

Abstract
Deficiencies in prolyl hydroxylase domain proteins (PHDs) may lead to the accumulation of hypoxia-inducible factor-α proteins, the latter of which activate local angiogenic responses by paracrine mechanisms. Here, we investigate whether a keratinocyte-specific PHD deficiency may promote vascular survival and growth in a distantly located ischemic tissue by a remote signaling mechanism. We generated mice that carry a keratinocyte-specific Phd2 knockout (kPhd2KO) and performed femoral artery ligation. Relative to wild-type controls, kPhd2KO mice displayed improved vascular survival and arteriogenesis in ischemic hind limbs, leading to the accelerated recovery of hindlimb perfusion and superior muscle regeneration. Similar protective effects were also seen in type 1 and type 2 diabetic mice. Molecularly, both abundance of hypoxia-inducible factor-1α protein and expression of vascular endothelial growth factor-A were increased in epidermal tissues of kPhd2KO mice, accompanied by increased plasma concentration of vascular endothelial growth factor-A. Contrary to kPhd2KO mice, which are PHD2 deficient in all skin tissues, localized kPhd2KO in hindlimb skin tissues did not have similar effects, excluding paracrine signaling as a major mechanism. Confirming the existence of remote effects, hepatocyte-specific Phd2 knockout also protected hind limbs from ischemia injury. These data indicate that vascular survival and growth in ischemia-injured tissue may be stimulated by suppressing PHD2 in a remotely located tissue and may provide highly effective angiogenesis therapies without the need for directly accessing target tissues.
Disrupted arterial blood flow leads to ischemia diseases such as myocardial infarction and gangrene, which are devastating disorders with high rates of morbidity and mortality.1–3 In recent years, the incidence of ischemia diseases has been on the rise because of the aging population and increased prevalence of diabetes mellitus.3–5 Thus, medical needs are urgent for novel vascular therapies directed at restoring blood flow to affected tissues.6–8 Some examples of angiogenesis therapies under development include local or systemic administration of angiogenic factors and cell therapies that use bone marrow– or peripheral blood–derived mononuclear cells.7 Despite intensive efforts, successful clinical applications remain to be found.8
Prolyl hydroxylase domain proteins (PHDs) may represent a novel class of targets for angiogenesis therapy.9–11 PHD1, PHD2, and PHD3 are Fe++ and 2-oxoglutarate–dependent dioxygenases present in both the cytoplasm and nuclei.12–16 In addition, a related transmembrane protein termed P4HTM is present in the endoplasmic reticulum.17,18 In adequately oxygenated cells, PHDs use oxygen as a substrate to hydroxylate two specific prolyl residues in hypoxia inducible factor (HIF)-α proteins, tagging them for von Hippel Lindau protein–dependent polyubiquitination and proteasomal degradation.12–14
HIF-1α and HIF-2α are transcription factors that accumulate under hypoxia and form transcriptionally active heterodimers with the HIF-1β subunit.19–24 HIF heterodimers activate the transcription of a large number of target genes which encode proteins involved in hypoxia adaptation, including glycolytic enzymes,25 erythropoietin,26–28 and angiogenic factors.20,29 A prototypic example of hypoxia-induced angiogenic factor is the vascular endothelial growth factor (VEGF)-A that mediates vascular endothelial cell (EC) survival, proliferation, and migration mostly by activating VEGF receptor-2.20,30
To date, angiogenesis in response to tissue hypoxia or HIF-α overexpression has been studied within the framework of paracrine signaling mechanisms wherein hypoxia-induced angiogenic factors interact with cognate cell surface receptors on nearby ECs.31,32 In this study, we demonstrate that keratinocyte-specific Phd2 knockout (kPhd2KO) promotes vascular survival, arteriogenic remodeling, and muscle regeneration in a mouse hindlimb ischemia model. We present compelling evidence that such effects are mediated by a humoral mechanism wherein elevated plasma concentration of VEGF-A may at least partially contribute to the beneficial effects of skin Phd2 knockout.
Materials and Methods
Mice
All animal procedures were approved by the Animal Care Committee at the University of Connecticut Health Center in compliance with Animal Welfare Assurance. Phd2f/f/KRT14CreERT, Phd2f/f/Phd3f/f/KRT14CreERT, and Phd1f/f/Phd2f/f/Phd3f/f/KRT14CreERT mice were generated by crossing floxed (f) Phd mice33 with a transgenic line expressing CreERT under the control of the human keratin 14 promoter (KRT14CreERT mice34; The Jackson Laboratory, Bar Harbor, ME; stock number 005107). Unless indicated otherwise, disruption of floxed alleles was induced at 6 to 7 weeks of age by oral gavage of tamoxifen (Sigma-Aldrich, St. Louis, MO), performed daily for 5 days at 80 mg/kg of body weight. After tamoxifen treatment, Phd2f/f/KRT14CreERT, Phd2f/f/Phd3f/f/KRT14CreERT, and Phd1f/f/Phd2f/f/Phd3f/f/KRT14CreERT mice were designated as kPhd2KO, kDKO, and kTKO mice, respectively. Phd2f/f mice were treated in parallel as de facto wild-type (WT) control.
To induce localized Phd2 knockout in hindlimb skin tissues, 4-hydroxytamoxifen (4-OHT; Sigma-Aldrich) was applied topically to left thigh skin. In brief, the left thigh was shaved, and a single dose of 4-OHT (2 μL at 5 mg/mL in dimethyl sulfoxide) was directly applied to the shaved skin surface. After 10 minutes, 4-OHT was completely cleaned away with saline. The resulting mice were designated as kPhd2KOlh mice to indicate localized knockout in the left hind limb.
Phd2f/f/AlbCre (AlbPhd2KO) mice were generated by crossing Phd2-floxed mice with AlbCre mice35 (The Jackson Laboratory; stock number 003574), which express constitutively active Cre under the control of the mouse albumin enhancer/promoter.
All genotypes were determined by PCR of ear punch DNA with the use of primers listed in Table 1.
Table 1.
PCR Primers for Genotyping
| Gene/transgene | Forward primer | Reverse primer |
|---|---|---|
| Phd1 | 5′-CTCTAACAAGCACAAACCATCATT-3′ | 5′-ACTGGTGAAGCCTGTAGCCTGTC-3′ |
| Phd2 | 5′-AACTCCGCCAAGCAGGTCAGAA-3′ | 5′-TCAACTCGAGCTGGAAACC-3′ |
| Phd3 | 5′-CTCTTCCTTCCTTCCTCTTAG-3′ | 5′-CCACGTTAACTCTAGAGCCACTGA-3′ |
| KRT14CreERT | 5′-GCGCCTGAGCTGCCCACACTA-3′ | 5′-CACCCCAACATCCCCATCAAAGAG-3′ |
| AlbCre | 5′-GGCTGGACCAATGTAAATA-3′ | 5′-GCTGAGTGGTTCGGGGAGTT-3′ |
FAL and Laser Doppler Imaging
Femoral artery ligation (FAL) was performed on the left leg of 10- to 12-week-old male mice or at indicated ages for specific experiments. Under anesthesia, the femoral artery was ligated distal to the inguinal ligament and proximal to the saphenous-popliteal bifurcation. After all side branches were occluded by electrical cautery, the femoral artery was separated from muscles, and the ligated segment was excised. Skin incisions were closed with a 6-0 nylon suture.
