | 607536 | KO: HH Regulates Kiss1 expression |
Redundancy KO: Genetic background effects |
14 | 78 | 92 |
| 2 | CXCR4 | chemokine (C-X-C motif) receptor 4 | 162643 | KO: ↓ GnRH neuron migration | Redundancy KO: Developmental lethality Linked to “WHIM” syndrome in humans |
28 | 105 | 133 |
| 3 | EBF2 | early B-cell factor 2 | 609934 | Human: part of HH-associated 8p12p21 deletions KO: ↓ GnRH neuron migration, HH |
Redundancy KO: Genetic background effects |
89 | 97 | 188 |
| 4 | EMX2 | empty spiracles homeobox 2 | 600035 | Human: deleted in KS patient KO: ↓ olfactory bulbs, skeletal and urogenital abnormalities |
Redundancy | 84 | 8 | 92 |
| 5 | FGF9 | fibroblast growth factor 9 | 600921 | Member of FGF family | Redundancy Linked to inherited “multiple synostosis” syndrome in humans |
53 | 37 | 90 |
| 6 | FGFR2 | fibroblast growth factor receptor 2 | 176943 | Member of FGFR family KO: renal abnormalities |
Redundancy KO: ♂-to-♀ sex reversal |
25 | 4 | 29 |
| 7 | MET | met proto-oncogene | 164860 | The MET ligand HGF regulates GnRH neuron migration in vitro and ex vivo KO of HGF processing enzymes: ↓ fertility |
Redundancy | 70 | 85 | 155 |
| 8 | MIR9-1 | microRNA 9-1 | 611186 | Zebrafish: homolog (miR9) regulates expression of FGF pathway members and promotes neurogenesis | Redundancy Essential for viability? |
73 | 55 | 128 |
| 9 | MIR9-2 | microRNA 9-2 | 611187 | 73 | 55 | 128 | ||
| 10 | MIR9-3 | microRNA 9-3 | 611188 | 73 | 55 | 128 | ||
| 11 | NDN | necdin | 602117 | Human: implicated in “Prader-Willi” syndrome KO: ↓ GnRH neuron content |
KO: Genetic background effects; minor ↓ in GnRH neurons; no infertility | 69 | 24 | 93 |
| 12 | NGN1 | neurogenin 1 | 601726 | KO: phenocopy of Prok2−/− and Prokr2−/− Regulates Prokr2 expression |
Redundancy | 68 | 25 | 93 |
| 13 | NHLH1 | nescient helix loop helix 1 | 162360 | Double KO with Nhlh2: further ↓ of GnRH neuron content; impaired migration | Redundancy | 67 | 23 | 90 |
| 14 | NHLH2 | nescient helix loop helix 2 | 162361 | KO: ↓ GnRH neuron content; ♂, HH; ♀, HH only when reared without ♂ | Regulates anterior pituitary size and Gnrhr expression | 67 | 23 | 90 |
| 15 | PROKR1 | prokineticin receptor 1 | 607122 | Receptor for PROK2 Expressed in olfactory bulb |
KO: normal olfactory bulbs, no HH | 91 | 2 | 93 |
| 16 | SDF1 | stromal cell-derived factor 1 | 600835 | Ligand for CXCR4 Regulates GnRH neuron migration ex vivo |
KO: Developmental lethality | 23 | 59 | 82 |
| 17 | TTF1 | thyroid transcription factor 1 | 600635 | Hypothalamic expression increases at onset of puberty Conditional KO: altered expression of puberty-related genes, ↓ fertility |
Constitutive KO: developmental lethality Linked to “brain-lung-thyroid” syndrome in humans |
2 | 75 | 77 |
| 18 | TYRO3 | TYRO3 protein tyrosine kinase | 600341 | Double KO with family member Axl: ↓ GnRH neuron content, ↓ migration, ↑ apoptosis |
Redundancy Single Tyro3 or Axl KO: no reproductive abnormalities |
22 | 24 | 46 |
| 19 | VAX1 | ventral anterior homeobox 1 | 604294 | Human: deleted in KS patient KO: ↓ olfactory bulbs, cleft palate, sterility |
Redundancy | 84 | 8 | 92 |
| 20 | VEGFA | vascular endothelial growth factor A | 192240 | Regulates GnRH neuron migration in vitro Receptor KO: ↓ GnRH neuron migration, ↓ fertility |
Redundancy Haploinsufficiency (in mice) |
93 | 2 | 95 |
KO, mouse knockout; HH, hypogonadotropic hypogonadism; KS, Kallmann syndrome; nIHH, normosmic idiopathic HH; WHIM, wart, hypogammaglobulinemia, infection, and myelokathexis syndrome.
