

Abstract
In the rodent nucleus accumbens (NAc), cocaine elevates levels of brain-derived neurotrophic factor (BDNF). Conversely, BDNF can augment cocaine-related behavioral responses. The latter could reflect enhancement of AMPA receptor (AMPAR) transmission, because AMPARs in the NAc mediate some cocaine-induced behaviors. Furthermore, in vitro studies in other cell types show that BDNF can promote AMPAR synaptic delivery. In this study, we investigated whether BDNF similarly promotes AMPAR trafficking in the adult rat NAc. After unilateral intracranial injection of BDNF into NAc core or shell, rats were killed at post-injection times ranging from 30 min to 3 days. NAc core or shell tissue from both injected and non-injected hemispheres was analyzed by Western blotting. A protein crosslinking assay was used to measure AMPAR surface expression. Assessment of tropomyosin receptor kinase B (TrkB) signaling demonstrated that injected BDNF was biologically active. BDNF injection into NAc core, but not NAc shell, led to a protein synthesis and extracellular signal-regulated kinase (ERK) dependent increase in cell surface GluA1 and a trend towards increased total GluA1. This was detected 30 min post-injection but not at longer time-points. GluA2 and GluA3 were unaffected, suggesting an effect of BDNF on homomeric GluA1 Ca2+-permeable AMPARs. These results demonstrate that exogenous BDNF rapidly increases AMPAR surface expression in the rat NAc core, raising the possibility of a relationship between increases in endogenous BDNF levels and alterations in AMPAR transmission observed in the NAc of cocaine-experienced rats.
Keywords: Ca2+ permeable AMPA receptor, cocaine, ERK, TrkB
Introduction
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is widely accepted as an important regulator of synaptic function and plasticity, including plasticity induced by drugs of abuse (Russo et al., 2009; Ghitza et al., 2010; McGinty et al., 2010). BDNF exerts its major biological functions through binding to tropomyosin receptor kinase B (TrkB), which is linked to a number of signal transduction cascades including mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3-K) and phospholipase C-γ pathways (Reichardt, 2006). BDNF can influence plasticity through multiple mechanisms. For example, it exerts both presynaptic and postsynaptic effects, and influences both early-phase and late-phase LTP (Bramham & Messaoudi, 2005; Waterhouse & Xu, 2009).
Regulation of AMPA receptor (AMPAR) trafficking plays an important role in synaptic plasticity and learning (Shepherd & Huganir, 2007; Kessels & Malinow, 2009; Keifer & Zheng, 2010). Recent work has highlighted the importance of AMPARs lacking the GluA2 subunit. These Ca2+-permeable AMPARs (CP-AMPARs) have high conductance and exhibit inward rectification due to voltage-dependent polyamine block (Cull-Candy et al., 2006; Liu & Zukin, 2007; Isaac et al., 2007). In vitro studies have shown that activation of BDNF-TrkB signaling increases synaptic delivery of CP-AMPARs (Caldeira et al., 2007; Li & Keifer, 2008; Li & Keifer, 2009). Whether BDNF similarly promotes CP-AMPAR trafficking in vivo is unknown.
Cocaine exposure can increase BDNF expression in addiction-related brain regions, including the nucleus accumbens (NAc) (Zhang et al., 2002; Grimm et al., 2003; Le Foll et al., 2005; Filip et al., 2006; Liu et al., 2006; Pu et al., 2006; Fumagalli et al., 2007; Graham et al., 2007; Im et al., 2010; Lu et al., 2010; Sadri-Vakili et al., 2010), and can also modulate TrkB expression and phosphorylation in the NAc (Freeman et al., 2003; Filip et al., 2006; Graham et al., 2009; Crooks et al., 2010). Elevation of BDNF levels in rodent NAc is associated with enhancement of cocaine-related behavioral responses, including locomotor sensitization, responding for conditioned reward, conditioned place preference, and cocaine seeking after withdrawal (Horger et al., 1999; Grimm et al., 2003; Graham et al., 2007; Bahi et al., 2008), although this may depend on whether BDNF elevation occurs in core or shell (see Discussion). While the underlying mechanism is unclear, it is intriguing to speculate that it may involve BDNF’s effects on AMPAR trafficking, based on the importance of AMPAR plasticity in the NAc for some behavioral consequences of repeated cocaine exposure (Wolf & Ferrario, 2010).
As a first step towards testing this hypothesis, we measured AMPAR surface expression after acute injection of BDNF into the NAc core or shell of adult rats. Cell surface levels of GluA1, but not other AMPAR subunits, were increased in the core 30 min after BDNF injection. This effect was dependent on protein synthesis and extracellular signal-regulated kinase (ERK) activity, dissipated at longer times after BDNF injection, and was not detected in the shell. Our results suggest that BDNF produces a rapid and transient increase in cell surface levels of CP-AMPARs in the NAc core.
Materials and Methods
Subjects
Male Sprague Dawley rats (270–320 g) were housed in groups of three with food and water available ad libitum. A 12 h/12 h light/dark cycle was used with the lights on at 7:00 A.M. All intracranial injections were performed between 8:00 A.M. and 4:00 P.M. All procedures were approved by the Institutional Animal Care and Use Committee of Rosalind Franklin University of Medicine and Science. Rats were allowed to acclimate 7–10 days prior to injection.
