

Abstract
Chronic heart failure is a worldwide cause of mortality and morbidity and is the final outcome of a number of different etiologies. This reflects both the complexity of the disease and our incomplete understanding of its underlying molecular mechanisms. One experimental approach to address this is to study subcellular organelles and how their functions are activated and synchronized under physiological and pathological conditions. In this review, we discuss the application of proteomic technologies to organelles and how this has deepened our perception of the cellular proteome and its alterations with heart failure. The use of proteomics to monitor protein quantity and post-translational modifications (PTMs) has revealed a highly intricate and sophisticated level of protein regulation. PTMs have the potential to regulate organelle function and interplay most likely by targeting both structural and signaling proteins throughout the cell, ultimately coordinating their responses. The potentials and limitations of current proteomic technologies are also discussed emphasizing that the development of novel methods will enhance our ability to further investigate organelles and decode intracellular communication.
Keywords: Proteomics, Heart Failure, Posttranslational Modifications, Organelle, Communication
INTRODUCTION
Chronic heart failure (HF) can be the outcome of several disease states such as hypertension, ischemic heart disease or valvular defects1. Despite these diverse etiologies, a number of features are frequently shared, including impaired Ca2+-handling, depressed contractility and altered energy metabolism. Each feature reflects functional changes in specific subcellular organelles such as sarcoplasmic reticulum (SR), sarcomeres and mitochondria2-4. Other common features of HF (such as the activation of intracellular signaling in response to increased adrenergic and neuro-hormonal stimulation, and perturbation of organelles’ spatial distribution) also imply an orchestrated action involving multiple organelles5-7. Yet it is unclear exactly how the various organelles coordinate their responses under both physiological and pathological conditions. Indeed, their dys-coordination could be considered another feature of the failing heart8.
Organellar interplay can occur via two routes: second messengers or structural connections (Figure 1). The concentration of second messengers (e.g. Ca2+, NO• and cAMP) fluctuates throughout the cell. It is their local levels that regulate the activity of downstream protein targets, either directly or indirectly, via signaling cascades; and a common final outcome is the post-translational modification (PTM) of their protein targets. These proteins can be localized to different organelles providing a means for widespread cellular action. A good example is Ca2+, which binds directly to both the sarco(endo) plasmic reticulum calcium-ATPase 2 (SERCA2) in the SR and troponin C (TnC) in the sarcomeres9, 10. Another example is cAMP which can activate protein kinase A (PKA), resulting in phosphorylation of proteins throughout the cardiomyocyte11.
Figure 1. A proteomic strategy to study organellar communication.

After organelle fractionation, proteomic methods including protein separation, quantification and identification are employed to study the different sub-proteomes. Integrated analysis of the data generated from several organelles can help highlighting common molecular components involved in the coordination of different cell compartments. MYO, myofilament; MT, mitochondrion; G, gap junction; D, desmosome.
Direct physical connection is the other mechanism for organelle interplay, occurring through the three major components of the cytoskeleton: microfilaments (actin), intermediate filaments (desmin which is specific to cardiomyocytes) and microtubules (tubulin)12. The roles of microtubules and the actin cytoskeleton in cellular plasticity, motility and protein trafficking are well known13, 14, as is the role of intermediate filaments in maintaining proper localization of different organelles15. However, novel functions for modified cytoskeletal proteins in diverse cellular functions are emerging16-19.
We postulate that PTMs are important in the development of HF as they likely mediate, and therefore regulate, organellar cross-talk. To test this hypothesis, organelles can be analyzed after sub-cellular fractionation using a variety of proteomic technologies. An additional advantage of this approach is the enrichment of low abundant, organelle-specific proteins. The integrated analysis of the data generated from several organelles can help highlight common molecular components involved in the coordination among different cell compartments.
In the present review, we describe how our knowledge of HF has benefitted from proteomics’ ability to scrutinize biological processes and reveal the new levels of complexity introduced by PTMs. We have also included novel analytical approaches, and discuss how these can be applied to specific biological questions regarding HF. These hands-on examples illustrate the contribution of proteomic approaches in moving the field forward.
Proteomics: When, Why and How
Historically, proteomics originated when separation technologies such as two-dimensional gel electrophoresis (2DE) and liquid chromatography (LC) were combined with mass spectrometry (MS). The need to increase our understanding of the “PROTEin complement of the genOME”, the PROTEOME, has driven the continuous development of these technologies. With the implementation and integration of various technological platforms it has become possible to separate, identify, characterize and quantify complex mixtures comprising hundreds to thousands of proteins or peptides20. These are key features to gather a more thorough understanding of intricate and elusive diseases such as heart failure. Table 1 gives an overview of the most common technologies, with their advantages, limits and possible biological applications.
Table 1.
Proteomic techniques
| Name | Advantages | Limits | Resolution | Quantification | Applications* |
|---|---|---|---|---|---|
| 1DE22,23,48,67,122,127,128,134,148,165 |
|
|
good | poor | D, ID |
| 2DE20,43,58,72,103,110,127,135,155,157,160 |
|
|
very good | very good | D, Q, ID, PTM |
| 1DLC§70,105,127,129,131,135,148 |
|
|
good | poor | D, ID |
| 2DLC65-67,106,135 |
|
|
good | N/A | D, ID, PTM |
| Tag-free83 |
|
|
N/A | good | Q |
| MS-tag52,53,72,86,148 |
|
|
N/A | very good | Q |
| MRM88,89 |
|
|
N/A | very good | Q‡, PTM |
D: creating tissue, cellular or organelle database; Q: protein quantification (‡relative and absolute); ID: protein identification; PTM: protein discovery and/or validation; N/A: non applicable; §mainly reverse-phase. Original articles cited in the text are also listed in the table, in bold the ones that used cardiac samples.
A few key concepts have emerged from the proteomic studies performed over the last sixteen years. First, even a single protein can be a complex mixture of its many modified forms. Through the specific measurement of a number of PTMs, and the determination of the actual modified amino acid residues, it has become clear that a protein can have multiple PTMs at multiple amino acid residues. Furthermore, different PTMs may compete with each other for a particular residue21. To date, over 400 different PTMs have been described, although far fewer have been documented in higher organisms such as mammals (see a detailed list at http://www.uniprot.org/docs/ptmlist). In the cardiac subproteome, phosphorylation is by far the best described PTM and most modified proteins have multiple phosphorylation sites (Figure 2A, Online Table I). To date, phosphorylation has been observed for 78%, 80% and 48% of cardiac proteins located in the SR, myofilaments and mitochondria, respectively (Figure 2B, Online Table II). However, the wide majority of the potential PTMs have not been fully investigated, in part due to a lack of technologies that specifically target them. Moreover, as a cautionary note, because the extent of PTMs occurrence is not clear and PTMs can modify the antigenic properties of proteins, quantification methods that rely solely on antibodies may lead to biased conclusions.
Figure 2. PTMs in heart proteins.
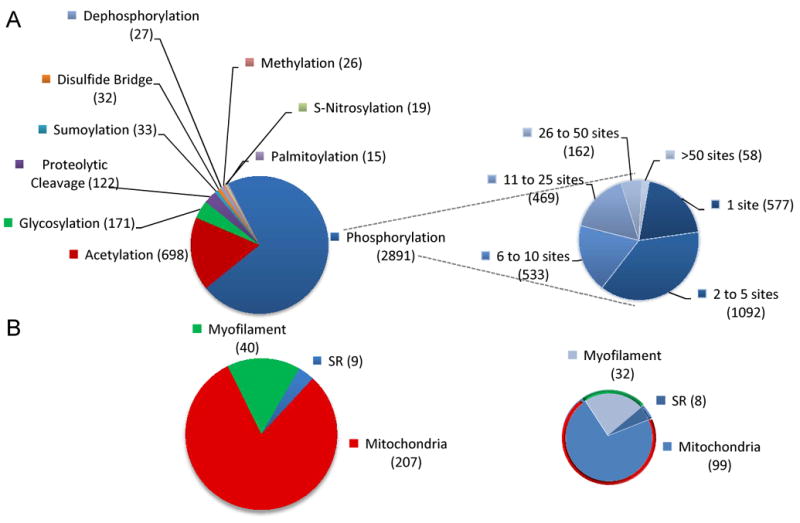
A) Proteins annotated as “heart” with their site specific PTMs* (left) with emphasis on phosphorylated proteins grouped based on the total number of phosphorylation sites (right). B) Proteins annotated as “heart” with their subcellular localization in either SR , sarcomeres or mitochondria; the total number (left) and the number of the ones that are phosphorylated (right) are provided, as reported in the Uniprot Protein Knowledgebase (UniprotKB). *Only PTMs that appeared in more than 15 entries in the Human protein reference database were considered.
Second, in multicellular organisms, different cell types share many of the same proteins (proteome redundancy). These common proteins are part of essential cellular systems and may reflect the minimum number of functional proteins required for cell survival. This implies that the unique attributes of a specific cell type must be derived from variations in protein quantity, isoform expressions (from splice variants or different genes) or PTMs. Furthermore, this suggests that the regulation of proteins at the time of transcription and translation, as well as protein complex assembly, play a major role in determining the exact composition of the proteome and thus the cellular fate. Data to support this concept arise from a number of detailed MS-based analyses, including work on mitochondria isolated from different tissues (heart, kidney, liver and brain)22, 23. These studies demonstrate that, at least two third of mitochondrial proteome are conserved across different tissues. As the levels of these shared proteins greatly vary from organ to organ23 it is possible that the quantity, rather than the type of protein within functional clusters discriminates among different tissues. However, protein isoforms are also important, as illustrated by the cell specificity of the Tn-complex (comprising TnT, TnC and TnI): the different cell-specific isoforms for each subunit constitute unique attributes of a cardiomyocyte. As well, various combinations of the subunits’ isoforms (slow and fast) differentiate cardiac and skeletal myocytes24.
Third, technological development is fundamental for scientific advancement. Improvements in each step of the proteomic pipeline have expanded the number and types of proteins (e.g. hydrophobic, basic, etc) that can be observed. Considerable contributions have arisen from methods that improved protein separation (e.g. 2DE25, 26), or enrichment of PTM peptides (e.g. O-linked beta-N-acetylglucosamine, O-GlcNAc27, 28 and phosphorylation29-31). Developments in mass spectrometers have also lead to enhanced speed, sensitivity and mass accuracy. When coupled with advanced bioinformatic techniques, this has resulted in reduced false discovery rates and improved protein identification.
Hundreds of review articles on proteomics and human diseases have been published, but not as many address the application of proteomic technologies in the elucidation of cardiac disease (selected reviews32-37). In the next section we will describe the different techniques commonly utilized in organelle proteomics, beginning with sub-cellular fractionation and following each step of the proteomic workflow through MS-based protein characterization.