Human genetics
Several unbiased genetic methodologies have been successfully applied to genomic DNA samples from GnRH-deficient patients to discover the affected genes: KAL1 (11, 12) and FGFR1 (20) were identified by mapping overlapping gross chromosomal rearrangements present in unrelated patients who presented with complex phenotypes suggestive of contiguous gene syndromes (in which two or more adjacent genes are deleted). KISS1R was nominated by two independent linkage studies, each focused on a large consanguineous family with isolated GnRH deficiency inherited in an autosomal recessive fashion (13, 14). TAC3 and TACR3 were identified through homozygosity mapping, i.e., the identification of genomic segments that were homozygous and identical by descent across a set of consanguineous families from endogamous Turkish and Kurdish populations (15). These breakthroughs demonstrate the power of unbiased, “candidate gene-independent” queries of the entire human genome to seek and find linkage to the disease phenotype. Thus, the application of human genetics to familial GnRH deficiency is a powerful method of uncovering genes for which there may be no prior incriminating clues from basic science studies.
Studying clear-cut phenotypes, such as the infertility that results from GnRH deficiency, can be an advantage in genetic investigations. However, the application of unbiased genetic approaches to GnRH deficiency is severely limited by both the rarity of the disease and its negative impact on family size. Successes (11–15, 20), although powerful and fortuitous, are unlikely to provide a complete and expeditious cataloging of all the genes responsible for isolated GnRH deficiency. Moreover, by relying solely on the occasional informative inbred patient/family for new candidate gene identification, these methods have not taken full advantage of the several large DNA banks from GnRH-deficient patients from more outbred populations that have been assembled by various investigators as primary resources for gene discovery.
Biological candidates
Some genes associated with GnRH deficiency were nominated as plausible candidates for genetic screening because of basic science-derived insights that directly testified to their roles in GnRH biology: GNRH1 and GNRHR were, of course, obvious candidates (17, 33, 34). NELF (35) had been found in cell-based studies to play a role in GnRH neuronal migration. Mouse knockouts of the PROK2 and PROKR2 homologs exhibited underdeveloped olfactory bulbs and reproductive organs as a result of hypogonadotropic hypogonadism caused by a disrupted GnRH neuron migratory path (36, 37). Finally, FGF8 was prioritized among the 22 ligands for fibroblast growth factor (FGF) receptors from in silico structural modeling data indicating that a specific FGFR1 mutation causing GnRH deficiency selectively impaired FGF8 binding (38). Thus, various criteria can be legitimately employed to nominate a gene as a candidate based on basic research, including membership in the same pathway as a known disease gene (such as FGF9, FGFR2, and PROKR1, Table 1), evidence of a role in the underlying biology from cell or organotypic culture systems (such as VEGFA, SDF1, or MET, Table 1), or description of a genetically modified model organism that phenocopies the human disease or some aspect(s) of it (such as CXCR4, NGN1, and NHLH2, Table 1) (39). The trouble is that such criteria are not foolproof. Many genes that present as perfectly plausible candidates turn out not to harbor pathogenic mutations in screened patient cohorts (Table 1 and Supporting Online Material). Moreover, the candidate gene approach has the additional caveat that nonhuman-derived information might erroneously disqualify actual disease genes from genetic screening. For example, Tacr3−/− mice exhibited no apparent reproductive phenotypes (40). However, this finding proved misleading as subsequent clinical investigations showed that patients with TAC3 or TACR3 mutations have GnRH deficiency (15, 16).