Hindlimb blood flow in the paw area was measured with a Moor Infrared Laser Doppler Imager (Moor Instruments, Wilmington, DE) immediately before and after FAL (pre and day 0, respectively) and at various time points thereafter (days 3, 7, 14, and 28).
Histological Analysis
Hindlimb muscle tissues were fixed for 24 hours in zinc fixative (100 mmol/L Tris-HCl, pH7.4, 0.5 mg/mL calcium acetate, 5 mg/mL zinc acetate, and 5 mg/mL zinc chloride), and equilibrated overnight in 30% sucrose. Fixed and equilibrated tissues were embedded in OTC compound and cut at 10 μm. Skin tissues were fixed in the same manner, embedded in paraffin after dehydration, and cut at 5 μm. Sections were used for H&E and Gomori trichrome staining, or immunohistochemical (IHC) and immunofluorescence (IF) staining. For IHC or IF staining, sections were subject to antigen retrieval by heating in Citrate Buffer and were blocked with Power Block Universal Blocking Reagent (BioGenex, San Roman, CA).
For IHC staining, sections were incubated with anti–HIF-1α (Novus Biologicals, Littleton, CO; NB100-134), anti-CD31 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-1506), or anti-CD45 (BioLegend, San Diego, CA; number 103101). Signals were detected by staining with biotinylated secondary antibodies in conjunction with avidin-biotin complex and diaminobenzidine kits (Vector Laboratories, Burlingame, CA). For IF staining, sections were incubated with each of the following pairs of primary and secondary antibodies: anti-CD31 (Santa Cruz Biotechnology; sc-1506) and Alexa Fluor 488–conjugated anti-goat IgG (Life Technologies, Carlsbad, CA; A-11055), anti-Ki67 (BioLegend; number 652401) and cyanine 3–conjugated anti-rat IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, PA; 712-166-153), and anti–activated caspase 3 (Abcam, Cambridge, MA; ab13847) and Alexa Fluor 594–conjugated anti-rabbit IgG (Life Technologies; A-11072). IHC- or IF-stained sections were visualized by bright field light microscopy or laser confocal microscopy, respectively, and quantified with the assistance of ImageJ software version 1.46e (NIH, Bethesda, MD).
For whole-mount staining, skin specimens were digested with 3 mg/mL dispase II (Roche Diagnostics, Mannheim, Germany), and dermal and epidermal layers were physically separated. Isolated dermal tissues were fixed in 4% paraformaldehyde for 1 hour and subsequently in acetone for 20 minutes. Fixed dermal tissues were double stained with Alexa Fluor 488–conjugated anti-mouse CD31 antibody (BioLegend; 102513) and allophycocyanin-conjugated anti-mouse CD11b antibody (BioLegend; 101211). Stained specimens were mounted on slides and examined by confocal laser microscopy. Results were quantified with the assistance of ImageJ software.
Molecular Analysis
RT-PCR and real-time quantitative PCR (qPCR) analyses were performed for RNA samples isolated from the epidermal layer of skin tissues. Primer sequences for RT-PCR and qPCR are listed in Tables 2 and 3, respectively. All qPCR signal intensities were normalized against that of beta-actin. VEGF-A protein levels in skin lysates and plasma were determined by adapting the manufacturer's instructions of a VEGF-A ELISA kit (R&D Systems, Emeryville, CA).
Table 2.
RT-PCR Primers
| mRNA | Forward primer | Reverse primer |
|---|---|---|
| Phd1 | 5′-ACCGCGCAGCATTCGTG-3′ | 5′-GGGGCTGGCCATTAGGTAGGTGTA-3′ |
| Phd2 | 5′-GCCCGGCTGCGAAACCATC-3′ | 5′-TCGCTCGCTCATCTGCATCAAAAT-3′ |
| Phd3 | 5′-CTGCGTGCTGGAGCGAGTCAA-3′ | 5′-TCATGTGGATTCCTGCGGTCTG-3′ |
Table 3.
qPCR Primers
| mRNA | Forward primer | Reverse primer |
|---|---|---|
| Phd1 | 5′-CATCAATGGGCGCACCA-3′ | 5′-GATTGTCAACATGCCTCACGTAC-3′ |
| Phd2 | 5′-TAAACGGCCGAACGAAAGC-3′ | 5′-GGGTTATCAACGTGACGGACA-3′ |
| Phd3 | 5′-CTATGTCAAGGAGCGGTCCAA-3′ | 5′-GTCCACATGGCGAACATAACC-3′ |
| VEGF-A | 5′-GCACATAGGAGAGATGAGCTTCC-3′ | 5′-CTCCGCTCTGAACAAGGCT-3′ |
| PlGF | 5′-CCTGTCTGCTGGGAACAACTCA-3′ | 5′-CACCTCATCAGGGTATTCATCCAAG-3′ |
| TNFα | 5′-AAGCCTGTAGCCCACGTCGTA-3′ | 5′-GGCACCACTAGTTGGTTGTCTTTG-3′ |
| Ang1 | 5′-CACATAGGGTGCAGCAACCA-3′ | 5′-CGTCGTGTTCTGGAAGAATGA-3′ |
| Ang2 | 5′-CATCTGCAAGTGTTCCCAGA-3′ | 5′-AAGTTGGAAGGACCACATGC-3′ |
| PDGFB | 5′-CAAAGGCAAGCACCGAAAGTTTA-3′ | 5′-CCGAATCAGGCATCGAGACA-3′ |
| SDF1 | 5′-CAGCCGTGCAACAATCTGAAG-3′ | 5′-CTGCATCAGTGACGGTAAACC-3′ |
| IL-1b | 5′-CTGTGACTCATGGGATGATGATG-3′ | 5′-GCCTGTAGTGCAGTTGTCTAAT-3′ |
| Ccl-2 | PPM03151F (Qiagen) | PPM03151F (Qiagen) |
| β-actin | 5′-GGCTGTATTCCCCTCCATCG-3′ | 5′-CCAGTTGGTAACAATGCCATGT-3′ |
Ang, angiopoietin 5; Ccl-2, chemokine (C-C motif) ligand 2; PlGF, placental growth factor; PDGFB, platelet-derived growth factor B; SDF1, stromal cell derived factor 1; TNFα, tumor necrosis factor α.
Induction of Diabetes
To induce type 1 diabetes, 6- to 7-week-old male Phd2f/f/KRT14CreERT and Phd2f/f mice were deprived of food for 4 hours and injected intraperitoneally with a single dose of streptozotocin (STZ; Sigma-Aldrich; 150 mg/kg in citrate buffer, pH 4.5). Four days after STZ injection, mice with ad libitum blood glucose concentrations >400 mg/dL were subject to further experimentation. Oral gavage of tamoxifen was performed 3 weeks after STZ injection, and FAL was performed after another 7 weeks.