Sub-Cellular Fractionation for Proteomics
Each separation technique used in proteomics has a limited capacity regarding the number and types of proteins that can be resolved. This technical limitation prevents the detection of low abundant proteins and cannot always be overcome by simply increasing the total protein load as this may paradoxically result in protein loss (e.g. by precipitation). Sensitivity and resolving power can instead be increased by focusing on a particular sub-proteome35, 37. This requires balancing two aspects: sample purity, and preservation of one protein’s PTM status. Importantly, regardless of the methods employed, it is difficult to isolate a pure organelle preparation. It is equally difficult to prove that a preparation is pure. This arises from the dilemma that a subproteome can comprise resident organellar proteins, those with multiple cellular localizations and contaminating proteins. Enrichment of a particular protein during the preparation can help, but validation through other techniques, such as microscopy, is ultimately required to confirm sub-cellular localization23.
When compared to traditional biochemical organelle purification methods, a major concern for proteomic studies is keeping modifications intact during fractionation. To avoid introducing artificial PTMs during sample processing, either pharmacological inhibitors (e.g. phosphatases, proteases inhibitors) or harsh denaturing conditions can be used35, 38. On the other hand, mild conditions are required when enzyme activity or protein-protein interactions (e.g. complexes) are being addressed39. For example, our group has optimized a fast and reproducible method (called IN Sequence) to fractionate frozen heart tissue for proteomic applications: dividing the cardiac proteome into separate myofilament- and cytosolic-enriched fractions40. This method was developed to deplete the high abundant myofilament subproteome, allowing enhanced observation of the cytosolic proteins and preserving the original endogenous PTM status. While this method is appropriate for proteomics, it leaves the myofilament subproteome denatured, inactive and not amenable for functional studies. Additional precautions may be required for some oxidative PTMs, where the procedures should be performed in the dark (e.g. S-nitrosylation41, 42). For PTMs that are extremely labile (due to chemical instability or activation of regulatory enzymes), the speed of the procedure will be a factor in protocol optimization. The decision on which approach to use depends on the biological question and the assays that will be performed downstream of the proteomic pipeline (such as enzyme activity and other functional assays).
Protein and Peptide Separation Techniques
Electrophoretic Methods: Classical and Non-classical 2DE
Two-dimensional gel electrophoresis (2DE), has been used for protein separation since the first proteomic study20. The 2DE protocol most widely used today was developed in the mid ‘70s by O’Farrell43. Two notable improvements since then are: the introduction of immobilized pH gradient strips for isoelectrofocusing (IEF)44 and fluorescent protein labeling45, which greatly reduce technical variability. In fact, researchers can now reproducibly separate thousands of protein forms and reliably quantify protein changes across different samples by combining these methods (Table 1)25, 36, 46.
In the first dimension of classical 2DE, proteins are separated according to their isoelectric points (pI) by IEF. Some PTMs, such as phosphorylation, acetylation, deamination, or alkylation can modify one protein’s pI, inducing a shift along the IEF axis47. In the second dimension, proteins are separated according to their size using polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulphate (SDS-PAGE). Other PTMs can modify protein size and thus, electrophoretic mobility in the second dimension; these include proteolytic cleavage, glycosylation and ubiquitination48. Phosphorylation can also slow down electrophoretic mobility, inducing an apparent alteration in protein size, most likely because of the interference with protein-SDS interactions49.
Recently, several “non-classical” 2DE techniques have been re-discovered and optimized combining different separation methods. One example is blue-native gel electrophoresis (BNE) where proteins are separated under non-denaturing conditions. Protein complexes can be separated by BNE followed by further separation under denaturing conditions, either by SDS-PAGE or classical 2DE. The initial BNE step contributes additional information regarding protein-protein interactions and the composition of protein complexes50.
2DE has the advantage of providing a quantification method based on the area/volume of a protein spot after gel staining. Spot matching and differential display analysis are carried out through semi-automated software analysis. Fluorescent stains and, in particular, the difference gel electrophoresis approach (DIGE45) offer a very reliable platform for quantifying changes in protein levels between different experimental conditions (e.g. disease vs. healthy). Moreover, the image analysis that used to be extremely laborious can now be contracted to companies that perform this routinely51. Despite 2DE’s advantages, a few considerations have to be mentioned. Each spot visualized can be a mixture of several proteins. In this case, another quantitative step is required to determine the specific protein responsible for the difference in spot intensity52, 53. As well, a single protein maybe presented as multiple spots, each representing different forms. Therefore, a 2DE gel containing 1,000 spots may only represent 300-400 different proteins54. These factors should be taken into account when protein identity and quantity changes are assigned based on 2DE.
One physical limitation of 2DE is the poor capacity for studying hydrophobic proteins, because anionic detergents cannot be used in the first dimension (IEF)55, 56. Although we57, 58 and others have shown previously that is possible to separate highly hydrophobic proteins on 2DE, alternative techniques such as liquid chromatography (LC) or SDS-PAGE may be a preferable strategy. 2DE is, however, currently the only method that provides a direct indication of many different PTMs in a single analysis.
Chromatographic Methods: 1 and 2DLC
Different LC techniques such as ion exchange, size exclusion, affinity and reversed-phase (RP), can be employed for the separation of complex protein and peptide mixtures based on their intrinsic properties (i.e. size, charge/pI, hydrophobicity and protein-binding properties40, 59, 60). In these methods, analytes are separated in solution according to differences in their partitioning between the stationary and mobile phases. The most widely used LC in proteomics is RP, either off- or on-line with MS. A wide selection of chromatographic materials for RPLC is available: the choice of stationary phase (particle size, pore size, surface area, and chemistry of the substrate surface), and, to a lesser degree, flow rate and buffer composition, can be optimized to improve resolution.
As with gel-based methods, the combination of different analytical strategies such as in multi-dimensional LC (nDLC) increases resolution59-62. The separation of peptides by two-dimensional LC (2DLC) after protein digestion is referred to as shotgun approach63, 64, as opposed to 2DLC systems that separate intact proteins, potentially providing additional information about protein isoforms and PTM status65-67. LC methods are advantageous due to minimal operator-dependence, although they require more complex MS capability and have a far greater MS workload than gel-based methods (Table 1). Similar to 2DE, LC methods do not allow for unambiguous assignment of quantity changes based on elution profile alone, and require protein labeling prior to MS (see below).
A specific LC technique that is worth mentioning is affinity chromatography. Affinity chromatography is based on reversible antigen-antibody interaction, and allows protein or protein complexes to be recovered with good purity and in some cases with retained biological activity68-70. The most common technical challenges with affinity chromatography are: determining which proteins are contaminants due to nonspecific binding; and distinguishing between the direct and indirect binding partners that form a protein complex. Different factors that can contribute to nonspecific binding are ionic or hydrophobic interactions. These can be limited by carefully optimizing both washing and binding conditions.
The use of multiple proteomic methods that combine different protein and/or peptide separation methods increases the overall coverage of the proteome71, 72. This is particularly important when studying species which have poor or underrepresented annotation, such as rabbit or dog73, 74. In these species, isolation of a higher number of peptides is required for exact protein identification and characterization. In fact, enhanced detection power increases the probability of matching a peptide sequence to a homolog in a more annotated species.
Protein Identification and Quantification Methods: Mass Spectrometry
Today, MS is the most common method for identification of proteins. MS has eclipsed other techniques, such as Edman’s degradation, due to its speed and level of detail (e.g. PTM characterization; for historical account of MS development see Zhou et al.75).
MS instruments consist of three modules: an ion source, a mass analyzer and a detector. Once ionized, particles are accelerated and sorted according to their mass to charge ratio (m/z) in the mass analyzer, before they are detected. Thanks to the development of soft ionization methods, such as matrix assisted laser desorption (MALDI) and electrospray ionization (ESI), it is now possible to ionize large biological molecules including peptides or even entire proteins. The impact of these technologies on modern science is fundamental and their inventors were awarded the Nobel Prize for chemistry in 200276. Common analyzers include time-of-flight (TOF), quadrupoles (Q) and ion traps. Each of these has been combined in tandem mass spectrometry (MS/MS or MS2) so that peptides can be further broken down using collision induced dissociation (CID). An advantage of MS2 is that it provides detailed chemical information about the amino acid sequence77 (Online Figure I). Because mass accuracy is best over the low mass range, analysis of peptides rather than intact proteins allows for superior mass detection and, consequently, improved amino acid sequence assignment (called a “bottom-up” analysis). This fact, along with other benefits of peptide-centric proteomics (which include better separation, ionization, and database searching, see above), has determined the larger use of bottom-up approaches. However, MS can also be used to characterize intact, non-digested proteins, with or without prior separation (also known as a “top-down” approach)78, 79. Due to the continuous improvements of MS technologies, it is foreseeable that top-down approaches will become increasingly popular in the future.
Several approaches based on MS have been developed for quantification. These can be done with or without adding a mass tag to the peptide (tag- vs. label-free methods, respectively)80. Label-free methods base quantification on the signal intensity originated in the MS (peak area) or on the number of times a peptide is observed81, 82. Tag methods require peptide labeling with isotopic tags83 via enzymatic (e.g. stable isotope labeling with amino acid in cell culture (SILAC) and 18O-labeling52, 84, 85) or chemical derivatization (e.g. isobaric tag for relative and absolute quantification (iTRAQ))86. SILAC can only be used in proliferating cells, but chemical derivatization methods are more flexible and allow labeling of protein extracted from tissue and body fluids. In chemical methods, samples are labeled using tags with the same molecular weight (isobaric). Once labeled peptides are introduced in the MS2, each tag releases a different fragment with a specific mass that can be tracked in the lower portion of the spectra (∼100 Da m/z). The intensity of the peaks generated by these reporter ions is proportional to the quantity of the protein in the original sample. Typically, at least four different tags are available, allowing multiple biological conditions to be analyzed simultaneously (multiplexing).
A newly adapted MS technology is multiple reaction monitoring (MRM) and its closely related method single/selective reactive monitoring (SRM)87, 88 (Online Figure I). MRM is a tandem MS technique that allows relative and absolute quantification of proteotypic peptides (i.e. peptides that have a unique amino acid sequence specific to a particular protein or isoform) and, hence, of proteins. In short, triple quadrupoles (Q3) MS can be used to select a specific peptide of interest (including modifications or polymorphism89, 90) based on its m/z, in quadrupole Q1. This parent ion is then fragmented in the Q2 and the daughter fragments (also called transition ions) are monitored and quantified in the Q3. The choice of the transition ions is critical as it allows monitoring a specific peptide, based on MS2 identification. Normally, few transitions need to be monitored in order to achieve reliable quantification (n≥5). In many cases, heavy isotope-labeled standard peptides are spiked in for absolute quantification, but relative quantification (sample A vs. B) is relatively straightforward. The biggest advantages with MRM are the unbiased quantification and the ability to distinguish between PTMs and other protein forms. This capacity is often missing when using antibodies88. In addition, multiplexing is straight-forward compared to ELISA and a relative large number of peptides can be quantified during a single run.