Several potential reasons account for the high error rate of this gene discovery strategy. First, members of the same protein family might be able to substitute for each other’s role in GnRH biology (functional redundancy) such that a deleterious mutation in any single gene is not by itself sufficient to cause GnRH deficiency. Genes with such potential functional redundancy are MIR9-1, -2, and -3, NHLH1 and NHLH2, or EMX2 and VAX1 (Table 1). Second, a GnRH-deficient phenocopy observed in a particular genetically modified mouse might depend on the presence or absence of unknown modifier mutations in other genes specific to its genetic background. This situation makes it difficult to predict the phenotypic outcome of mutations in the human homolog. Examples of such genes include CRTC1, EBF2, and NDN (Table 1). Third, a gene with a role in GnRH biology may also be required for viability during embryonic development or neonatal life. In that case, mutations in this gene in humans of pubertal age are unlikely to be found. For example, targeted disruptions of the mouse homologs of SDF1 and CXCR4 are homozygous lethal, and the mouse homolog of VEGFA is haploinsufficient (i.e., lack of one gene copy causes lethality) (Table 1 and Supporting Online Material). Fourth, mutations in genes that have roles in GnRH biology might nonetheless be associated with diseases whose clinical spectra do not include GnRH deficiency. For example, in humans, mutations in CXCR4, FGF9, and TTF1 are associated with genetic syndromes that do not show clinical overlap with IHH (Table 1). Lastly, given the rapidly expanding knowledge about the complexity and interconnectivity of signaling pathways, it is very difficult to select one among many plausible candidates within a given pathway. Thus, although educated guesses might work well for ligand-receptor pairs (GNRH1-GNRHR and FGF8-FGFR1 being good examples), upstream regulators and downstream effectors of known disease genes are riskier bets. The lack of mutations to date in NGN1 (upstream of prokineticin 2), TTF1 (upstream of GnRH as well as of kisspeptin, the ligand for KISS1R), and CRTC1 (upstream of kisspeptin) demonstrates this caveat (Table 1 and Supporting Online Material).
In rare occasions, GnRH deficiency is described in a syndromic context such as part of a broader clinical genetic condition, e.g., Lawrence-Moon-Biedl syndrome, Prader-Willi syndrome, or myotonic muscular dystrophy. In these settings, the mapping of the gene(s) responsible for these syndromes offers the possibility to characterize not only the gene’s role in GnRH biology, but also its mutational status in GnRH-deficient patients and thus the degree of clinical and genetic overlap between the syndrome and isolated GnRH deficiency. Both unbiased human genetics and basic science-driven approaches have been successful in such cases (Fig. 2): the first mutations in DAX1 (41, 42) and CHD7 (43, 44) in GnRH-deficient patients were mapped using human genetics. LEP (45) and LEPR (46) were nominated as good biological candidates based on the corresponding knockout mouse phenotypes. Notably, PC1 was highlighted by associating the endocrine features of the syndrome with the known biochemical role of the gene in processing prohormones (47, 48). Although these genetic syndromes have contributed substantially to understanding GnRH biology by revealing new pathways involved in its regulation, they are even rarer occurrences than isolated GnRH deficiency, and thus the caveats of human genetic methods are even more relevant.
ROADBLOCKS AND THE ROAD BEYOND
A broader exploration of both unbiased human genetics and biological candidate gene approaches to gene discovery in GnRH deficiency and other rare genetic diseases is presently hindered by barriers that must be overcome in order to fully elucidate their genetics.
A game of chance and numbers
The larger the size of the well-phenotyped population with a human disease model assembled, the greater the likelihood of finding new disease genes. This general principle applies to virtually all gene discovery strategies because numbers increase not only the chance of encountering the odd patient/family that is uniquely informative for unbiased human genetics, but also the ability to screen sufficient numbers of unrelated patients to identify genes that are mutated in small percentages (i.e., 1 to 2%) of the disease population. GNRH1 is the most illustrative case in point: Despite being the best biologically nominated candidate gene for a disease of “GnRH deficiency,” it took 12 years from the discovery of GNRHR mutations and several hundreds of screened subjects to document GNRH1 mutations in just a handful of patients with isolated GnRH deficiency (33, 34).
Consequently, unless there is strong faith in a candidate, it is unlikely that sufficient numbers of patients will be screened before declaring the gene a “negative” one. A corollary to this principle is that a “false negative” call made based on a relatively small screened cohort can, in turn, divert screening efforts away from a given candidate and its associated signaling pathway, despite the possibility that other genes in the same network may represent rich veins of pathogenic mutations. As a general strategy to deal with this “numbers” issue, we routinely screen DNA from at least 90 to 100 patients for each candidate gene (Table 1). Of course, it would be preferable to screen 400 to 500 patients to increase the probability of detecting mutations in genes that are affected at low frequency. Unfortunately, the cost of DNA sequencing is currently a limiting factor. As sequencing costs decline over time, thus permitting more candidates to be screened for the same or smaller expenditure, it will be possible to overcome this limitation. At present, however, the plethora of attractive biological candidates for screening often forces us to drop or shelve the “lead candidate” in a pathway if no mutations are detected in the first 100 screened patients.