Type 2 diabetes-like symptoms were induced by high-fat diet (HFD; 60/Fat Adjusted Calorie Diet; Harlan Laboratories, Inc., Indianapolis, IN; catalog number TD.06414). Specifically, tamoxifen-treated Phd2f/f (WT) and kPhd2KO male mice were maintained on HFD for 4 months, starting from 7 weeks of age, and were subjected to FAL afterward.
Statistical Analysis
Statistical differences were analyzed by two-tailed Student's t-tests or two-way analysis of variance (ANOVA). Data are shown as means ± SEM. All n values refer to the number of mice per group. P < 0.05 was considered to be statistically significant. Perfusion mode values were identified by plotting histogram of mice in different ranges of left-to-right perfusion ratio. Correlation between perfusion at day 3 and gastrocnemius muscle weight at day 28 was evaluated by calculating coefficient of determination R2 value of a linear-fitted curve.
Results
Accelerated Recovery of Hindlimb Perfusion in kPhd2KO Mice after FAL
Keratinocyte specific Phd2 knockout was induced by oral gavage of tamoxifen in 6- to 7-week-old Phd2f/f/KRT14CreERT mice. Efficient epidermal Phd2 disruption was confirmed by genomic PCR and RT-PCR (Supplemental Figure S1, A and B). In kPhd2KO mice, Phd2-specific primers detected only the deleted allele, whereas Phd1- or Phd3-specific primers detected only WT bands. According to qPCR, epidermal Phd2 mRNA in kPhd2KO mice was reduced to 7.8% of floxed (WT) controls, whereas Phd3 mRNA was significantly increased (Supplemental Figure S1C). No targeted Phd2 band (del) was present in the hindlimb skeletal muscle and a variety of other organs and tissues, including the heart, lung, kidney, liver, and spleen (Supplemental Figure S2). Thus, in kPhd2KO mice, efficient Phd2 knockout occurred exclusively in the epidermal tissues.
To assess the effect of epidermal PHD2 deficiency on arteriogenesis in muscular tissues, we induced hindlimb ischemia by FAL in the left leg of 10-week-old Phd2f/f (WT) and kPhd2KO mice and monitored blood flow by laser Doppler imaging over a 4-week time frame. Hindlimb perfusion recovered significantly faster in the kPhd2KO group, with the left-to-right hindlimb perfusion ratio reaching 0.65 by day 28 after FAL, whereas WT mice had an average ratio of 0.33 (Figure 1, A and B).
Figure 1.

Accelerated recovery of hindlimb perfusion in kPhd2KO mice. A and B: Measurement of blood perfusion by laser Doppler imaging in WT (white bars) and kPhd2KO (black bars) mice. C: Left-to-right perfusion ratios for individual mice are plotted for indicated time points, with average values for each group indicated at the top. D: Correlation analysis between left-to-right perfusion ratio on day 3 and gastrocnemius muscle weight on day 28. E: Histogram for perfusion ratio distribution on day 3. Values below the x axis are different ranges of left-to-right perfusion ratios, with modal (most frequent) ranges indicated by arrows. n = 10 (A, B, and D, WT); n = 7 (A, B, and D, kPhd2KO); n = 21 (C and E, WT); n = 15 (C and E, kPhd2KO). ∗P < 0.05 by Student's t-tests (C); ††P < 0.01 by ANOVA (A and B). Day 0, immediately after FAL; Pre, immediately before FAL.
To determine whether improved perfusion occurred as an early time point after FAL, we compared left-to-right perfusion ratios at day 3 after FAL by a dot plotting method. As shown in Figure 1C, although WT and kPhd2KO mice had the same low perfusion ratio of 0.07 immediately after FAL (day 0), kPhd2KO mice recovered much faster, with perfusion ratio reaching 0.15 by day 3, whereas the average perfusion ratio in the WT group was significantly lower (Figure 1C). Hindlimb perfusion on day 3 was well correlated to calf muscle weight on day 28 (R2 = 0.6426) (Figure 1D), indicating that higher perfusion rates at earlier stages supported subsequent muscle regeneration.
However, perfusion values did vary substantially among different mice within the same group. Hence, we generated a histogram by plotting number of mice versus different perfusion ranges. This effort identified perfusion ranges that contained the largest number of mice for each group (mode values), which were 0.06 to 0.10 in WT and 0.16 to 0.20 in kPhd2KO mice (Figure 1E). In subsequent studies, mice within modal ranges were further examined by histological and molecular approaches.
Improved Microvascular Survival, Arteriogenesis, and Hindlimb Muscle Protection in kPhd2KO Mice
We performed histological analyses of adductor muscle cross-sections 3 days after FAL. By anti–active caspase 3 IHC staining, widespread myocyte apoptosis was detected in the WT group but not in kPhd2KO mice (Figure 2, A and B). Similar results were found for capillary ECs by double IF staining for CD31 and active caspase 3 (Figure 2, C and D). Because regeneration of ischemia-injured tissues typically involves active cell proliferation to replace lost tissues, we also examined cell proliferation in hindlimb tissues by anti-CD31 and anti-Ki67 IF staining. Proliferative ECs were abundantly present in WT mice (Figure 2, E and F), which was suggestive of an adaptive response to severe tissue ischemia and capillary loss. In kPhd2KO mice, however, hindlimb tissues were mostly quiescent, and proliferative cells, including proliferative ECs, were scarce. When examined at day 28 after FAL, kPhd2KO mice were also found to display superior features over WT controls. In histological sections of semimembranosus muscles, arterial lumens were significantly larger in kPhd2KO mice (Figure 2, G and H). These findings indicated that keratinocyte-specific Phd2 knockout protected thigh microvessels and muscle tissues from ischemia injury and improved subsequent arteriogenesis.
Figure 2.

Reduced ischemia injury and improved arterial growth in thigh muscles of kPhd2KO mice. A and B: Detection of apoptotic myocytes (brown) by anti–activated caspase 3 IHC staining. Adductor muscle cross-sections used in this assay were prepared 3 days after FAL. Images in the WT group were taken from necrotic areas. In kPhd2KO mice, necrotic areas were not found in adductor muscle sections. C and D: Anti-CD31 and anti–activated caspase 3 double IF staining of adductor muscles 3 days after FAL. Images in WT group were taken and quantified from areas adjacent to severely necrotic tissues. Images in kPhd2KO sections were taken from random areas. E and F: Anti-CD31 and anti-Ki67 IF staining of adductor muscle sections 3 days after FAL. Double-positive cells are indicated by arrows. Images were taken from similar areas as described for C and D. G: H&E-stained sections of semimembranosus muscles on day 28, with arterial lumens indicated by arrows. H: Measurements of arterial lumen diameters. n = 7 (A, B, G, and H); n = 3 (C–F). ∗P < 0.05, ∗∗P < 0.01 (B, D, F, and H) by Student's t-tests. Scale bars: 100 μm (A and B); 20 μm (C–F); 10 μm (G).