PTM Characterization
As mentioned earlier “a protein” can be a complex and highly dynamic mixture of its modified forms. Important aspects of one protein’s function (such as activity, localization, half-life and ability to form complexes) are all regulated by PTMs, and thus it is crucial that they be characterized. MS, alone or in combination with separation and/or enrichment techniques, currently represents the preferred method for PTM characterization. Most PTMs are recognizable in a MS spectrum through specific mass signatures. For instance, phosphorylation typically induces an increase in mass of 80 Da due to the weight of the phosphate group. A more common observation is the occurrence of neutral loss: the loss of a phosphate group alone (80 Da) or plus water (80+18=98 Da)91 (for a review on phospho-proteomics see92, 93).
Two main factors limit the detection of peptides containing PTMs. One is the stoichiometric relation between the modified peptide and all other peptides in the samples. In other words, because peptide detection is a competitive process and the amount of a peptide with a single type of modification is relatively low, more abundant, variously unmodified and modified peptides are likely to be detected first. In some cases this limitation can be overcome by enriching for the specific modified peptides downstream of protein digestion (Figure 1). Such is the case for phospho-peptides. There are a few protocols available, based either on the affinity of phospho-peptides for positively charged metal ions94, 95 or on other LC methods; RPLC96 and hydrophilic interaction chromatography97. Another consideration of PTM analyses is related to the modification’s stability. For example, the detection of phospho-peptides by MS is challenged by the lability of the phosphate bond during CID. Phospho-esters are weaker than peptide bonds; therefore they tend to break during fragmentation. For this reason a method that catalyzes the preferential fragmentation of peptide bonds with improved detection of phospho-peptides in the MS has been developed (electron transfer dissociation)91. Similar considerations can be made for other PTMs, although phosphorylation is currently the most studied98. Until recently, a technological challenge was site-specific quantification of a given PTM. With MRM, it is now possible to measure the relative and absolute abundance of PTMs at each site within and across different proteins89, 90.
Bioinformatics
Bioinformatics is integral to proteomics. In fact, bioinformatic tools are needed to assist almost every step of the proteomic workflow. This includes the quantitative analysis of proteins and peptides after separation, as well as the MS data handling99. Bioinformatic tools are also utilized for the prediction of protein characteristics (such as pI and MW values, PTM consensus sequence, etc.), and several algorithms are publicly available (e.g. www.expasy.net). Also freely accessible are a number of curated protein databases (see Online Table III). While these databases are essential for proteomic studies, it is important to mention that they are still under development and therefore far from being exhaustive. The number of proteins assigned to a specific tissue or organelle (Figure 2) depends on the way these are annotated in the database. Also the total number of residues modified by a given PTM in a protein (Figure 2A, right pie chart, Online Table I) will vary depending on the annotation criteria ( Online Table I). For instance, modifications can be assigned to a given protein entry based on the consensuses sequence of the modifying enzyme or if the modification has been observed in an isoform.
It was the establishment of the first genomic databases, as a basis for the creation of protein databases that allowed proteomics to develop. Nowadays these protein databases are routinely mined by algorithms that can match experimental MS spectra to protein sequences fragmented in silico, in a semi-automated fashion100. Algorithms, like OMSA, Mascot, Sequest, X!tandem are able to extract the numerical values of peptide masses from MS spectra and then search against protein databases to assign peptides to a protein entry. In all cases, manual inspection of the spectra is warranted, especially when there is an indication of a potential modification. In fact, being programmed “to retrieve” (e.g. assign a peptide to a protein), these algorithms show an inherent bias for false positive identifications. Another issue associated with database-based identification is the fact that the databases can have multiple entries for a given protein (database redundancy). This can inflate the list of assigned entries. To overcome this issue and ensure single assignments, protein clustering algorithms can be helpful identifying sequence similarities based on a high degree of global (across the whole protein) or local (short protein domains) homology. Moreover, the correct assignment of a particular protein isoform or taxonomy (species) often requires extensive sequence coverage. This is because an amino acid sequence that is unique for a particular isoform/taxonomy must be assigned. On one hand, database redundancy and poor sequence quality can generate false positive identifications; on the other hand, the poor annotation of some species (e.g. dog or rabbit) can decrease the probability of identifying a protein correctly (false negative). These and other issues with database search were recently identified by the Human Proteome Organization as the major cause of lab-to-lab variability in reporting protein identification101. Even though databases are likely to improve in the future along with MS performance, awareness and education on these issues will be crucial for the generation of reliable data.
DIVIDE AND CONQUER: ONE SUBPROTEOME AT A TIME
The different proteomic techniques described above, from cellular fractionation to detailed protein characterization by MS, constitute a powerful tool in studying such a complex disease as HF. Even deeper insight on the disease can be gained by dividing the cardiomyocyte into separate fractions, and analyzing the resulting subproteomes, rather than studying the whole tissue. In this section we will provide examples of how proteomics has been successfully employed to increase our understanding of the role of sarcoplasmic reticulum, myofilament and mitochondria in relation to cardiac disease.
The Sarcoplasmic Reticulum – Control of Calcium and Beyond
The SR organelle, appearing as an intracellular membrane network, serves as the main storage for intracellular Ca2+ in cardiomyocytes. It has been the dogma that release of Ca2+ from SR, in a Ca2+-sensitive manner, is essential and necessary for normal cardiac function102. Thus, the observed reduction in cardiac force in HF patients has been partly explained by a reduction in the amounts and activity of Ca2+ cycling proteins. Particular focus has been targeted on two SR proteins: SERCA and its regulator, phospholamban (PLB). SERCA is the main regulator of Ca2+ up-take during cardiomyocyte relaxation, and its activity is tightly modulated through interaction with PLB9.
When studying SR proteins, the initial challenges are the organelle’s extensive membrane network and its connection to other cellular compartments, which complicate the isolation of a pure structure. Also the hydrophobic properties and relatively low abundance of the proteins comprising the SR can make them difficult to study. A number of general strategies have been developed for the enrichment of membrane proteins and these are applicable to SR proteins. These methods include fractionation by density gradients and/or differential centrifugation steps. To prevent precipitation of the hydrophobic proteins, they can be solubilized using high concentrations of detergents. This can be followed by electrophoretic separation and MS identification103, 104. Another approach is to avoid the gel separation step by employing shotgun methods in the presence of MS-friendly surfactants that are compatible with in solution enzyme digestion and MS identification (e.g. RapigestTM105). The first study to identify SERCA using 2DE, successfully combined centrifugation enrichment with diheptanoyl-phosphatidylcholine solubilization105. Although the report showed that it is possible to achieve considerable enrichment of the SR, and that the SERCA protein can be separated by 2DE, it also illustrated the current challenge separating SR from other membrane organelles. Most preparations contain other membrane proteins (in particular from mitochondria103, 105, 106) and this complicates the goal of a precise identification of the SR sub-proteome. Due to the ambiguous definition of the SR sub-proteome, leading to a poor implementation of the term “sarcoplasmic reticulum”, the proteins from this organelle are underrepresented in different databases (e.g. the ryanodine receptor is not annotated to the SR in the UniprotKB, Figure 2B, Online Table II).
A central role for SERCA2 in cardiac disease was questioned in last year’s study by Andersson and colleagues107. This study showed that a cardiac-specific, inducible SERCA2 KO mouse survived for several weeks even with no detectible levels of SERCA2 protein in the cardiomyocytes (based on western blotting). Surprisingly, under these conditions the SR still contributed with Ca2+ to the cardiomyocyte contractions. This result implies either that the heart uses an alternative mechanism for cycling Ca2+ into the SR or that a small, non-immuno-reactive fraction of SERCA2 is present. As mentioned, due to the problems in obtaining pure SR, it remains debatable which other SR proteins might be up-regulated or modified as a consequence of severe SERCA2 depletion. This emphasizes the need for an accurate description of the SR sub-proteome and precisely how it changes with heart disease. An alternative strategy is to specifically target SERCA2 using affinity chromatography combined with targeted MS measurements (e.g. MRM).
Several genetically modified mouse models targeting expression of SERCA and PLB have been constructed to study the overall biological effects on Ca2+-cycling disruption (SERCA conditional knockout (KO)107; PLB KO and over-expression108). As expected, significant effects on cardiac contractility due to altered Ca2+-homeostasis are commonly observed in these models. Interestingly, by applying different proteomic approaches (e.g. 2DE, gel-free, MS2), it is clear that alterations in Ca2+-handling induces phenotypical effects beyond the contractile apparatus. These include changes in cytoskeleton protein composition, membrane receptors and intracellular signaling pathways, and energy metabolism109, 110.
The Sarcomeric Organelle – Power is Nothing without Control
Sarcomeric proteins are generally thought of as the molecular motors in the cardiomyocyte, generating force and conducting cellular shortening. Briefly, Ca2+ released from SR binds to TnC which alters the interaction between the thin (actin, tropomyosin, and troponin - consisting of TnI, TnT and TnC) and thick (myosin comprising myosin heavy chain and two light chains, and myosin binding protein C) filaments leading to muscle contraction10. Recently, it has been proposed that sarcomeres should be regarded as a cellular organelle because of its close involvement in structural, electrical, and metabolic aspects of cardiomyocyte dynamics111. In HF, this reduction in cardiac pump function can be related to changes in the SR but also to sarcomeric function10. While numerous mutations in the sarcomeric proteins causing familial hypertrophic and dilated cardiomyopathies have been described112, the importance of PTMs for sarcomeric proteins is also well established113.
In the Tn complex, TnI is the inhibitory subunit111 and its function is highly regulated by phosphorylation. The importance of Ser23 and Ser24 in controlling cardiac contractility and myofilaments responsiveness to Ca2+ has been extensively studied114. Recently, several other phosphorylation residues were identified, and currently there are seven known sites in humans (Ser23, Ser24, Ser31, Ser42, Ser44, Thr141 and Ser150)115; some of these are also important in the control of cardiac dynamics114. In contrast, when exploring the basal in vivo phosphorylation of TnI in a murine model, only the Ser22/23 sites (equivalent to the human Ser23/24) were found to be modified116. The biological importance of these TnI phospho-sites is under debate115. Our group has developed MRM assays to specifically quantify and monitor the phosphorylation status of six of the identified TnI phosphor-sites to determine the stoichiometric ratios of each residue in vivo.
One contribution of proteomic technologies to basic science is the rapid discovery of new PTMs, including the modified residues. An example, which was recently identified in the sacromeric proteins, is acetylglucoseamine at Ser and Thr residues (O-linked beta-N-acetylglucosamine, O-GlcNAc). When first discovered, identification of O-GlcNAcylation was challenging and time consuming117. However, today’s enrichment methods, in combination with sensitive MS, have established widespread occurrence of this PTM21. Recently, O-GlcNAcylation was implicated in myofilament contractile alterations118. Ramirez-Correa and colleagues discovered myofilament Ca2+ desensitizing when exposing cardiac TX-100-skinned muscle fibers to GlcNAc (without disturbing the phosphorylation status of Ser23/24). Using MS-based methods, the group indentified 32 O-GlcNAcylation sites in the myofilament subproteome, one being Ser150 of TnI. Knowing that there is extensive crosstalk between O-GlcNAcylation and phosphorylation21, it will be interesting to see how the alternate modification at Ser150 contributes to the biological function of TnI and the regulation of muscle contractile function.