The trouble with “pay-to-play” technologies
The identification of new gene mutations relies totally on sequencing the DNA of well phenotyped patients with the disorder, and thus the substantial cost of clinical, biochemical, and radiological investigation of the patients must be added to the sequencing cost. In addition, the realization that isolated GnRH deficiency can be an oligogenic disease (49, 50) necessitates the sequencing of all known genes in each patient to understand the full genotype-phenotype relationships, thus dramatically raising the expenditure. In addition, the disease’s genetic and clinical heterogeneity makes it important not only to assemble large patient cohorts, but also to characterize each patient and his/her extended family clinically in as much phenotypic detail as possible in terms of both reproductive and non-reproductive associated phenotypes. Large and well characterized cohorts of ethnically matched unaffected individuals are indispensable for evaluating the potential importance of rare sequence variants identified in patients. Thus, the burden and expense of phenotyping and genotyping the control individuals parallels that of the patient cohorts. As we have been continuously enrolling new patients into our genetic research program over the last 20 years, we have incurred corresponding “opportunity-costs” in screening large numbers of biologically plausible candidate genes.
The “next generation” is now!
DNA sequencing costs are currently a rate-limiting factor. However, the diverse emerging “next generation” sequencing technologies promise both dramatically increased throughput and substantially lowered expenditure (51). Such methods will facilitate not only the deep re-sequencing of attractive genes in large patient cohorts, but also sequencing the whole exome—all gene exons, which represent about 1% of the entire genome—and then the whole genome from select patients. In fact, whole-exome sequencing has already proven successful for rapidly mapping mutations for some rare genetic diseases (52, 53). The prospect of applying such methods to isolated GnRH deficiency signifies that the scientific benefit from continuing to recruit and determine the phenotype of patients might soon outweigh the associated sequencing expenditures. Even though it is not currently economically feasible to sequence all the known disease genes as well as all good biological candidates in each patient, this possibility will clearly become practicable in the next few years. In addition to next-generation sequencing, other new genome-wide DNA analysis methods are already being applied to genetic diseases and will soon be broadly employed in genetic studies of GnRH deficiency. These include comparative genome hybridization arrays for the detection of copy number variation (CNVs, i.e., deletions, duplications, etc.) (54), and single-nucleotide polymorphism genotyping arrays for both CNV detection and homozygosity mapping (55). Indeed, homozygosity mapping using these new arrays recently led to the discovery of TAC3 and TACR3 as genes associated with GnRH deficiency (15).
Changing models of clinical genetic research
The advent of these powerful new tools for DNA analysis will clearly change the way in which genetic research on mono/oligogenic diseases like GnRH deficiency is done (Fig. 3). We envision that this application will increase the productivity of gene discovery efforts dramatically over the next few years and that these efforts will be driven primarily by unbiased human genetics, i.e., CNV studies, homozygosity mapping, and whole-exome/genome sequencing. From our perspective, these methods will be applied first to outstanding GnRH patients/families, then to select groups of patients, and eventually to entire patient cohorts. Unavoidably, the sheer volume of data will produce long lists of rare sequence variants and corresponding candidate disease genes. These candidates will then need to be filtered by cross-referencing basic science-derived biological information, including mRNA and protein expression profiles, mouse knockout models, cell culture studies, signaling pathway analyses, etc. In this manner, the candidate genes will be prioritized according to the weight of evidence supporting a role in GnRH biology and then further tested in order of priority for loss-of-function mutations in large populations of IHH patients. When unequivocal evidence incriminating a gene is not obtained, this information will be used for further bioinformatic filtering and re-prioritizing the list of candidates. Given the volume and complexity of the data, bioinformatics tools will be indispensable for the analysis of the raw genetic data, including the identification of rare sequence variants and the assessment of their potential functional importance. The comparison of the rare sequence variants found in patients to an equally well phenotyped and genotyped control population that is ethnically matched will be crucial. Moreover, the genetic results will be integrated with biological information from the literature and from genetic, transcriptomic, and proteomic databases regarding each candidate gene. Together, this ensemble will serve to provide a stringent test of the plausibility of each candidate, elucidate the respective signaling pathway(s), and prioritize the list of candidate genes for validation by parallel investigations in humans, model organisms, and in vitro cellular systems. The transition from the traditional to this emerging model of genetic research in GnRH deficiency will proceed in a gradual manner: biologically plausible candidate genes derived from various methods will continue to be screened even in the absence of prior support from human genetics; at the same time, the application of the new genetic tools will steadily expand. Ultimately, however, as the use of these genetic platforms becomes cheaper and the tools and rules for their application are better validated and clarified (with the unavoidable trials and errors), a complete move away from the old paradigm will take place. To stay abreast of these developments, translational investigators committed to genetic research will need to evolve and adapt accordingly, by adopting the emerging technologies and forming the necessary partnerships with their peers in basic science and bioinformatics.