Differential Skin Angiogenesis among WT, kPhd2KO, kDKO, and kTKO Mice
In addition to PHD2, PHD1 and PHD3 are also known to participate in the regulation of HIF-α stability.16,36 To evaluate the effects of keratinocyte-specific Phd2/Phd3 double knockout and Phd1/Phd2/Phd3 triple knockout, we generated kDKO and kTKO mice by oral gavage of Phd2f/f/Phd3f/f/KRT14CreERT and Phd1f/f/Phd2f/f/Phd3f/f/KRT14CreERT mice, respectively (Supplemental Figure S1, A–C). In contrast to kPhd2KO mice that had indistinguishable external appearance from WT mice, both kDKO and kTKO mice displayed conspicuous skin redness (Figure 3A). In mice without any surgical operation, hindlimb perfusion in kDKO and kTKO but not kPhd2KO mice were significantly elevated from WT levels, likely reflecting increased perfusion in the skin (Figure 3B). Indeed, anti-CD31 IF staining of skin tissues showed multifold increases in vascular branching points in kDKO or kTKO mice (Figure 3C). Furthermore, significant infiltration of CD11b+ macrophages was found only in kDKO and kTKO mice (Figure 3C). Quantitative data of the above-mentioned observations are presented in Figure 3, D–F.
Figure 3.

Differential skin angiogenesis among WT, kPhd2KO, kDKO, and kTKO mice without operation by FAL. A: Paw redness in kDKO and kTKO, but not WT and kPhd2KO, mice 4 weeks after tamoxifen treatment. B: Laser Doppler imaging of paws. C: Whole-mount anti-CD31 (green) and anti-CD11b (red) double IF staining of the dermal layer of skin tissues. D: Blood perfusion values in different groups of mice relative to WT, the latter of which was set at onefold. E: Number of CD31+ vascular branching points per high power field. F: CD11b+ cells per unit area. n = 6 (A, B, and D); n = 3 (C, E, and F). ∗P < 0.05, ∗∗P < 0.01 (D–F) by Student's t-tests. Scale bars: 100 μm (C).
Facilitated Muscle Regeneration in kPhd2KO But Not kDKO or kTKO Mice
At 28 days after FAL, capillaries had clearly different distribution patterns between gastrocnemius muscle sections in WT and kPhd2KO mice. In WT mice, capillaries were irregularly clustered within necrotic tissues (Figure 4A). By contrast, capillaries in kPhd2KO mice were regularly intercalated between fully intact myocytes (Figure 4A). Thus, although the overall capillary densities were similar between WT and kPhd2KO mice, the distribution pattern in the latter appeared much healthier (Figure 4B). It is important to note that in mice without any surgical operation, vascular morphology in hindlimb muscles was similar between WT and kPhd2KO groups (Supplemental Figure S3, A–D), excluding the possibility that differential vascular morphologies were already present before surgery.
Figure 4.

Differential calf muscle regeneration and toe protection among WT, kPhd2KO, kDKO, and kTKO mice. All sections were from gastrocnemius muscles 28 days after FAL. A and B: Anti-CD31 staining and quantification of CD31+ capillaries. C and D: H&E-stained sections and quantification of areas occupied by healthy (non-necrotic) muscle bundles. Necrotic muscle tissues were excluded from quantification. Examples of non-necrotic muscle bundles are demarcated by dotted lines in C. E and F: Gomori trichrome-stained gastrocnemius muscle sections and quantification of fibrotic area (blue). G and H: Anti-CD45–stained sections and number of CD45+ cells (brown dots) per high power field. I: Paw and foot necrosis 28 days after FAL; blackened nails or nail loss are indicated by arrows. J: Necrosis scores from day 0 to day 28. n = 7 to 10 (A–J). ∗P < 0.05 (F and H), ∗∗P < 0.01 (D) by Student's t-tests; ††P < 0.01 (J) by ANOVA). Scale bars: 15 μm (A); 50 μm (C and G); 100 μm (E).
In H&E-stained cross-sections of the calf (gastrocnemius) muscle, intact muscle fibers occupied just more than one-half of the total areas in WT mice, with the rest of the area containing necrotic tissues. In kPhd2KO mice, intact muscle structures cover essentially the entire section areas (Figure 4, C and D). In sections stained with Gomori trichrome, 12.5% of total tissue area was found to be collagen-positive fibrotic scars in WT mice, whereas the corresponding value in kPhd2KO mice was only 1.1% (Figure 4, E and F). By anti-CD45 IHC staining, massive leukocyte infiltration was found in WT but not kPhd2KO mice (Figure 4, G and H).
In agreement with improved vascular and muscular integrity, there was virtually no foot necrosis in kPhd2KO mice that had undergone FAL. For quantification, we determined foot necrosis scores, defined as the number of toes with blackened or lost nails. As shown in Figure 4, I and J, the WT group had an average necrosis score of 2 between days 7 and 28, whereas the corresponding score in kPhd2KO mice remained at 0.
In contrast, kDKO and kTKO mice that had undergone FAL failed to display improved vascular and muscle integrity, displaying irregularly arranged microvessels in necrotic muscle tissues, conspicuous amounts of nonmuscular and fibrotic tissues, as well as massive leukocyte infiltration into ischemia-damaged hindlimb tissues (Figure 4, A–H). Consistent with these findings, toes in kDKO and kTKO mice were not resistant to ischemic damage (Figure 4, I and J).
Molecular Analysis and Evidence for a Remote Signaling Mechanism
To understand molecular mechanisms that regulate vascular growth, we investigated HIF-1α protein levels and angiogenic factor expression in epidermal tissues. IHC staining of epidermal sections indicated that HIF-1α protein levels progressively increased in the order of WT < kPhd2KO < kDKO < kTKO (Figure 5, A and B). qPCR also ranked epidermal VEGF-A mRNA abundance in the order of WT (1.0-fold) < kPhd2KO (2.3-fold) < kDKO (5.9-fold) < kTKO (18.8-fold) (Figure 5C). In addition to VEGF-A, we also performed qPCR for another 8 RNA species that encode various angiogenic factors, but none of them were increased in kPhd2KO mice, although placental growth factor was increased in kTKO mice (Figure 5C). VEGF-A ELISA of skin tissue lysates confirmed the same order of VEGF-A protein abundance as its mRNA levels, with WT having the lowest and kTKO having the highest levels of VEGF-A (Figure 5D). We also quantified plasma VEGF-A levels by ELISA. As shown in Figure 5E, circulating VEGF-A in kPhd2KO mice was increased by approximately 43.3% than in WT controls. Surprisingly, no increased VEGF-A was detected in plasma samples from kDKO or kTKO mice (Figure 5E).
Figure 5.

Molecular analysis for HIF-1α protein levels and angiogenic factor expression. A and B: Anti–HIF-1α IHC staining of epidermis sections and quantification of positive HIF-1α areas. C: qPCR of epidermal RNA from WT, kPhd2KO, kDKO, and kTKO mice 4 weeks after tamoxifen administration. All data, including WT values, were normalized against β-actin and shown as relative values to WT. D: VEGF-A ELISA for total skin lysates from WT, kPhd2KO, kDKO, and kTKO mice. Shown are picograms of VEGF-A per 50 mg total skin protein. E: Quantification of plasma VEGF-A in WT and kPhd2KO, kDKO, and kTKO mice by ELISA. n = 4 to 5 (A and B); n = 3 (C and E, kTKO); n = 4 (D and E, kDKO); n = 14 (E, WT), n = 5 (E, kPhd2KO). ∗P < 0.05, ∗∗P < 0.01 (B–E) by Student's t-tests. Scale bar = 10 μm.