Mitochondrial Proteomics: PTM Switches to Control Energy Production
Because the heart consumes more ATP than any organ of the body, it is particularly rich in mitochondria119. During the development of HF, both the energy demands and metabolism change, showing a remarkable decrease in oxidative phosphorylation and a shift towards glucose over fatty acid utilization120. Interestingly, during HF therapy like cardiac resynchronization (CRT), coordinated changes in the mitochondrial proteome suggest a reversal to a healthier phenotype. These alterations include proteins controlling metabolism and ATP production, establishing the importance of mitochondria in modulating the development of HF58.
The extensive application of proteomics to mitochondria also has historical and practical reasons. The study of mitochondria has exploited protein biochemistry for many decades, and proteomics itself can be considered an evolution of protein biochemistry. Furthermore, methods to enrich and purify the mitochondria are also several decades old, representing one of the most long-lived examples of proteome sub-fractionation121. Proteomic studies have certainly increased our understanding of mitochondria by providing evidence for the large and unexpected occurrence of PTMs.
The human mitochondrial proteome was originally characterized by Taylor et al., who initially described 651 mitochondrial and mitochondrial associated proteins122. Now, curated databases for mitochondrial proteins are also available123, including one recently compiled by Mootha and colleagues, who assigned 1,098 mitochondrial proteins (MitoCharta) by combining extensive MS characterization with microscopy and computation to confirm mitochondrial protein localization23. Unfortunately there is a lack of equivalent databases for the less annotated species that are used in cardiac research (e.g. rat, canine, porcine). Moreover, according to Forner et al.,22 who recently compared the proteome of mitochondria extracted from different rat organs (skeletal muscle, heart and liver), mitochondrial proteins can be annotated for other organelles (but not mitochondria) in widely used databases. The Authors also pointed out that mitochondrial proteomes from different organs are similar, at least qualitatively, with one third of the proteins identified being expressed exclusively in one organ. However, what seem to make a functional difference are the levels of expression of certain proteins.
Due to the high complexity of the mitochondrial proteome, and its inclusion of proteins with highly diverse properties, the proteomic approach for its study must be carefully selected. For instance, membrane proteins, which are more basic124 and overrepresented in the mitochondria122, are normally poorly separated by 2DE56. We reported previously that ∼1,000 mitochondrial protein forms, purified from the heart tissue in an animal model of HF, can be separated using 2DE58. These include basic membrane proteins such as Prohibitin 2 (pI=10.04) and VDACs (pI≥7,48), which could be nicely resolved through improved sample loading using paper-bridge57. Other groups used LC-MS2 in combination with 1DE22 or iTRAQ125 to compare the proteins from cardiac mitochondria under different conditions.
Targeted strategies can be used to focus on a specific portion of the mitochondria, such as the respiratory chain. The respiratory chain consists of five protein complexes (I-V); the first four generate the proton gradient that is used by complex V to synthesize ATP126. At least three of the five complexes in the chain have been investigated using proteomics127, 128. This is not surprising, as some of them could be considered a proteome on their own. For instance, complex I is constituted by 45 subunits129. Moreover this complexity is further increased by PTMs130-132. Recently, Palmisano and colleagues were able to identify phosphorylation sites in at least five subunits of complex I by combining BNE and phospho-peptide enrichment through titanium dioxide35, 130. We demonstrated complex V (ATP-synthase) phosphorylation and its effects on ATP synthesis using, respectively, phospho-peptide enrichment and mutation analysis combined with functional assays58, 133. Recently, Ping et al. identified 61 phosphorylation sites in all major mitochondrial pathways including 10 for the electron transport chain and four for complex V, in the murine heart134.
Other mitochondrial compartments can be enriched to peer deeper and deeper into this proteome and its modifications135-137 (Figure 3). The very notion that mitochondrial proteins can be modified is a fairly recent advent and it opens the way to a more accurate description of the regulation of energy production in the normal and failing hearts. PTMs, among others, govern the way proteins interact to form supramolecular complexes, and their characterization is crucial.
Figure 3. Proteomic to study PTM regulation of protein-protein interaction.
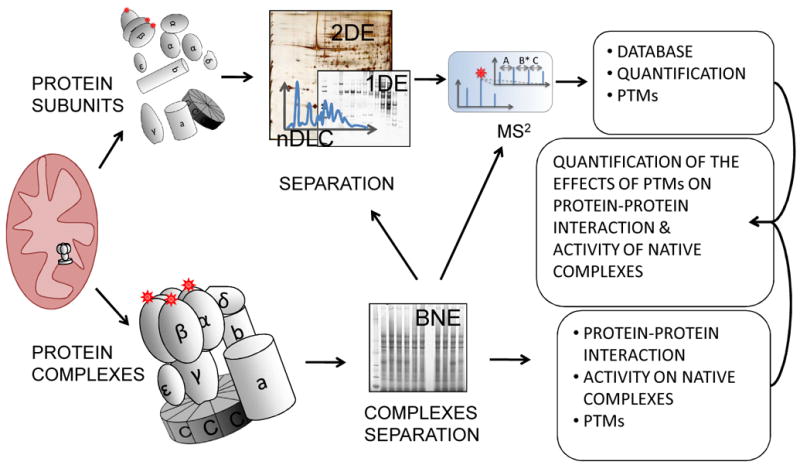
Proteins and protein complexes can be alternatively separated from organelles, such as mitochondria. As an example, schematically presented, ATP-synthase (complex V, adapted from Elston T, et al., Nature. 1998, 29;391:510-3)can be denatured into its individual subunits or separated as a whole. The first approach is more suitable to monitor PTMs, the latter provides information on complex composition. The integrated analysis of the two strategies can indicate the role of PTMs in complex formation.
Sub-proteomics and Cellular Interplay: Putting humpty dumpty Back Together
As described so far, isolating and studying single organelles is a good approach to uncover substantial information regarding their regulation and interplay. However, organelles can be tied together by either oscillations in the levels of second messengers or physically, through structural proteins. In the heart, perturbation of the complex and coordinated interplay between different cellular compartments might be an underlying sign of disease.
Second Messengers and Proteomics: Identifying the Targets
In this section we discuss the involvement of the second messengers Ca2+, NO•, cGMP and cAMP in organelle communication in relation to HF. In healthy cardiomyocytes, contraction and relaxation are triggered by a transient rise and decline in the concentration of intracellular Ca2+; which cycles between sarcolemma, SR and sarcomeres10. Ca2+ enters the cell through ion channels in the sarcolemma, triggering further release of Ca2+ from the SR. This occurs where Ca2+ channels in the plasma membrane and SR membrane are in close proximity. A common feature in HF is that this proximity becomes disorganized, which inhibits efficient SR-Ca2+ release and results in weakened cardiac contractions7. It has also become evident that mitochondria can provide a significant storage of Ca2+ and that mitochondrial Ca2+ uptake increases ATP production138. Thus, on a beat-to-beat basis, Ca2+ both induces muscle contractions and increases ATP production to meet the needs of heightened energy demand. This supports the idea that Ca2+ is an important factor in organellar interplay (Figure 4). Ca2+ can also be stored intra-cellularly in microdomains, where, unlike in the cytosol, levels do not fluctuate during the action potential139. This pool of Ca2+ can bind to and activate cytosolic enzymes (e.g calmodulin and calcineurin), thereby inducing Ca2+-dependent signaling cascades. One important aspect involved in cardiac disease is the pro-hypertrophic response mediated by the Ca2+-calcineurin dependent signaling pathway140.
Figure 4. Organelle Dyscoordination in Heart Failure.
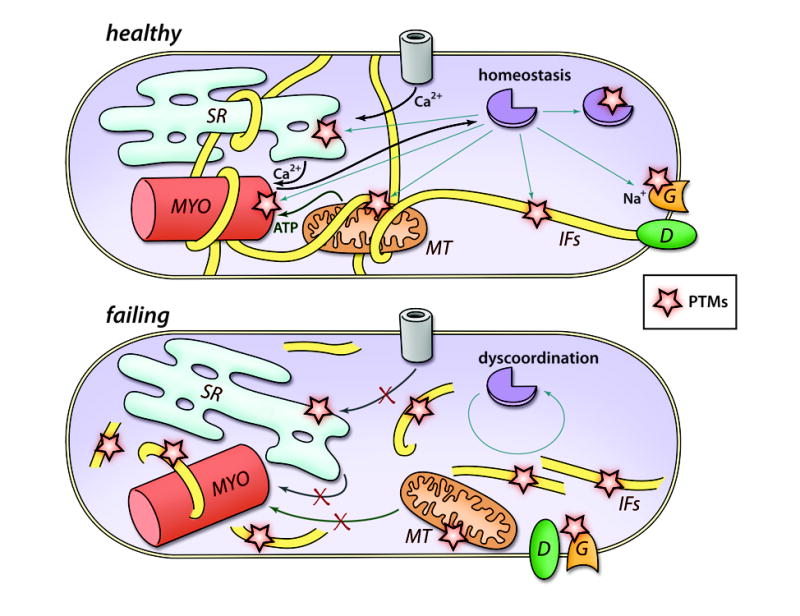
Schematic presentation of features in a failing cardiomyocyte: increased neurohormonal stimulation and mechanical stress affects cellular contractility, energy metabolism, conduction, intracellular signaling and cell structure mislocalization. We propose that dys-regulation of various organelles could be considered another feature of the failing heart. We further hypothesize that this is regulated through changes in the homeostasis of secondmessengers, resulting in dynamic regulation of PTMs, and by the disruption of cytoskeletal structures such as IFs. MYO, myofilament; MT, mitochondrion; G, gap junction; D, desmosome.
In contrast to Ca2+, NO• diffuses across the sarcolemma. NO• can directly alter protein function by covalently modifying cysteine thiols to generate S-nitrosothiol (SNO). Recently, Lima and colleagues suggested that a majority of the NO• effects are mediated by this PTM141. Because of its labile nature, considerable work has been invested in the improvement of proteomic techniques to study SNO42. It is important to develop robust technology for the identification and quantification of SNO, as its presumed role as a regulator in cardiovascular disease is growing141. Recently, impaired S-nitrosylation of the ryanodine receptor was shown to induce Ca2+ leak from the SR and thus contribute to cardiac dysfunction142.