Fig. 3.
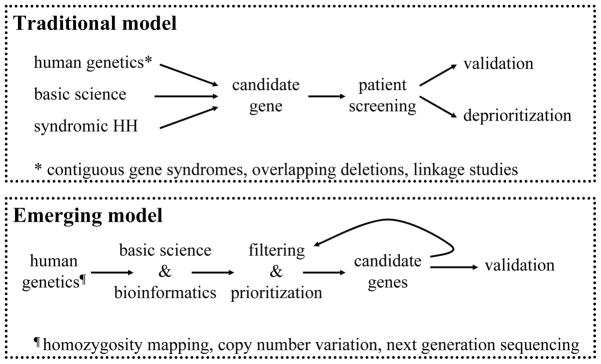
Changing models of clinical genetic research.
CREDIT: C. BICKEL/SCIENCE TRANSLATIONAL MEDICINE
A CALL FOR INTERNATIONAL COOPERATION
The first 20 years of genetic studies on isolated GnRH deficiency have yielded momentous insights into the biology of GnRH and the causes of its dysregulation (6, 7, 10). The next 20 years should herald the thorough genetic elucidation of this disease, the complete expansion of its phenotypic spectrum, and the translation of these advancements into diagnostic, prognostic, and therapeutic innovations. Specific actions can be taken by the research community to expedite this process. The most important would be to broaden the international cooperation among clinical investigators and geneticists studying this condition. Most of the progress in this field has been pioneered by a handful of research groups that operate in relative isolation from each other. Although this high-level competition has fostered scientific progress, it has also prevented the creation of a research consortium similar to those established for other genetic diseases like cystic fibrosis. World-wide cooperation on this matter could greatly facilitate the exchange of scientific ideas. As an example, each group could publicly disclose “negative” gene screening results as we are doing herein (Table 1 and Supporting Online Material). This information can help others to avoid repetition in gene screening and better allocate their limited resources. This shared information will ultimately facilitate the screening of more candidate genes, and help elucidate more of the puzzle about the regulation of puberty than each group could do on its own. It would be a rather simple matter for leading groups in IHH genetics to commit to reporting such data in back-to-back papers in an agreed upon journal every couple of years.
Taking international cooperation to a next level could lead to the establishment of a “global IHH cohort” containing a few thousand DNA samples and associated phenotypes from ethnically diverse patients and control subjects. Such a cohort could be run under accepted rules for communal governance and resource utilization with an online database that would facilitate access to both DNAs for gene screening and clinical information for genotype-phenotype correlations. As GWASs have shown, consortia of this type can have great discovery power and scientific success in clinical genetic research (56, 57). Such a development could also boost awareness of translational research on GnRH biology, thus increasing patient referrals by clinicians as well as self-referrals of individual patients and organized patient groups. The conduct of translational medicine in GnRH deficiency and other genetic diseases with broad implications for human biology and clinical practice “takes a village.” Now is the time to move to the next level and embrace “globalization.”
The principles discussed here are broadly applicable to human genetic research. It is increasingly appreciated that most diseases with a genetic component (including common ones like diabetes, hypertension, and other major public health issues) are linked to rare mutations, which can even be “private” to individual patients or families (58). The genetic elucidation of GnRH deficiency and other diseases of the reproductive system will continue to provide valuable paradigms on which to model the complex architectures of multifactorial disorders.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Eunice Kennedy Shriver NICHD/NIH through cooperative agreement U54 HD028138 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research; ARRA supplement U54 HD028138-19S1; RO1 grant HD015788-23; RO1 grant HD056264; and ARRA supplement HD056264-02S1.
Footnotes
Competing interests: None.