We wondered if improved hindlimb recovery in kPhd2KO mice was due to paracrine actions of angiogenic factors secreted from adjacent PHD2-deficient hindlimb skins. To address this issue, we induced Phd2 disruption in left hindlimb skin tissues by topical application of 4-OHT to Phd2f/f/KRT14CreERT mice (Supplemental Figure S4A), resulting in kPhd2KOlh mice whereby lh refers to left hind limb. PCR analysis revealed essentially complete Phd2 knockout in left hindlimb skin tissues in kPhd2KOlh mice but not from untreated skins anywhere else (Supplemental Figure S4B). After FAL, kPhd2KOlh mice failed to show any improved perfusion over WT mice (Figure 6, A and B), suggesting that paracrine mechanisms played negligible roles in accelerating arteriogenesis in kPhd2KO mice and that a remote effect might be involved.
Figure 6.
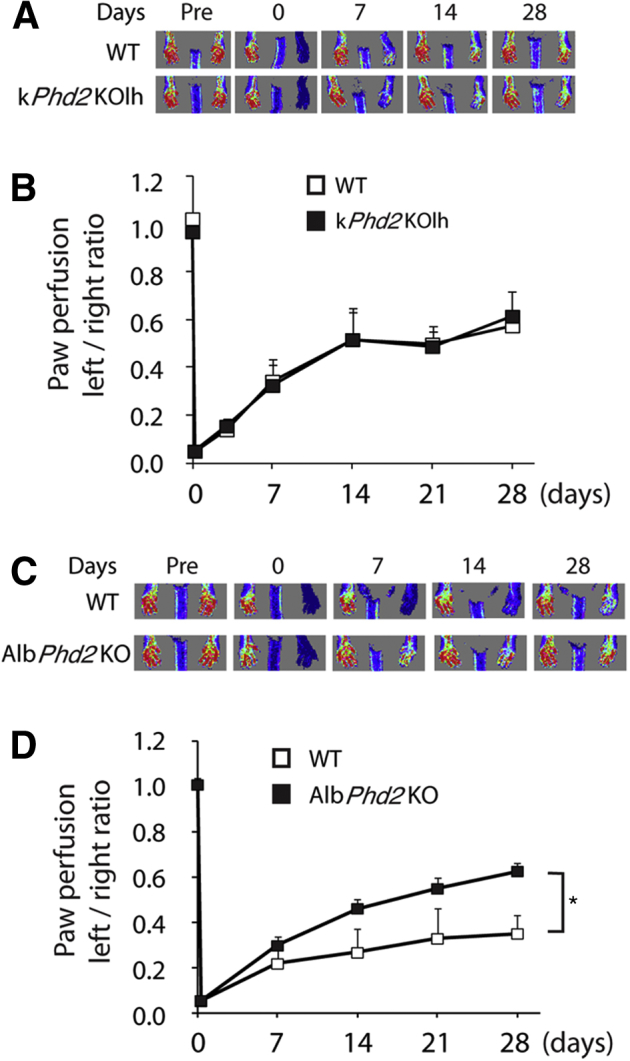
Evidence for the existence of a remote signaling mechanism. A: Measurement of paw perfusion by laser Doppler imaging in WT and kPhd2KOlh mice, both topically treated with 4-OHT on the left thigh. B: Left-to-right perfusion ratios in WT and kPhd2KOlh mice. C: Laser Doppler imaging for WT and AlbPhd2KO mice. D: Left-to-right perfusion ratios in WT and AlbPhd2KO mice. n = 5 (A, WT); n = 6 (A, kPhd2KOlh; C, AlbPhd2KO); n = 8 (C, WT). ∗P < 0.05 (B and D, by ANOVA). Day 0, immediately after FAL; Pre, immediately before FAL.
To further test the possible existence of a remote signaling mechanism, we investigated whether the recovery of hindlimb perfusion could be accelerated by PHD2 deficiency in a distantly located organ. We chose to perform this test in AlbPhd2KO mice, which were PHD2 deficient in hepatocytes (Supplemental Figure S5, A–C). Note the presence of the del band only in the liver (Supplemental Figure S5B). When housed under normal conditions, AlbPhd2KO mice were indistinguishable from floxed mice, and their liver histology, as well as vascular morphology, was not grossly changed (Supplemental Figure S5, D and E). After FAL, hindlimb perfusion recovered substantially faster in AlbPhd2KO mice (Figure 6, C and D). Given the vital function of the liver itself, it is unlikely that hepatic PHD2 per se would be a therapeutic target for angiogenesis; nonetheless, our findings indicate the existence of a remote trans-tissue mechanism whereby PHD2 deficiency in a specific tissue may improve vascular and tissue repair in a distant organ.
PHD2-Deficient Skin Protects Hind Limb under Diabetic Conditions
One of the most common pathological conditions that leads to vascular destruction and tissue ischemia is diabetes.37–39 To test if kPhd2KO improves ischemic tissue perfusion under diabetic conditions, we induced diabetes in WT and kPhd2KO mice with STZ. After FAL, hindlimb perfusion recovery was significantly faster in kPhd2KO mice (Figure 7A), accompanied by complete lack of toe necrosis (Figure 7B). Besides improving hindlimb perfusion and protecting toes from necrotic damages, kPhd2KO also helped maintain normal structural organization of epidermal tissues under prolonged hyperglycemia, whereas WT, kDKO, and kTKO mice all developed hyperkeratosis under similar conditions (Supplemental Figure S6).
Figure 7.

Accelerated perfusion recovery and superior toe protection in diabetic kPhd2KO mice. A: Laser Doppler images and quantification of left-to-right perfusion ratio in STZ-treated WT and kPhd2KO mice. B: Foot necrosis scores for STZ-treated WT and kPhd2KO mice between days 0 and 28 after FAL. C: Laser Doppler images and quantification of left-to-right perfusion ratios in HFD-treated WT and kPhd2KO mice. D: Foot necrosis scores in HFD-treated WT and kPhd2KO mice. n = 6 (A, WT); n = 7 (A, kPhd2KO; B, WT); n = 8 (B–D, kPhd2KO); n = 9 (C and D, WT). ∗P < 0.05. Day 0, immediately after FAL; Pre, immediately before FAL.
Because vascular repair is frequently compromised in patients with type 2 diabetes,40,41 we asked if keratinocyte-specific PHD2 deficiency might be beneficial in a mouse model of type 2 diabetes. We induced type 2 diabetes in kPhd2KO mice by HFD (Supplemental Figure S7, A and B) and performed FAL. As shown in Figure 7, C and D, kPhd2KO mice demonstrated accelerated perfusion recovery and negligible toe necrosis.