Another important feature of NO• is the induction of another second messenger, cGMP, through activation of the soluble guanylyl cyclase. cGMP activates PKG, similar to cAMP activation of PKA; both induce signaling cascades, resulting in altered cardiomyocyte growth and function11, 143. Interestingly, both PKG and PKA signaling cascades converge in the phosphorylation of several proteins (e.g. both enzymes specifically induce phosphorylation of Ser 23/24 in TnI115, 144). With respect to organelle communication, PKA and PKG phosphorylate proteins that are located in different cellular compartments: SR (PKA/PKG: Ryanodine receptor and PLB); myofilaments (PKA/PKG:TnI, PKA: myosin binding protein C); mitochondria (PKA/PKG: mitochondrial ATP regulated potassium channel); and sarcolemma (PKA/PKG: L-type Ca2+-channel)11, 145-147. Phosphorylation via these kinase signaling cascades can be important in transmitting signals throughout the cell and coordinating the action of several organelles. Recently, an impressive 20,443 phospho peptides where quantified in a single study148. Although this was done in HeLa cells, similar levels will most likely occur in cardiomyocytes. Other PTMs that have been successfully enriched for are acetylation, metylation, ubiquitylation and glycosylation149; but such investigation in the cardiac system remains recent.
Organelles Physical Connection – One Ring to Rule Them All
Organelles can also communicate through the physical connection mediated by the cytoskeleton. For instance, the many interactions between intermediate filaments (IFs) and all major cell compartments make them strong candidates to mediate organelles communication15. In the heart, IFs are constituted by desmin and have been found to regulate the function of myofilaments150, 151 mitochondria15 and gap junctions152. The central role of IFs in HF is corroborated by recent proteomic studies that investigated all major sub-proteomes of the cardiac cell and found that modified desmin forms were altered as a common denominator. The increase of desmin PTM-forms with disease is a consistent observation from proteomic studies that investigated various human and animal models of HF, so it is highly unlikely that its alterations are non-specific58, 153-162.
Desmin co-enriches with the myofilament fraction of the proteome as IFs are “wrapped” around the sarcomeres and keep them aligned150. Desmin mutations cause contractile failure and it was recently postulated that the mis-alignment of the sarcomeres that arises when desmin cytoskeleton is disrupted may represent a cause of contractile failure itself150, 162 (Figure 4). Our group has recently reported that the levels of PTM-forms of desmin are altered in the mitochondrial fraction of a canine model of HF compared both to shams and animals rescued by CRT58. This observation opens an interesting line of investigation, as desmin is known to regulate mitochondrial positioning and behavior through, albeit, an unknown mechanism5, 155. One potential explanation is that desmin, maintaining the spatial organization between sarcomeres and mitochondria, ensures that energy production and consumption are properly coupled (Figure 4). As well, the integrity of IFs cytoskeleton plays a major role in cardiac conduction as gap-junction localization at the intercalated discs is ensured by desmin through the interaction with the desmosomes152 (Figure 1 and 4). Lateralization of gap-junctions, which are observed in the failing heart, may be a cause of arrhythmias, and can be induced by mutation of desmin, or of desmosomal proteins, at the sites of interaction with the IFs19. Moreover, a number of novel functions have been attributed to the IFs in the heart including the regulation of the cardiac commitment of stem cells16, of autophagy17, and more recently of translation18. We believe that PTMs finely regulate these functions of the IFs.
Quantification of desmin in human HF, using immunostaining techniques, has generated controversial results as different groups reported either an increase or the absence of desmin in the cardiomyocytes of HF patients compared to healthy subjects163, 164. On the other hand, desmin quantification by 2DE has shown that multiple PTM-forms of the protein are increased during human HF160. We reported the increase of modified forms of the protein in cultured cardiomyocytes that become hypertrophied with endothelin-1 treatment155. Therefore we postulate that the discrepancy generated by immuno-detection studies may be due to PTMs. This exemplifies how proteomics could help in deciphering the molecular reasons for this controversy, and most importantly the biological value of these modifications. A detailed characterization and quantification of desmin PTM species would not only help elucidate the functions of the IFs in the heart, but could also highlight novel candidate biomarkers for cardiac disease157.
Several groups have reported changes in desmin modified forms with disease, in vivo, and Capetanaki et al. established a causal link between desmin modification and the formation of aggregates in a transgenic mouse model of HF15. The increase of an “acidic” form of desmin by 2DE was reported in the same study. However, despite the existence of several reported in vitro desmin modifications, desmin PTMs have been characterized in detail in vivo only recently. We identified the first phosphorylation sites of desmin in the heart, and linked them to HF in both a canine model and in humans157. Desmin has also been found phosphorylated or acetylated in non-cardiac cells and tissue109, 148, 165; this was unexpected, as desmin is thought to be specifically expressed in myocytes. The discoveries of desmin PTMs open new and exciting lines of investigation to address the functional roles of these modifications, particularly in HF.
In conclusion, the disruption of IF networks affects the function of at least three of the most important cell compartments in the heart, and possibly regulates many other aspects of cell function as well. Desmin phosphorylation and proteolysis have been long-known regulators of its assembly and function in vitro166, 167, yet without site information, in vivo, progress has been hampered. Further study of the biological role played by these PTMs will be helpful in elucidating the mechanisms that lead to IFs lattice modification during HF and its effect on organellar interplay.
CONCLUSIONS AND FUTURE PERSPECTIVES
There is a need to develop novel, experimental approaches to study HF. We have reviewed how proteomic studies and technologies have enhanced our understanding and revealed new levels of complexity in HF at the cellular, organelle and protein levels. Integration of data obtained from different sub-proteomes and system-wide proteomic studies is important in understanding cellular phenotypes. We have emphasized the need to understand the complex and coordinated interplay between organelles. We hypothesize that the mechanisms involved in synchronizing different cellular compartments may be disrupted with HF. Furthermore, we propose that the regulation and end effectors of this communication reside, at least in part, with the induction of PTMs of target proteins. The quantification and characterization of proteome changes are a necessity, and will drive the development and adaption of new proteomic approaches. In this way, we will eventually understand the global communication that drives the cellular phenotype.
Supplementary Material
Acknowledgments
We thank Christopher Murray for critical comments to the manuscript and Lars Husberg for his data mining support of the HPRD.
Non-standard Abbreviations and Acronyms
- 1/2/nDLC
One/two/multi-dimensional liquid chromatography
- 1/2DE
One/two dimensional electrophoresis
- BNE
Blue native electrophoresis
- CID
Collision induced dissociation
- CRT
Cardiac resynchronization therapy
- DIGE
Difference gel electrophoresis
- ESI
Electrospray ionization
- HF
Heart failure
- IEF
Isoelectrofocusing
- IFs
Intermediate filaments
- MALDI
Matrix assisted laser desorption
- MS2
Tandem mass spectrometry
- O-GlcNAc
O-linked beta-N-acetylglucosamine
- Q
Quadrupole
- pI
Isoelectric point
- PLB
Phospholamban
- PTMs
Posttranslational modifications
- RP
Reversed-phase
- SDS-PAGE
Sodium dodecyl sulphate polyacrylamide gel electrophoresis
- SERCA2
Sarco(endo) plasmic reticulum calcium-ATPase 2
- SILAC
Stable isotope labeling with amino acid in cell culture
- S/MRM
Single (selective)/ multiple reaction monitoring
- SR
Sarcoplasmic reticulum
- Tn
Troponin
- TOF
Time-of-flight
Footnotes
SOURCES OF FOUNDING
J.V.E.: NHLBI - HV-10-05 (2); NHLBI- P50 HL 084946-01; and the Clinical Translational Science Award at Johns Hopkins University
C.H.: Oslo University Hospital – Ullevaal, Oslo, Norway
G.A.: NIH Program Project Grant PO1-HL077180 and Compagnia di San Paolo, Turin, Italy
DISCLOSURES
None
References
- 1.Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: Cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- 2.Houser SR, Margulies KB. Is de pressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358. doi: 10.1161/01.RES.0000060027.40275.A6. [DOI] [PubMed] [Google Scholar]
- 3.Lehnart SE, Maier LS, Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev. 2009;14:213–224. doi: 10.1007/s10741-009-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capetanaki Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc Med. 2002;12:339–348. doi: 10.1016/s1050-1738(02)00184-6. [DOI] [PubMed] [Google Scholar]
- 6.Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: Architectural integration of energy production and utilization. Circ Res. 2001;89:153–159. doi: 10.1161/hh1401.093440. [DOI] [PubMed] [Google Scholar]
- 7.Louch WE, Sejersted OM, Swift F. There goes the neighborhood: Pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J Biomed Biotechnol. 2010;2010:503906. doi: 10.1155/2010/503906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, Tappia PS. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res. 2009;81:429–438. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- 9.Periasamy M, Bhupathy P, Babu GJ. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res. 2008;77:265–273. doi: 10.1093/cvr/cvm056. [DOI] [PubMed] [Google Scholar]
- 10.Hamdani N, Kooij V, van Dijk S, Merkus D, Paulus WJ, Remedios CD, Duncker DJ, Stienen GJ, van der Velden J. Sarcomeric dysfunction in heart failure. Cardiovasc Res. 2008;77:649–658. doi: 10.1093/cvr/cvm079. [DOI] [PubMed] [Google Scholar]
- 11.El-Armouche A, Eschenhagen T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev. 2009;14:225–241. doi: 10.1007/s10741-008-9132-8. [DOI] [PubMed] [Google Scholar]