REFERENCES AND NOTES
- 1.Nathan BM, Palmert MR. Regulation and disorders of pubertal timing. Endocrinol Metab Clin North Am. 2005;34:617–641. ix. doi: 10.1016/j.ecl.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Grumbach MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab. 2005;90:3122–3127. doi: 10.1210/jc.2004-2465. [DOI] [PubMed] [Google Scholar]
- 3.So much more to know. Science. 2005;309:78–102. doi: 10.1126/science.309.5731.78b. [DOI] [PubMed] [Google Scholar]
- 4.Conn PM, Crowley WF., Jr Gonadotropin-releasing hormone and its analogues. N Engl J Med. 1991;324:93–103. doi: 10.1056/NEJM199101103240205. [DOI] [PubMed] [Google Scholar]
- 5.Seminara SB, Hayes FJ, Crowley WF., Jr Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations. Endocr Rev. 1998;19:521–539. doi: 10.1210/edrv.19.5.0344. [DOI] [PubMed] [Google Scholar]
- 6.Jameson JL. Rites of passage through puberty: a complex genetic ensemble. Proc Natl Acad Sci U S A. 2007;104:17247–17248. doi: 10.1073/pnas.0708636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5:569–576. doi: 10.1038/nrendo.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santoro N, Butler JP, Filicori M, Crowley WF., Jr Alterations of the hypothalamic GnRH interpulse interval sequence over the normal menstrual cycle. Am J Physiol. 1988;255:E696–701. doi: 10.1152/ajpendo.1988.255.5.E696. [DOI] [PubMed] [Google Scholar]
- 9.Waldstreicher J, Santoro NF, Hall JE, Filicori M, Crowley WF., Jr Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab. 1988;66:165–172. doi: 10.1210/jcem-66-1-165. [DOI] [PubMed] [Google Scholar]
- 10.Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB, Crowley WF., Jr Human GnRH deficiency: A unique disease model to unravel the ontogeny of GnRH neurons. Neuroendocrinology. doi: 10.1159/000314193. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, Carrozzo R, Maestrini E, Pieretti M, Taillon-Miller P, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. doi: 10.1038/353529a0. [DOI] [PubMed] [Google Scholar]
- 12.Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, Wunderle V, Millasseau P, Le Paslier D, Cohen D, Caterina D, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–435. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- 13.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O’Rahilly S, Carlton MB, Crowley WF, Jr, Aparicio SA, Colledge WH. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 15.Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O’Rahilly S, Semple RK. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–358. doi: 10.1038/ng.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, Abreu AP, Carroll J, Trarbach E, Silveira LF, Costa EM, de Mendonca BB, de Castro M, Lofrano A, Hall JE, Bolu E, Ozata M, Quinton R, Amory JK, Stewart SE, Arlt W, Cole TR, Crowley WF, Kaiser UB, Latronico AC, Seminara SB. TAC3/TACR3 Mutations Reveal Preferential Activation of Gonadotropin-Releasing Hormone Release by Neurokinin B in Neonatal Life Followed by Reversal in Adulthood. J Clin Endocrinol Metab. doi: 10.1210/jc.2009-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–1602. doi: 10.1056/NEJM199711273372205. [DOI] [PubMed] [Google Scholar]
- 18.Caron P, Chauvin S, Christin-Maitre S, Bennet A, Lahlou N, Counis R, Bouchard P, Kottler ML. Resistance of hypogonadic patients with mutated GnRH receptor genes to pulsatile GnRH administration. J Clin Endocrinol Metab. 1999;84:990–996. doi: 10.1210/jcem.84.3.5518. [DOI] [PubMed] [Google Scholar]
- 19.Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, Azcona C, MacColl GS, Jacobs HS, Conway GS, Besser M, Stanhope RG, Bouloux PM. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 2001;55:163–174. doi: 10.1046/j.1365-2265.2001.01277.x. [DOI] [PubMed] [Google Scholar]
- 20.Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 21.Pitteloud N, Acierno JS, Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF., Jr Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2006;103:6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manolio TA. Cohort studies and the genetics of complex disease. Nat Genet. 2009;41:5–6. doi: 10.1038/ng0109-5. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 24.Hegele RA. Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet. 2009;10:109–121. doi: 10.1038/nrg2481. [DOI] [PubMed] [Google Scholar]
- 25.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 27.Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, Attanasio M, Utsch B, Antignac C, Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 28.Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet. 2004;13(Spec1):R65–71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
- 29.Badano JL, Katsanis N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet. 2002;3:779–789. doi: 10.1038/nrg910. [DOI] [PubMed] [Google Scholar]