Discussion
A Possible Positive Feedback Cycle between Microvascular Survival and Arteriogenic Remodeling
Here, we provide evidence that keratinocyte-specific PHD2 deficiency may promote EC and myocyte viability and may accelerate vascular growth and muscle regeneration. Reduced number of proliferative and apoptotic ECs as early as 3 days after FAL raised the possibility that keratinocyte-specific PHD2 deficiency protected hindlimb microvascular ECs from ischemia-inflicted apoptosis. Dramatically improved microvascular viability early after surgery might accelerate subsequent arteriogenic remodeling by making microvascular building blocks more readily available. In turn, accelerated arteriogenic remodeling because of superior microvascular viability may moderate hindlimb ischemia, which may further improve EC viability. Thus, a positive feedback cycle might ensue, leading to the formation of larger arterial branches and improved myocyte survival and regeneration.
A Potential Role of Plasma VEGF-A
The precise mediator for the observed protective effects remains to be confirmed, but our data partially support a role for increased plasma VEGF-A. VEGF-A has established roles in angiogenesis, EC survival, and arteriogenesis, and its effects on vascular development in mouse embryos are known to be dose sensitive.42–44 Increased plasma concentration of VEGF-A in kPhd2KO mice agrees well with improved vascular integrity and accelerated perfusion recovery.
The failure of kDKO and kTKO to reduce ischemia damage correlates with the lack of increased plasma VEGF-A in these mice. It is unclear why there was a discrepancy between VEGF-A expression data in the skin and ELISA data for plasma samples, but dramatically increased microvessels in kDKO and kTKO mice could potentially bind up more VEGF-A from the circulation. Increased skin angiogenesis might also adversely affect hindlimb blood flow and therefore interfere with tissue repair. This possibility was illuminated by an earlier study showing that robust skin angiogenesis reduced perfusion within other organs such as kidney and liver by diverting blood to the skin.45 Although these are plausible explanations, possibilities of HIF-independent effects are not ruled out, such as augmentation in the activation and/or expression of NF-кB and IL-8.46 Further investigations are needed to pinpoint the precise mechanisms.
Evidence for Remote Signaling
In a previous study, we demonstrated that global Phd2 knockout led to robust angiogenesis throughout different tissues.9 In the present study, we show two independent examples, in the skin and liver, respectively, that nonischemic PHD2-deficient tissues failed to display substantial vascular growth. Although some angiogenesis was observed in the skin, it was modest compared with what was observed in global Phd2 knockout mice. This controversy might be explained by hypothesizing that much of angiogenesis after global Phd2 knockout was due to systemic effects mediated by plasma VEGF-A, which was robustly increased. However, kDKO and kTKO clearly activated paracrine angiogenic mechanisms in skin tissues.
If we hypothesize that angiogenesis in global PHD2-deficient mice was mediated by plasma VEGF-A, a reasonable question is why similar outcomes were not found in kPhd2KO mice, whose plasma VEGF-A was also increased. In this regard, it may be noted that the extent of plasma VEGF-A increase was different between the two mouse lines. In mice globally deficient for PHD2, plasma VEGF-A levels increased by roughly twofold, whereas the corresponding increase in kPhd2KO mice was a relatively modest 43.3%.
Two lines of direct evidence support the existence of a humoral mechanism in kPhd2KO mice. First, localized Phd2 knockout in hindlimb skin tissues failed to accelerate perfusion recovery after FAL. Second, hepatocyte-specific Phd2 knockout not only conferred improved hindlimb perfusion after FAL but also protected toes from necrotic damages. These findings clearly demonstrated the existence of a remote signaling mechanism.
Concluding Remarks
In summary, the main contribution of this study is the demonstration that Phd2 knockout in skin tissues may protect vascular integrity and may improve perfusion in ischemic hind limbs. These findings raise the possibility that skin PHD2 may be a novel therapeutic target which can be manipulated at a conveniently located tissue to accelerate vascular repair and tissue regeneration in hard-to-access organs or tissues, without undesirable neoangiogenesis elsewhere. Given that ischemia diseases often occur in vital organs that are accessed only through highly invasive surgical procedures, the remotely protective effects defined in this study may be of significant therapeutic potentials.
Footnotes
Supported by NIH grant 5R01EY019721 (G.-H.F.) and American Heart Association Scientist Development grant 11SDG7580014 (K.T.).
Disclosures: None declared.
Supplemental Data
KRT14CreERT-mediated knockout of Phd1, Phd2, and Phd3. A: PCR of genomic DNA from epidermal tissues of WT, kPhd2KO, kDKO, and kTKO mice. Template targets are labeled on the left. Data show essentially complete disruption of floxed alleles. For example, in kPhd2KO mice, Phd2-specific primers detected only the deleted allele, whereas Phd1- or Phd3-specific primers detected only WT bands. B: RT-PCR for Phd1-, Phd2-, and Phd3-encoded mRNAs. Total RNAs from epidermal tissues were used. C: qPCR for mRNA encoded by Phd1, Phd2, and Phd3. n = 3 mice per data set. ∗P < 0.05, ∗∗P < 0.01 by Student's t-test versus the corresponding column in the WT group. Del, Phd alleles after KRT14CreERT-mediated deletion of floxed exons; flox, Phd alleles before KRT14CreERT-mediated deletion of floxed exons.
Epidermis-specific deletion of floxed exon 2 in kPhd2KO mice. Genomic DNA samples were isolated from various tissues as indicated and analyzed by PCR with Phd2-specific primers. The del band is visible only in the epidermis lane. Del, Phd alleles after KRT14CreERT-mediated deletion of floxed exons; flox, Phd alleles before KRT14CreERT-mediated deletion of floxed exons.
Lack of apparent vascular defects in nonischemic gastrocnemius muscles of kPhd2KO mice. A: Representative images of anti-CD31 IHC staining for gastrocnemius muscle sections of mice without any surgical operation. B: Quantification of number of capillaries per myocyte. C: Anti-SMA IF staining for nonischemic gastrocnemius muscles. D: Quantification of SMA+ vessels per 0.25 mm2. n = 5 (B and D). Scale bars: 30 μm (A); 200 μm (C). SMA, smoth muscle actin.
Localized Phd disruption by topical application of 4-OHT. A: Lower body of a Phd2f/f/KRT14CreERT mouse, showing the location of 4-OHT application to the shaved area. B: PCR of epidermal DNA from various skin tissues.
Hepatocyte-specific Phd2 disruption in AlbPhd2KO (Phd2f/f/AlbCre) mice. A: PCR of genomic DNA from various tissues. B: RT-PCR used Phd2-specific primers and total RNA samples from different tissues. C: Anti-PHD2 Western blot analysis of liver nuclear extracts. D: H&E-stained liver sections. E: Anti-CD31 IHC-stained liver sections, showing similarly distributed vascular structures. Scale bars: 50 μm (D and E). Del, the Phd2 allele after the deletion of the floxed exon; flox, floxed Phd2 alleles; n.s., a nonspecific band used as a loading control.
Normal skin morphology in kPhd2KO mice under diabetic conditions. Skin tissues were dissected from 12-week-old mice, which were either not diabetic or diabetic after STZ treatment. Histological sections were prepared from skin tissues and analyzed by H&E staining. In nondiabetic mice, epidermal morphology appeared normal in all different genotypes. In STZ-induced diabetic mice, epidermal surfaces in WT, kDKO, and kTKO mice became rough and lumped, a feature characteristic of hyperkeratosis. By contrast, epidermal surfaces remained relatively smooth in kPhd2KO mice. n = 3 to 4. Scale bar = 10 μm.