- 12.Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res. 2000;45:273–278. doi: 10.1016/s0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- 13.Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leach RN, Desai JC, Orchard CH. Effect of cytoskeleton disruptors on L-type ca channel distribution in rat ventricular myocytes. Cell Calcium. 2005;38:515–526. doi: 10.1016/j.ceca.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res. 2007;313:2063–2076. doi: 10.1016/j.yexcr.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 16.Hollrigl A, Hofner M, Stary M, Weitzer G. Differentiation of cardiomyocytes requires functional serine residues within the amino-terminal domain of desmin. Differentiation. 2007;75:616–626. doi: 10.1111/j.1432-0436.2007.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol. 2010;11:75–81. doi: 10.1038/nrm2818. [DOI] [PubMed] [Google Scholar]
- 19.Saffitz JE. Adhesion molecules: Why they are important to the electrophysiologist. J Cardiovasc Electrophysiol. 2006;17:225–229. doi: 10.1111/j.1540-8167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 20.Wasinger VC, Cordwell SJ, Cerpa-Poljak A, Yan JX, Gooley AA, Wilkins MR, Duncan MW, Harris R, Williams KL, Humphery-Smith I. Progress with gene-product mapping of the mollicutes: Mycoplasma genitalium. Electrophoresis. 1995;16:1090–1094. doi: 10.1002/elps.11501601185. [DOI] [PubMed] [Google Scholar]
- 21.Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. FEBS Lett. 2010;584:2526–2538. doi: 10.1016/j.febslet.2010.04.044. [DOI] [PubMed] [Google Scholar]
- 22.Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics. 2006;5:608–619. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomes AV, Venkatraman G, Davis JP, Tikunova SB, Engel P, Solaro RJ, Potter JD. Cardiac troponin T isoforms affect the ca(2+) sensitivity of force development in the presence of slow skeletal troponin I: Insights into the role of troponin T isoforms in the fetal heart. J Biol Chem. 2004;279:49579–49587. doi: 10.1074/jbc.M407340200. [DOI] [PubMed] [Google Scholar]
- 25.Gorg A, Drews O, Luck C, Weiland F, Weiss W. 2-DE with IPGs. Electrophoresis. 2009;30(Suppl 1):S122–32. doi: 10.1002/elps.200900051. [DOI] [PubMed] [Google Scholar]
- 26.Righetti PG. Electrophoresis: The march of pennies, the march of dimes. J Chromatogr A. 2005;1079:24–40. doi: 10.1016/j.chroma.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 27.Wells L, Hart GW. O-GlcNAc turns twenty: Functional implications for post-translational modification of nuclear and cytosolic proteins with a sugar. FEBS Lett. 2003;546:154–158. doi: 10.1016/s0014-5793(03)00641-0. [DOI] [PubMed] [Google Scholar]
- 28.Zachara NE. Detecting the “O-GlcNAc-ome”; detection, purification, and analysis of O-GlcNAc modified proteins. Methods Mol Biol. 2009;534:251–279. doi: 10.1007/978-1-59745-022-5_19. [DOI] [PubMed] [Google Scholar]
- 29.Nita-Lazar A, Saito-Benz H, White FM. Quantitative phosphoproteomics by mass spectrometry: Past, present, and future. Proteomics. 2008;8:4433–4443. doi: 10.1002/pmic.200800231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemeer S, Heck AJ. The phosphoproteomics data explosion. Curr Opin Chem Biol. 2009;13:414–420. doi: 10.1016/j.cbpa.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 31.Grimsrud PA, Swaney DL, Wenger CD, Beauchene NA, Coon JJ. Phosphoproteomics for the masses. ACS Chem Biol. 2010;5:105–119. doi: 10.1021/cb900277e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards AV, White MY, Cordwell SJ. The role of proteomics in clinical cardiovascular biomarker discovery. Mol Cell Proteomics. 2008;7:1824–1837. doi: 10.1074/mcp.R800007-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Ping P. Getting to the heart of proteomics. N Engl J Med. 2009;360:532–534. doi: 10.1056/NEJMcibr0808487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Didangelos A, Simper D, Monaco C, Mayr M. Proteomics of acute coronary syndromes. Curr Atheroscler Rep. 2009;11:188–195. doi: 10.1007/s11883-009-0030-x. [DOI] [PubMed] [Google Scholar]
- 35.Agnetti G, Kane LA, Guarnieri C, Caldarera CM, Van Eyk JE. Proteomic technologies in the study of kinases: Novel tools for the investigation of PKC in the heart. Pharmacol Res. 2007;55:511–522. doi: 10.1016/j.phrs.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faber MJ, Agnetti G, Bezstarosti K, Lankhuizen IM, Dalinghaus M, Guarnieri C, Caldarera CM, Helbing WA, Lamers JM. Recent developments in proteomics: Implications for the study of cardiac hypertrophy and failure. Cell Biochem Biophys. 2006;44:11–29. doi: 10.1385/CBB:44:1:011. [DOI] [PubMed] [Google Scholar]
- 37.Schoenhoff FS, Fu Q, Van Eyk JE. Cardiovascular proteomics: Implications for clinical applications. Clin Lab Med. 2009;29:87–99. doi: 10.1016/j.cll.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leach C, Shenolikar S, Brautigan DL. Phosphorylation of phosphatase inhibitor-2 at centrosomes during mitosis. J Biol Chem. 2003;278:26015–26020. doi: 10.1074/jbc.M300782200. [DOI] [PubMed] [Google Scholar]
- 39.Bisetto E, Di Pancrazio F, Simula MP, Mavelli I, Lippe G. Mammalian ATPsynthase monomer versus dimer profiled by blue native PAGE and activity stain. Electrophoresis. 2007;28:3178–3185. doi: 10.1002/elps.200700066. [DOI] [PubMed] [Google Scholar]
- 40.Neverova I, Van Eyk JE. Role of chromatographic techniques in proteomic analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815:51–63. doi: 10.1016/j.jchromb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Sanchez LM, Muntane J, de la Mata M, Rodriguez-Ariza A. Unraveling the S-nitrosoproteome: Tools and strategies. Proteomics. 2009;9:808–818. doi: 10.1002/pmic.200800546. [DOI] [PubMed] [Google Scholar]
- 43.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 44.Bjellqvist B, Ek K, Righetti PG, Gianazza E, Gorg A, Westermeier R, Postel W. Isoelectric focusing in immobilized pH gradients: Principle, methodology and some applications. J Biochem Biophys Methods. 1982;6:317–339. doi: 10.1016/0165-022x(82)90013-6. [DOI] [PubMed] [Google Scholar]
- 45.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 46.Gianazza E, Righetti PG. Immobilized pH gradients. Electrophoresis. 2009;30(Suppl 1):S112–21. doi: 10.1002/elps.200800641. [DOI] [PubMed] [Google Scholar]
- 47.Halligan BD, Ruotti V, Jin W, Laffoon S, Twigger SN, Dratz EA. ProMoST (protein modification screening tool): A web-based tool for mapping protein modifications on two-dimensional gels. Nucleic Acids Res. 2004;32:W638–44. doi: 10.1093/nar/gkh356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shirai A, Matsuyama A, Yashiroda Y, Hashimoto A, Kawamura Y, Arai R, Komatsu Y, Horinouchi S, Yoshida M. Global analysis of gel mobility of proteins and its use in target identification. J Biol Chem. 2008;283:10745–10752. doi: 10.1074/jbc.M709211200. [DOI] [PubMed] [Google Scholar]
- 49.Dorfleutner A, Cho Y, Vincent D, Cunnick J, Lin H, Weed SA, Stehlik C, Flynn DC. Phosphorylation of AFAP-110 affects podosome lifespan in A7r5 cells. J Cell Sci. 2008;121:2394–2405. doi: 10.1242/jcs.026187. [DOI] [PubMed] [Google Scholar]
- 50.Burre J, Wittig I, Schagger H. Non-classical 2-D electrophoresis. Methods Mol Biol. 2009;564:33–57. doi: 10.1007/978-1-60761-157-8_3. [DOI] [PubMed] [Google Scholar]
- 51.Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA, Chien KR. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 2010;106:695–704. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- 52.Miyagi M, Rao KC. Proteolytic 18O-labeling strategies for quantitative proteomics. Mass Spectrom Rev. 2007;26:121–136. doi: 10.1002/mas.20116. [DOI] [PubMed] [Google Scholar]
- 53.Righetti PG, Sebastiano R, Citterio A. Isotope-coded two-dimensional maps: Tagging with deuterated acrylamide and 2-vinylpyridine. Methods Mol Biol. 2008;424:87–99. doi: 10.1007/978-1-60327-064-9_8. [DOI] [PubMed] [Google Scholar]
- 54.Friedman DB. Quantitative proteomics for two-dimensional gels using difference gel electrophoresis. Methods Mol Biol. 2007;367:219–239. doi: 10.1385/1-59745-275-0:219. [DOI] [PubMed] [Google Scholar]
- 55.Santoni V, Molloy M, Rabilloud T. Membrane proteins and proteomics: Un amour impossible? Electrophoresis. 2000;21:1054–1070. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1054::AID-ELPS1054>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 56.Rabilloud T. Membrane proteins and proteomics: Love is possible, but so difficult. Electrophoresis. 2009;30(Suppl 1):S174–80. doi: 10.1002/elps.200900050. [DOI] [PubMed] [Google Scholar]
- 57.Kane LA, Yung CK, Agnetti G, Neverova I, Van Eyk JE. Optimization of paper bridge loading for 2-DE analysis in the basic pH region: Application to the mitochondrial subproteome. Proteomics. 2006;6:5683–5687. doi: 10.1002/pmic.200600267. [DOI] [PubMed] [Google Scholar]
- 58.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. doi: 10.1161/CIRCGENETICS.109.871236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horvatovich P, Hoekman B, Govorukhina N, Bischoff R. Multidimensional chromatography coupled to mass spectrometry in analysing complex proteomics samples. J Sep Sci. 2010;33:1421–1437. doi: 10.1002/jssc.201000050. [DOI] [PubMed] [Google Scholar]
- 60.Sandra K, Moshir M, D'hondt F, Tuytten R, Verleysen K, Kas K, Francois I, Sandra P. Highly efficient peptide separations in proteomics. part 2: Bi- and multidimensional liquid-based separation techniques. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1019–1039. doi: 10.1016/j.jchromb.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 61.Zhang X, Fang A, Riley CP, Wang M, Regnier FE, Buck C. Multi-dimensional liquid chromatography in proteomics--a review. Anal Chim Acta. 2010;664:101–113. doi: 10.1016/j.aca.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen H, Li X, Bieberich CJ, Frey DD. Reducing sample complexity in proteomics by chromatofocusing with simple buffer mixtures. Methods Mol Biol. 2008;424:187–203. doi: 10.1007/978-1-60327-064-9_16. [DOI] [PubMed] [Google Scholar]
- 63.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 64.Motoyama A, Yates JR., 3rd Multidimensional LC separations in shotgun proteomics. Anal Chem. 2008;80:7187–7193. doi: 10.1021/ac8013669. [DOI] [PubMed] [Google Scholar]
- 65.Chong BE, Yan F, Lubman DM, Miller FR. Chromatofocusing nonporous reversed-phase high-performance liquid chromatography/electrospray ionization time-of-flight mass spectrometry of proteins from human breast cancer whole cell lysates: A novel two-dimensional liquid chromatography/mass spectrometry method. Rapid Commun Mass Spectrom. 2001;15:291–296. doi: 10.1002/rcm.227. [DOI] [PubMed] [Google Scholar]
- 66.Sheng S, Chen D, Van Eyk JE. Multidimensional liquid chromatography separation of intact proteins by chromatographic focusing and reversed phase of the human serum proteome: Optimization and protein database. Mol Cell Proteomics. 2006;5:26–34. doi: 10.1074/mcp.T500019-MCP200. [DOI] [PubMed] [Google Scholar]