- 30.Pitteloud N, Durrani S, Raivio T, Sykiotis GP. Complex Genetics in Idiopathic Hypogonadotropic Hypogonadism. Front Horm Res. 2010;39:142–153. doi: 10.1159/000312700. [DOI] [PubMed] [Google Scholar]
- 31.Nachtigall LB, Boepple PA, Pralong FP, Crowley WF., Jr Adult-onset idiopathic hypogonadotropic hypogonadism--a treatable form of male infertility. N Engl J Med. 1997;336:410–415. doi: 10.1056/NEJM199702063360604. [DOI] [PubMed] [Google Scholar]
- 32.Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF, Jr, Pitteloud N. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med. 2007;357:863–873. doi: 10.1056/NEJMoa066494. [DOI] [PubMed] [Google Scholar]
- 33.Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, Lombes M, Millar RP, Guiochon-Mantel A, Young J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–2748. doi: 10.1056/NEJMoa0900136. [DOI] [PubMed] [Google Scholar]
- 34.Chan YM, de Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, Tsiaras S, Gaspert A, Lavoie HB, Wu CH, Crowley WF, Jr, Amory JK, Pitteloud N, Seminara SB. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2009;106:11703–11708. doi: 10.1073/pnas.0903449106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miura K, Acierno JS, Jr, Seminara SB. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH) J Hum Genet. 2004;49:265–268. doi: 10.1007/s10038-004-0137-4. [DOI] [PubMed] [Google Scholar]
- 36.Ng KL, Li JD, Cheng MY, Leslie FM, Lee AG, Zhou QY. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science. 2005;308:1923–1927. doi: 10.1126/science.1112103. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto S, Yamazaki C, Masumoto KH, Nagano M, Naito M, Soga T, Hiyama H, Matsumoto M, Takasaki J, Kamohara M, Matsuo A, Ishii H, Kobori M, Katoh M, Matsushime H, Furuichi K, Shigeyoshi Y. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc Natl Acad Sci U S A. 2006;103:4140–4145. doi: 10.1073/pnas.0508881103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dattani MT. The candidate gene approach to the diagnosis of monogenic disorders. Horm Res. 2009;71(Suppl 2):14–21. doi: 10.1159/000192431. [DOI] [PubMed] [Google Scholar]
- 40.Kung TT, Crawley Y, Jones H, Luo B, Gilchrest H, Greenfeder S, Anthes JC, Lira S, Wiekowski M, Cook DN, Hey JA, Egan RW, Chapman RW. Tachykinin NK3-receptor deficiency does not inhibit pulmonary eosinophilia in allergic mice. Pharmacol Res. 2004;50:611–615. doi: 10.1016/j.phrs.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, Lalli E, Moser C, Walker AP, McCabe ER, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature. 1994;372:635–641. doi: 10.1038/372635a0. [DOI] [PubMed] [Google Scholar]
- 42.Muscatelli F, Strom TM, Walker AP, Zanaria E, Recan D, Meindl A, Bardoni B, Guioli S, Zehetner G, Rabl W, et al. Mutations in the DAX–1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- 43.Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 44.Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2008;83:511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–215. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- 46.Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, Lebouc Y, Froguel P, Guy-Grand B. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 47.O’Rahilly S, Gray H, Humphreys PJ, Krook A, Polonsky KS, White A, Gibson S, Taylor K, Carr C. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995;333:1386–1390. doi: 10.1056/NEJM199511233332104. [DOI] [PubMed] [Google Scholar]
- 48.Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 49.Dode C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, Lespinasse J, Lienhardt-Roussie A, Mathieu M, Moerman A, Morgan G, Murat A, Toublanc JE, Wolczynski S, Delpech M, Petit C, Young J, Hardelin JP. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. doi: 10.1371/journal.pgen.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZ, Li WP, Maccoll G, Eliseenkova AV, Olsen SK, Ibrahimi OA, Hayes FJ, Boepple P, Hall JE, Bouloux P, Mohammadi M, Crowley W. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 2007;117:457–463. doi: 10.1172/JCI29884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 52.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shinawi M, Cheung SW. The array CGH and its clinical applications. Drug Discov Today. 2008;13:760–770. doi: 10.1016/j.drudis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 55.Gibbs JR, Singleton A. Application of genome-wide single nucleotide polymorphism typing: simple association and beyond. PLoS Genet. 2006;2:e150. doi: 10.1371/journal.pgen.0020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360:1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 58.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.