Induction of type 2 diabetes in WT and kPhd2KO mice by HFD. A: Body weights of WT and kPhd2KO mice before and after 3 months of HFD feeding. B: Sixteen-hour fasting blood glucose levels in WT and kPhd2KO before and after HFD feeding. n = 11 (A and B). ∗∗P < 0.01.
References
- 1.Criqui M.H., Denenberg J.O., Langer R.D., Fronek A. The epidemiology of peripheral arterial disease: importance of identifying the population at risk. Vasc Med. 1997;2:221–226. doi: 10.1177/1358863X9700200310. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch A.T., Criqui M.H., Treat-Jacobson D., Regensteiner J.G., Creager M.A., Olin J.W., Krook S.H., Hunninghake D.B., Comerota A.J., Walsh M.E., McDermott M.M., Hiatt W.R. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA. 2001;286:1317–1324. doi: 10.1001/jama.286.11.1317. [DOI] [PubMed] [Google Scholar]
- 3.Roger V.L., Go A.S., Lloyd-Jones D.M., Benjamin E.J., Berry J.D., Borden W.B. Executive summary: heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125:188–197. doi: 10.1161/CIR.0b013e3182456d46. [DOI] [PubMed] [Google Scholar]
- 4.Emerging Risk Factors Collaboration. Seshasai S.R., Kaptoge S., Thompson A., Di Angelantonio E., Gao P., Sarwar N., Whincup P.H., Mukamal K.J., Gillum R.F., Holme I., Njolstad I., Fletcher A., Nilsson P., Lewington S., Collins R., Gudnason V., Thompson S.G., Sattar N., Selvin E., Hu F.B., Danesh J. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011;364:829–841. doi: 10.1056/NEJMoa1008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aronow H. Peripheral arterial disease in the elderly: recognition and management. Am J Cardiovasc Drugs. 2008;8:353–364. doi: 10.2165/0129784-200808060-00002. [DOI] [PubMed] [Google Scholar]
- 6.Ouma G.O., Jonas R.A., Usman M.H., Mohler E.R., 3rd Targets and delivery methods for therapeutic angiogenesis in peripheral artery disease. Vasc Med. 2012;17:174–192. doi: 10.1177/1358863X12438270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Germani A., Di Campli C., Pompilio G., Biglioli P., Capogrossi M.C. Regenerative therapy in peripheral artery disease. Cardiovasc Ther. 2009;27:289–304. doi: 10.1111/j.1755-5922.2009.00105.x. [DOI] [PubMed] [Google Scholar]
- 8.Jones W.S., Annex B.H. Growth factors for therapeutic angiogenesis in peripheral arterial disease. Curr Opin Cardiol. 2007;22:458–463. doi: 10.1097/HCO.0b013e328236741b. [DOI] [PubMed] [Google Scholar]
- 9.Takeda K., Cowan A., Fong G.H. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation. 2007;116:774–781. doi: 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- 10.Loinard C., Ginouves A., Vilar J., Cochain C., Zouggari Y., Recalde A., Duriez M., Levy B.I., Pouyssegur J., Berra E., Silvestre J.S. Inhibition of prolyl hydroxylase domain proteins promotes therapeutic revascularization. Circulation. 2009;120:50–59. doi: 10.1161/CIRCULATIONAHA.108.813303. [DOI] [PubMed] [Google Scholar]
- 11.Takeda Y., Costa S., Delamarre E., Roncal C., Leite de Oliveira R., Squadrito M.L., Finisguerra V., Deschoemaeker S., Bruyere F., Wenes M., Hamm A., Serneels J., Magat J., Bhattacharyya T., Anisimov A., Jordan B.F., Alitalo K., Maxwell P., Gallez B., Zhuang Z.W., Saito Y., Simons M., De Palma M., Mazzone M. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479:122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Epstein A.C., Gleadle J.M., McNeill L.A., Hewitson K.S., O'Rourke J., Mole D.R., Mukherji M., Metzen E., Wilson M.I., Dhanda A., Tian Y.M., Masson N., Hamilton D.L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P.H., Pugh C.W., Schofield C.J., Ratcliffe P.J. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 13.Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J.M., Lane W.S., Kaelin W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 14.Jaakkola P., Mole D.R., Tian Y.M., Wilson M.I., Gielbert J., Gaskell S.J., Kriegsheim A., Hebestreit H.F., Mukherji M., Schofield C.J., Maxwell P.H., Pugh C.W., Ratcliffe P.J. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 15.Lieb M.E., Menzies K., Moschella M.C., Ni R., Taubman M.B. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80:421–426. doi: 10.1139/o02-115. [DOI] [PubMed] [Google Scholar]
- 16.Appelhoff R.J., Tian Y.M., Raval R.R., Turley H., Harris A.L., Pugh C.W., Ratcliffe P.J., Gleadle J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 17.Koivunen P., Tiainen P., Hyvarinen J., Williams K.E., Sormunen R., Klaus S.J., Kivirikko K.I., Myllyharju J. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem. 2007;282:30544–30552. doi: 10.1074/jbc.M704988200. [DOI] [PubMed] [Google Scholar]
- 18.Oehme F., Ellinghaus P., Kolkhof P., Smith T.J., Ramakrishnan S., Hutter J., Schramm M., Flamme I. Overexpression of PH-4, a novel putative proline 4-hydroxylase, modulates activity of hypoxia-inducible transcription factors. Biochem Biophys Res Commun. 2002;296:343–349. doi: 10.1016/s0006-291x(02)00862-8. [DOI] [PubMed] [Google Scholar]
- 19.Semenza G.L. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8:588–594. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 20.Forsythe J.A., Jiang B.H., Iyer N.V., Agani F., Leung S.W., Koos R.D., Semenza G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang B.H., Rue E., Wang G.L., Roe R., Semenza G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 22.Tian H., Hammer R.E., Matsumoto A.M., Russell D.W., McKnight S.L. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng J., Zhang L., Drysdale L., Fong G.H. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci U S A. 2000;97:8386–8391. doi: 10.1073/pnas.140087397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maltepe E., Schmidt J.V., Baunoch D., Bradfield C.A., Simon M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 25.Semenza G.L., Jiang B.H., Leung S.W., Passantino R., Concordet J.P., Maire P., Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 26.Semenza G.L., Wang G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pugh C.W., Tan C.C., Jones R.W., Ratcliffe P.J. Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:10553–10557. doi: 10.1073/pnas.88.23.10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza G.L., Nejfelt M.K., Chi S.M., Antonarakis S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:5680–5684. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly B.D., Hackett S.F., Hirota K., Oshima Y., Cai Z., Berg-Dixon S., Rowan A., Yan Z., Campochiaro P.A., Semenza G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 30.Waltenberger J., Claesson-Welsh L., Siegbahn A., Shibuya M., Heldin C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 31.Anisimov A., Tvorogov D., Alitalo A., Leppanen V.M., An Y., Han E.C., Orsenigo F., Gaal E.I., Holopainen T., Koh Y.J., Tammela T., Korpisalo P., Keskitalo S., Jeltsch M., Yla-Herttuala S., Dejana E., Koh G.Y., Choi C., Saharinen P., Alitalo K. Vascular endothelial growth factor-angiopoietin chimera with improved properties for therapeutic angiogenesis. Circulation. 2013;127:424–434. doi: 10.1161/CIRCULATIONAHA.112.127472. [DOI] [PubMed] [Google Scholar]