- 67.Gundry RL, Tchernyshyov I, Sheng S, Tarasova Y, Raginski K, Boheler KR, Van Eyk JE. Expanding the mouse embryonic stem cell proteome: Combining three proteomic approaches. Proteomics. 2010 doi: 10.1002/pmic.201000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wingren C, James P, Borrebaeck CA. Strategy for surveying the proteome using affinity proteomics and mass spectrometry. Proteomics. 2009;9:1511–1517. doi: 10.1002/pmic.200800802. [DOI] [PubMed] [Google Scholar]
- 69.Uhlen M. Affinity as a tool in life science. BioTechniques. 2008;44:649–654. doi: 10.2144/000112803. [DOI] [PubMed] [Google Scholar]
- 70.Forner F, Furlan S, Salvatori S. Mass spectrometry analysis of complexes formed by myotonic dystrophy protein kinase (DMPK) Biochim Biophys Acta. 2010;1804:1334–1341. doi: 10.1016/j.bbapap.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 71.Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, Van Eyk JE. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr Protoc Mol Biol. 2009 doi: 10.1002/0471142727.mb1025s88. Chapter 10: Unit10.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fu Z, Wang M, Gucek M, Zhang J, Wu J, Jiang L, Monticone RE, Khazan B, Telljohann R, Mattison J, Sheng S, Cole RN, Spinetti G, Pintus G, Liu L, Kolodgie FD, Virmani R, Spurgeon H, Ingram DK, Everett AD, Lakatta EG, Van Eyk JE. Milk fat globule protein epidermal growth factor-8: A pivotal relay element within the angiotensin II and monocyte chemoattractant protein-1 signaling cascade mediating vascular smooth muscle cells invasion. Circ Res. 2009;104:1337–1346. doi: 10.1161/CIRCRESAHA.108.187088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liska AJ, Shevchenko A. Expanding the organismal scope of proteomics: Cross-species protein identification by mass spectrometry and its implications. Proteomics. 2003;3:19–28. doi: 10.1002/pmic.200390004. [DOI] [PubMed] [Google Scholar]
- 74.Wright JC, Beynon RJ, Hubbard SJ. Cross species proteomics. Methods Mol Biol. 2010;604:123–135. doi: 10.1007/978-1-60761-444-9_9. [DOI] [PubMed] [Google Scholar]
- 75.Zhou M, Veenstra T. Mass spectrometry: M/z 1983-2008. BioTechniques. 2008;44:667–8. 670. doi: 10.2144/000112791. [DOI] [PubMed] [Google Scholar]
- 76.Tabet JC, Rebuffat S. Nobel prize 2002 for chemistry: Mass spectrometry and nuclear magnetic resonance. Med Sci (Paris) 2003;19:865–872. doi: 10.1051/medsci/20031989865. [DOI] [PubMed] [Google Scholar]
- 77.Pisitkun T, Hoffert JD, Yu MJ, Knepper MA. Tandem mass spectrometry in physiology. Physiology (Bethesda) 2007;22:390–400. doi: 10.1152/physiol.00025.2007. [DOI] [PubMed] [Google Scholar]
- 78.Raczynska ED, Gal JF, Maria PC, Zientara K, Szelag M. Application of FT-ICR-MS for the study of proton-transfer reactions involving biomolecules. Anal Bioanal Chem. 2007;389:1365–1380. doi: 10.1007/s00216-007-1508-4. [DOI] [PubMed] [Google Scholar]
- 79.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat Methods. 2007;4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Linscheid MW, Ahrends R, Pieper S, Kuhn A. Liquid chromatography-mass spectrometry-based quantitative proteomics. Methods Mol Biol. 2009;564:189–205. doi: 10.1007/978-1-60761-157-8_11. [DOI] [PubMed] [Google Scholar]
- 81.Wang M, You J, Bemis KG, Tegeler TJ, Brown DP. Label-free mass spectrometry-based protein quantification technologies in proteomic analysis. Brief Funct Genomic Proteomic. 2008;7:329–339. doi: 10.1093/bfgp/eln031. [DOI] [PubMed] [Google Scholar]
- 82.Aye TT, Scholten A, Taouatas N, Varro A, Van Veen TA, Vos MA, Heck AJ. Proteome-wide protein concentrations in the human heart. Mol Biosyst. 2010 doi: 10.1039/c004495d. [DOI] [PubMed] [Google Scholar]
- 83.Kline KG, Sussman MR. Protein quantitation using isotope-assisted mass spectrometry. Annu Rev Biophys. 2010;39:291–308. doi: 10.1146/annurev.biophys.093008.131339. [DOI] [PubMed] [Google Scholar]
- 84.Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Methods. 2003;29:124–130. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 85.Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, Cahill DJ, Emili A, Fitzgerald DJ, Maguire PB. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. 2004;103:2096–2104. doi: 10.1182/blood-2003-08-2804. [DOI] [PubMed] [Google Scholar]
- 86.Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;7:340–350. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- 87.Yocum AK, Chinnaiyan AM. Current affairs in quantitative targeted proteomics: Multiple reaction monitoring-mass spectrometry. Brief Funct Genomic Proteomic. 2009;8:145–157. doi: 10.1093/bfgp/eln056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cox DM, Zhong F, Du M, Duchoslav E, Sakuma T, McDermott JC. Multiple reaction monitoring as a method for identifying protein posttranslational modifications. J Biomol Tech. 2005;16:83–90. [PMC free article] [PubMed] [Google Scholar]
- 89.Domanski D, Murphy LC, Borchers CH. Assay development for the determination of phosphorylation stoichiometry using multiple reaction monitoring methods with and without phosphatase treatment: Application to breast cancer signaling pathways. Anal Chem. 2010;82:5610–5620. doi: 10.1021/ac1005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blackburn K, Goshe MB. Challenges and strategies for targeted phosphorylation site identification and quantification using mass spectrometry analysis. Brief Funct Genomic Proteomic. 2009;8:90–103. doi: 10.1093/bfgp/eln051. [DOI] [PubMed] [Google Scholar]
- 91.Boersema PJ, Mohammed S, Heck AJ. Phosphopeptide fragmentation and analysis by mass spectrometry. J Mass Spectrom. 2009;44:861–878. doi: 10.1002/jms.1599. [DOI] [PubMed] [Google Scholar]
- 92.Paradela A, Albar JP. Advances in the analysis of protein phosphorylation. J Proteome Res. 2008;7:1809–1818. doi: 10.1021/pr7006544. [DOI] [PubMed] [Google Scholar]
- 93.Rogers LD, Foster LJ. Phosphoproteomics--finally fulfilling the promise? Mol Biosyst. 2009;5:1122–1129. doi: 10.1039/b905580k. [DOI] [PubMed] [Google Scholar]
- 94.Thingholm TE, Larsen MR. The use of titanium dioxide micro-columns to selectively isolate phosphopeptides from proteolytic digests. Methods Mol Biol. 2009;527:57–66. doi: 10.1007/978-1-60327-834-8_5. xi. [DOI] [PubMed] [Google Scholar]
- 95.Block H, Maertens B, Spriestersbach A, Brinker N, Kubicek J, Fabis R, Labahn J, Schafer F. Immobilized-metal affinity chromatography (IMAC): A review. Methods Enzymol. 2009;463:439–473. doi: 10.1016/S0076-6879(09)63027-5. [DOI] [PubMed] [Google Scholar]
- 96.Gevaert K, Vandekerckhove J. Reverse-phase diagonal chromatography for phosphoproteome research. Methods Mol Biol. 2009;527:219–27. doi: 10.1007/978-1-60327-834-8_16. ix. [DOI] [PubMed] [Google Scholar]
- 97.McNulty DE, Annan RS. Hydrophilic interaction chromatography for fractionation and enrichment of the phosphoproteome. Methods Mol Biol. 2009;527:93–105. doi: 10.1007/978-1-60327-834-8_8. x. [DOI] [PubMed] [Google Scholar]
- 98.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 99.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, Eng JK, Martin DB, Nesvizhskii AI, Aebersold R. A guided tour of the trans-proteomic pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Creasy DM, Cottrell JS. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics. 2002;2:1426–1434. doi: 10.1002/1615-9861(200210)2:10<1426::AID-PROT1426>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 101.Bell AW, Deutsch EW, Au CE, Kearney RE, Beavis R, Sechi S, Nilsson T, Bergeron JJ. HUPO Test Sample Working Group. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat Methods. 2009;6:423–430. doi: 10.1038/nmeth.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 103.Babu GJ, Wheeler D, Alzate O, Periasamy M. Solubilization of membrane proteins for two-dimensional gel electrophoresis: Identification of sarcoplasmic reticulum membrane proteins. Anal Biochem. 2004;325:121–125. doi: 10.1016/j.ab.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 104.Helbig AO, Heck AJ, Slijper M. Exploring the membrane proteome--challenges and analytical strategies. J Proteomics. 2010;73:868–878. doi: 10.1016/j.jprot.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 105.Donoghue PM, Hughes C, Vissers JP, Langridge JI, Dunn MJ. Nonionic detergent phase extraction for the proteomic analysis of heart membrane proteins using label-free LC-MS. Proteomics. 2008;8:3895–3905. doi: 10.1002/pmic.200800116. [DOI] [PubMed] [Google Scholar]
- 106.Pan Y, Kislinger T, Gramolini AO, Zvaritch E, Kranias EG, MacLennan DH, Emili A. Identification of biochemical adaptations in hyper- or hypocontractile hearts from phospholamban mutant mice by expression proteomics. Proc Natl Acad Sci U S A. 2004;101:2241–2246. doi: 10.1073/pnas.0308174101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andersson KB, Birkeland JA, Finsen AV, Louch WE, Sjaastad I, Wang Y, Chen J, Molkentin JD, Chien KR, Sejersted OM, Christensen G. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol. 2009;47:180–187. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 108.Haghighi K, Gregory KN, Kranias EG. Sarcoplasmic reticulum ca-ATPase-phospholamban interactions and dilated cardiomyopathy. Biochem Biophys Res Commun. 2004;322:1214–1222. doi: 10.1016/j.bbrc.2004.07.164. [DOI] [PubMed] [Google Scholar]
- 109.Pan C, Olsen JV, Daub H, Mann M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol Cell Proteomics. 2009;8:2796–2808. doi: 10.1074/mcp.M900285-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Husberg C, Agnetti G, Nygaard S, Andersson KB, Christensen G, Van Eyk J. Abstract 2944: Activation of heat shock proteins and shifts in cytoskeleton composition in a cardiac-specific SERCA2 KO mouse. Circulation. 2009;120:S717. [Google Scholar]
- 111.Solaro RJ, Sheehan KA, Lei M, Ke Y. The curious role of sarcomeric proteins in control of diverse processes in cardiac myocytes. J Gen Physiol. 2010;136:13–19. doi: 10.1085/jgp.201010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. 2008;77:659–666. doi: 10.1093/cvr/cvm084. [DOI] [PubMed] [Google Scholar]
- 113.Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: The essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94:146–158. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- 114.Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem Biophys Res Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Solaro RJ, van der Velden J. Why does troponin I have so many phosphorylation sites? fact and fancy. J Mol Cell Cardiol. 2010;48:810–816. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ayaz-Guner S, Zhang J, Li L, Walker JW, Ge Y. In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry. 2009;48:8161–8170. doi: 10.1021/bi900739f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- 118.Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: A novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–1358. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 120.Lopaschuk GD, Rebeyka IM, Allard MF. Metabolic modulation: A means to mend a broken heart. Circulation. 2002;105:140–142. [PubMed] [Google Scholar]