- 32.Reinmuth N., Liu W., Jung Y.D., Ahmad S.A., Shaheen R.M., Fan F., Bucana C.D., McMahon G., Gallick G.E., Ellis L.M. Induction of VEGF in perivascular cells defines a potential paracrine mechanism for endothelial cell survival. FASEB J. 2001;15:1239–1241. doi: 10.1096/fj.00-0693fje. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K., Ho V.C., Takeda H., Duan L.J., Nagy A., Fong G.H. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vasioukhin V., Degenstein L., Wise B., Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A. 1999;96:8551–8556. doi: 10.1073/pnas.96.15.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Postic C., Shiota M., Niswender K.D., Jetton T.L., Chen Y., Moates J.M., Shelton K.D., Lindner J., Cherrington A.D., Magnuson M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 36.Minamishima Y.A., Kaelin W.G., Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science. 2010;329:407. doi: 10.1126/science.1192811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abaci A., Oguzhan A., Kahraman S., Eryol N.K., Unal S., Arinc H., Ergin A. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–2242. doi: 10.1161/01.cir.99.17.2239. [DOI] [PubMed] [Google Scholar]
- 38.Waltenberger J. Impaired collateral vessel development in diabetes: potential cellular mechanisms and therapeutic implications. Cardiovasc Res. 2001;49:554–560. doi: 10.1016/s0008-6363(00)00228-5. [DOI] [PubMed] [Google Scholar]
- 39.Ruiter M.S., van Golde J.M., Schaper N.C., Stehouwer C.D., Huijberts M.S. Diabetes impairs arteriogenesis in the peripheral circulation: review of molecular mechanisms. Clin Sci (Lond) 2010;119:225–238. doi: 10.1042/CS20100082. [DOI] [PubMed] [Google Scholar]
- 40.Thangarajah H., Yao D., Chang E.I., Shi Y., Jazayeri L., Vial I.N., Galiano R.D., Du X.L., Grogan R., Galvez M.G., Januszyk M., Brownlee M., Gurtner G.C. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc Natl Acad Sci U S A. 2009;106:13505–13510. doi: 10.1073/pnas.0906670106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hazarika S., Dokun A.O., Li Y., Popel A.S., Kontos C.D., Annex B.H. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. 2007;101:948–956. doi: 10.1161/CIRCRESAHA.107.160630. [DOI] [PubMed] [Google Scholar]
- 42.Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C., Declercq C., Pawling J., Moons L., Collen D., Risau W., Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 43.Ferrara N., Carver-Moore K., Chen H., Dowd M., Lu L., O'Shea K.S., Powell-Braxton L., Hillan K.J., Moore M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 44.Ferrara N., Gerber H.P., LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 45.Boutin A.T., Weidemann A., Fu Z., Mesropian L., Gradin K., Jamora C., Wiesener M., Eckardt K.U., Koch C.J., Ellies L.G., Haddad G., Haase V.H., Simon M.C., Poellinger L., Powell F.L., Johnson R.S. Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell. 2008;133:223–234. doi: 10.1016/j.cell.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan D.A., Kawahara T.L., Sutphin P.D., Chang H.Y., Chi J.T., Giaccia A.J. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell. 2009;15:527–538. doi: 10.1016/j.ccr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
KRT14CreERT-mediated knockout of Phd1, Phd2, and Phd3. A: PCR of genomic DNA from epidermal tissues of WT, kPhd2KO, kDKO, and kTKO mice. Template targets are labeled on the left. Data show essentially complete disruption of floxed alleles. For example, in kPhd2KO mice, Phd2-specific primers detected only the deleted allele, whereas Phd1- or Phd3-specific primers detected only WT bands. B: RT-PCR for Phd1-, Phd2-, and Phd3-encoded mRNAs. Total RNAs from epidermal tissues were used. C: qPCR for mRNA encoded by Phd1, Phd2, and Phd3. n = 3 mice per data set. ∗P < 0.05, ∗∗P < 0.01 by Student's t-test versus the corresponding column in the WT group. Del, Phd alleles after KRT14CreERT-mediated deletion of floxed exons; flox, Phd alleles before KRT14CreERT-mediated deletion of floxed exons.
Epidermis-specific deletion of floxed exon 2 in kPhd2KO mice. Genomic DNA samples were isolated from various tissues as indicated and analyzed by PCR with Phd2-specific primers. The del band is visible only in the epidermis lane. Del, Phd alleles after KRT14CreERT-mediated deletion of floxed exons; flox, Phd alleles before KRT14CreERT-mediated deletion of floxed exons.
Lack of apparent vascular defects in nonischemic gastrocnemius muscles of kPhd2KO mice. A: Representative images of anti-CD31 IHC staining for gastrocnemius muscle sections of mice without any surgical operation. B: Quantification of number of capillaries per myocyte. C: Anti-SMA IF staining for nonischemic gastrocnemius muscles. D: Quantification of SMA+ vessels per 0.25 mm2. n = 5 (B and D). Scale bars: 30 μm (A); 200 μm (C). SMA, smoth muscle actin.
Localized Phd disruption by topical application of 4-OHT. A: Lower body of a Phd2f/f/KRT14CreERT mouse, showing the location of 4-OHT application to the shaved area. B: PCR of epidermal DNA from various skin tissues.
Hepatocyte-specific Phd2 disruption in AlbPhd2KO (Phd2f/f/AlbCre) mice. A: PCR of genomic DNA from various tissues. B: RT-PCR used Phd2-specific primers and total RNA samples from different tissues. C: Anti-PHD2 Western blot analysis of liver nuclear extracts. D: H&E-stained liver sections. E: Anti-CD31 IHC-stained liver sections, showing similarly distributed vascular structures. Scale bars: 50 μm (D and E). Del, the Phd2 allele after the deletion of the floxed exon; flox, floxed Phd2 alleles; n.s., a nonspecific band used as a loading control.
Normal skin morphology in kPhd2KO mice under diabetic conditions. Skin tissues were dissected from 12-week-old mice, which were either not diabetic or diabetic after STZ treatment. Histological sections were prepared from skin tissues and analyzed by H&E staining. In nondiabetic mice, epidermal morphology appeared normal in all different genotypes. In STZ-induced diabetic mice, epidermal surfaces in WT, kDKO, and kTKO mice became rough and lumped, a feature characteristic of hyperkeratosis. By contrast, epidermal surfaces remained relatively smooth in kPhd2KO mice. n = 3 to 4. Scale bar = 10 μm.
Induction of type 2 diabetes in WT and kPhd2KO mice by HFD. A: Body weights of WT and kPhd2KO mice before and after 3 months of HFD feeding. B: Sixteen-hour fasting blood glucose levels in WT and kPhd2KO before and after HFD feeding. n = 11 (A and B). ∗∗P < 0.01.