- 121.Neubert D, Wojtczak AB, Lehninger AL. Purification and enzymatic identity of mitochondrial contraction-factors I and II. Proc Natl Acad Sci U S A. 1962;48:1651–1658. doi: 10.1073/pnas.48.9.1651. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 123.Elstner M, Andreoli C, Ahting U, Tetko I, Klopstock T, Meitinger T, Prokisch H. MitoP2: An integrative tool for the analysis of the mitochondrial proteome. Mol Biotechnol. 2008;40:306–315. doi: 10.1007/s12033-008-9100-5. [DOI] [PubMed] [Google Scholar]
- 124.Schwartz R, Ting CS, King J. Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 2001;11:703–709. doi: 10.1101/gr.gr-1587r. [DOI] [PubMed] [Google Scholar]
- 125.Kavazis AN, Alvarez S, Talbert E, Lee Y, Powers SK. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol Heart Circ Physiol. 2009;297:H144–52. doi: 10.1152/ajpheart.01278.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rich PR. The molecular machinery of keilin's respiratory chain. Biochem Soc Trans. 2003;31:1095–1105. doi: 10.1042/bst0311095. [DOI] [PubMed] [Google Scholar]
- 127.Fearnley IM, Carroll J, Walker JE. Proteomic analysis of the subunit composition of complex I (NADH:Ubiquinone oxidoreductase) from bovine heart mitochondria. Methods Mol Biol. 2007;357:103–125. doi: 10.1385/1-59745-214-9:103. [DOI] [PubMed] [Google Scholar]
- 128.Schilling B, Murray J, Yoo CB, Row RH, Cusack MP, Capaldi RA, Gibson BW. Proteomic analysis of succinate dehydrogenase and ubiquinol-cytochrome c reductase (complex II and III) isolated by immunoprecipitation from bovine and mouse heart mitochondria. Biochim Biophys Acta. 2006;1762:213–222. doi: 10.1016/j.bbadis.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 129.Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 130.Palmisano G, Sardanelli AM, Signorile A, Papa S, Larsen MR. The phosphorylation pattern of bovine heart complex I subunits. Proteomics. 2007;7:1575–1583. doi: 10.1002/pmic.200600801. [DOI] [PubMed] [Google Scholar]
- 131.Pocsfalvi G, Cuccurullo M, Schlosser G, Scacco S, Papa S, Malorni A. Phosphorylation of B14.5a subunit from bovine heart complex I identified by titanium dioxide selective enrichment and shotgun proteomics. Mol Cell Proteomics. 2007;6:231–237. doi: 10.1074/mcp.M600268-MCP200. [DOI] [PubMed] [Google Scholar]
- 132.Gottlieb RA. Identification of targets of phosphorylation in heart mitochondria. Methods Mol Biol. 2007;357:127–137. doi: 10.1385/1-59745-214-9:127. [DOI] [PubMed] [Google Scholar]
- 133.Kane LA, Youngman MJ, Jensen RE, Van Eyk JE. Phosphorylation of the F(1)F(o) ATP synthase beta subunit: Functional and structural consequences assessed in a model system. Circ Res. 2010;106:504–513. doi: 10.1161/CIRCRESAHA.109.214155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P, Wang W, Young GW, Wang Y, Korge P, Lotz C, Doran P, Liem DA, Apweiler R, Weiss JN, Duan H, Ping P. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McDonald T, Sheng S, Stanley B, Chen D, Ko Y, Cole RN, Pedersen P, Van Eyk JE. Expanding the subproteome of the inner mitochondria using protein separation technologies: One- and two-dimensional liquid chromatography and two-dimensional gel electrophoresis. Mol Cell Proteomics. 2006;5:2392–2411. doi: 10.1074/mcp.T500036-MCP200. [DOI] [PubMed] [Google Scholar]
- 136.McDonald TG, Van Eyk JE. Mitochondrial proteomics. undercover in the lipid bilayer. Basic Res Cardiol. 2003;98:219–227. doi: 10.1007/s00395-003-0417-8. [DOI] [PubMed] [Google Scholar]
- 137.Distler AM, Kerner J, Hoppel CL. Proteomics of mitochondrial inner and outer membranes. Proteomics. 2008;8:4066–4082. doi: 10.1002/pmic.200800102. [DOI] [PubMed] [Google Scholar]
- 138.Lukyanenko V, Chikando A, Lederer WJ. Mitochondria in cardiomyocyte Ca2+ signaling. Int J Biochem Cell Biol. 2009;41:1957–1971. doi: 10.1016/j.biocel.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 140.Fiedler B, Wollert KC. Interference of antihypertrophic molecules and signaling pathways with the Ca2+-calcineurin-NFAT cascade in cardiac myocytes. Cardiovasc Res. 2004;63:450–457. doi: 10.1016/j.cardiores.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 141.Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gonzalez DR, Treuer AV, Castellanos J, Dulce RA, Hare JM. Impaired S-nitrosylation of the ryanodine receptor due to xanthine oxidase activity contributes to calcium leak in heart failure. J Biol Chem. 2010 doi: 10.1074/jbc.M110.154948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: Insights from genetically modified mice. Circ Res. 2003;93:907–916. doi: 10.1161/01.RES.0000100390.68771.CC. [DOI] [PubMed] [Google Scholar]
- 144.Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA. PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic Res Cardiol. 2010;105:337–347. doi: 10.1007/s00395-010-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Burley DS, Ferdinandy P, Baxter GF. Cyclic GMP and protein kinase-G in myocardial ischaemia-reperfusion: Opportunities and obstacles for survival signaling. Br J Pharmacol. 2007;152:855–869. doi: 10.1038/sj.bjp.0707409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: Role of CaMKII. J Mol Cell Cardiol. 2010;48:322–330. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nediani C, Raimondi L, Borchi E, Cerbai E. No/ros generation and Nitroso/redox imbalance in heart failure: From molecular mechanisms to therapeutic implications. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3198. [DOI] [PubMed] [Google Scholar]
- 148.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 149.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 150.Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: Malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. doi: 10.1172/JCI38027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Francalanci P, Gallo P, Bernucci P, Silver MD, d'Amati G. The pattern of desmin filaments in myocardial disarray. Hum Pathol. 1995;26:262–266. doi: 10.1016/0046-8177(95)90055-1. [DOI] [PubMed] [Google Scholar]
- 152.Gard JJ, Yamada K, Green KG, Eloff BC, Rosenbaum DS, Wang X, Robbins J, Schuessler RB, Yamada KA, Saffitz JE. Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc Res. 2005;67:539–547. doi: 10.1016/j.cardiores.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 153.Maloyan A, Gulick J, Glabe CG, Kayed R, Robbins J. Exercise reverses preamyloid oligomer and prolongs survival in {alpha}B-crystallin-based desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2007;104:5995–6000. doi: 10.1073/pnas.0609202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Otten E, Asimaki A, Maass A, van Langen IM, Wal AV, de Jonge N, van den Berg MP, Saffitz JE, Wilde AA, Jongbloed JD, van Tintelen JP. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm. 2010 doi: 10.1016/j.hrthm.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 155.Agnetti G, Bezstarosti K, Dekkers DH, Verhoeven AJ, Giordano E, Guarnieri C, Caldarera CM, Van Eyk JE, Lamers JM. Proteomic profiling of endothelin-1-stimulated hypertrophic cardiomyocytes reveals the increase of four different desmin species and alpha-B-crystallin. Biochim Biophys Acta. 2008;1784:1068–1076. doi: 10.1016/j.bbapap.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Panagopoulou P, Davos CH, Milner DJ, Varela E, Cameron J, Mann DL, Capetanaki Y. Desmin mediates TNF-alpha-induced aggregate formation and intercalated disk reorganization in heart failure. J Cell Biol. 2008;181:761–775. doi: 10.1083/jcb.200710049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Agnetti G, Halperin-Kuhns V, Guo Y, Sheng S, Fu Z, Nicolini F, Gherli T, Guarnieri C, Caldarera CM, Kass DA, Tomaselli GF, Van Eyk JE. Abstract 1809: Novel human and canine desmin phosphorylations as potential regulators of oligomer formation and disease in heart failure. Circulation. 2009;120:S559–c. [Google Scholar]
- 158.Faber MJ, Dalinghaus M, Lankhuizen IM, Bezstarosti K, Dekkers DH, Duncker DJ, Helbing WA, Lamers JM. Proteomic changes in the pressure overloaded right ventricle after 6 weeks in young rats: Correlations with the degree of hypertrophy. Proteomics. 2005;5:2519–2530. doi: 10.1002/pmic.200401313. [DOI] [PubMed] [Google Scholar]
- 159.Huang X, Li J, Foster D, Lemanski SL, Dube DK, Zhang C, Lemanski LF. Protein kinase C-mediated desmin phosphorylation is related to myofibril disarray in cardiomyopathic hamster heart. Exp Biol Med (Maywood) 2002;227:1039–1046. doi: 10.1177/153537020222701113. [DOI] [PubMed] [Google Scholar]
- 160.Corbett JM, Why HJ, Wheeler CH, Richardson PJ, Archard LC, Yacoub MH, Dunn MJ. Cardiac protein abnormalities in dilated cardiomyopathy detected by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis. 1998;19:2031–2042. doi: 10.1002/elps.1150191123. [DOI] [PubMed] [Google Scholar]
- 161.Rappaport L, Contard F, Samuel JL, Delcayre C, Marotte F, Tome F, Fardeau M. Storage of phosphorylated desmin in a familial myopathy. FEBS Lett. 1988;231:421–425. doi: 10.1016/0014-5793(88)80863-9. [DOI] [PubMed] [Google Scholar]
- 162.Bar H, Goudeau B, Walde S, Casteras-Simon M, Mucke N, Shatunov A, Goldberg YP, Clarke C, Holton JL, Eymard B, Katus HA, Fardeau M, Goldfarb L, Vicart P, Herrmann H. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat. 2007;28:374–386. doi: 10.1002/humu.20459. [DOI] [PubMed] [Google Scholar]
- 163.Di Somma S, Di Benedetto MP, Salvatore G, Agozzino L, Ferranti F, Esposito S, La Dogana P, Scarano MI, Caputo G, Cotrufo M, Santo LD, de Divitiis O. Desmin-free cardiomyocytes and myocardial dysfunction in end stage heart failure. Eur J Heart Fail. 2004;6:389–398. doi: 10.1016/j.ejheart.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 164.Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klovekorn WP, Schlepper M, Schaper W, Schaper J. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86:846–853. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- 165.Han G, Ye M, Liu H, Song C, Sun D, Wu Y, Jiang X, Chen R, Wang C, Wang L, Zou H. Phosphoproteome analysis of human liver tissue by long-gradient nanoflow LC coupled with multiple stage MS analysis. Electrophoresis. 2010;31:1080–1089. doi: 10.1002/elps.200900493. [DOI] [PubMed] [Google Scholar]
- 166.Nelson WJ, Traub P. Proteolysis of vimentin and desmin by the Ca2+-activated proteinase specific for these intermediate filament proteins. Mol Cell Biol. 1983;3:1146–1156. doi: 10.1128/mcb.3.6.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Inagaki M, Gonda Y, Matsuyama M, Nishizawa K, Nishi Y, Sato C. Intermediate filament reconstitution in vitro. the role of phosphorylation on the assembly-disassembly of desmin. J Biol Chem. 1988;263:5970–5978. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.