BDNF intracranial injections
Rats were anesthetized with a ketamine-xylazine cocktail (65mg/kg and 20mg/kg, respectively) and mounted onto a stereotaxic frame. A Hamilton syringe was slowly lowered and either vehicle (sterile PBS-saline, 1:1, v/v) or BDNF (0.75μg/0.5μl or 0.25μg/0.5μl, dissolved in vehicle solution; recombinant human BDNF, 248-BD, R&D Systems, Minneapolis, MN, USA) was unilaterally microinjected into NAc core or shell (0.1μl/min). Injectors were left in place for 2 min after the injection. The doses of BDNF were chosen based on previous studies (Lu et al., 2004). The coordinates for NAc core were: anteroposterior (AP) +1.2mm; lateral (L) +2.6mm (6° angle), and dorsoventral (DV) −7.0mm (Paxinos & Watson, 1998). The coordinates for NAc shell were: AP +1.4mm; L +1.5mm (6° angle), and DV −7.0mm. BDNF or vehicle was infused into a randomly assigned hemisphere. The other hemisphere served as a non-injected control. Rats were decapitated at different post-injection times (10 min, 30 min, 3 h, 24 h, and 3 days). To determine if BDNF’s effect on GluA1 at the 30 min time-point required protein synthesis or ERK activation, anisomycin (150 mg/kg, i.p., A9789, Sigma, St. Louis, MO, USA) or SL327 (30 mg/kg, i.p., Asc-082, Ascent Scientific, Princeton, NJ, USA) was administered ~30 min before microinjection of BDNF into the core. Systemic injection of anisomycin has been used by several groups to produce a long-lasting reduction in protein synthesis (e.g., Lattal & Abel, 2001; Tronel et al., 2005; Bernardi et al., 2007). Approximately 90% inhibition is produced with the dose and timing used in our study (see Flood et al., 1973). The dose of the ERK inhibitor, SL327, and the timing of its injection were chosen based on previous studies (e.g., Selcher et al., 1999; Wang et al., 2003; Ferguson & Robinson, 2004; Valjent et al., 2006a, b).
Protein crosslinking and immunoblotting
Rats were decapitated at different times after intracranial injection of BDNF as described above. NAc core and shell subregions were rapidly dissected from a 2 mm coronal section obtained using a brain matrix as described previously (McCutcheon et al., 2011). During the dissection, we verified that infusion sites were located within the boundaries of our standard core and shell dissections. For all time-points except 10 min, NAc core or shell tissue was collected and processed for BS3 [Bis(Sulfosuccinimidyl) suberate] crosslinking and Western blotting as described previously to distinguish surface-expressed and intracellular AMPAR subunits (Boudreau & Wolf, 2005; Ferrario et al., 2010). Tissue from the 10 min time-point was homogenized without crosslinking. The following primary antibodies were used: GluA1 (PA1-37776, 1:1000, Thermoscientific, Rockford, IL, USA), GluA2 (L21/32, 1:200, UC Davis/NIH NeuroMab Facility, Davis, CA, USA), GluA2/3 (AB1506, 1:1000, Millipore, Billerica, MA, USA), BDNF (sc-546, 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), pTrkB (Tyr 706/707; 4621, 1:1000, Cell Signaling, Danvers, MA, USA), TrkB (07-225, 1:2000, Millipore), pERK (4377, 1:1000, Cell Signaling) and ERK (4695, 1:1000, Cell Signaling). GAPDH (glyceraldehyde-3-phosphate dehydrogenase, CB1001, 1:10000, Calbiochem, San Diego, CA, USA) was used as a loading control. Results from injected hemispheres (I) are normalized to the non-injected hemisphere (N). Non-injected hemispheres for BDNF and vehicle rats did not differ significantly for any measure (data not shown).
Statistical Analysis
Quantity One analysis software (BioRad, Hercules, CA) was used to quantify bands of interest. A background value was obtained and diffuse densities for surface and intracellular bands in each lane were determined. Total protein levels were determined by summing surface and intracellular values. Surface, intracellular, and total protein values were then normalized to a loading control (GADPH), except for pTrkB and pERK, which were normalized to total TrkB and ERK levels determined with a phosphorylation independent antibody. Unpaired t-tests (two-tailed) were conducted to compare protein levels between non-injected and injected hemispheres within each experimental group. Significance was set at p<0.05.
Results
Effects of BDNF in NAc Core
To verify that injected BDNF was biologically active, we first measured the activation of TrkB receptors and extracellular signal-regulated kinase (ERK) 10 min after injection of the mature form of BDNF (14kDa; 0.75 μg/0.5 μL) into the NAc core. As expected, a robust increase in mature BDNF protein was detected in the injected hemisphere (Fig. 1, BDNF). At the same time, phosphorylation of the TrkB receptor and both isoforms of ERK were significantly elevated in the BDNF-injected hemisphere (I) compared to the non-injected hemisphere (N) (Fig. 1, pTrkB and pERK). The TrkB phospho-specific antibody used in our studies detects phosphorylation of Tyr706/707, which is required for TrkB activation (see Discussion). These results show that injected BDNF activates TrkB-related signaling cascades in the NAc core.
Figure 1.
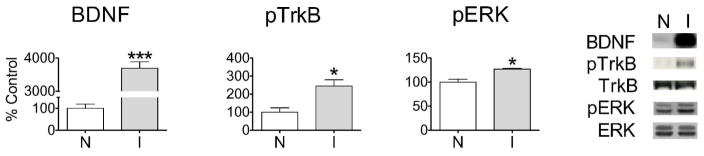
BDNF is biologically active after intracranial injection into the NAc core. Rats (n=3) received a unilateral injection of BDNF (0.75 μg/0.5 μL) and were killed 10 min later. Compared to the non-injected hemisphere (N), the injected hemisphere (I) showed elevated levels of BDNF protein (t4=18.95, ***p<0.0005), phosphorylated TrkB (pTrkB) ((t4=3.34, *p<0.05) and phosphorylated ERK (pERK) (t4=4.25, *p<0.05). pERK levels are expressed as the sum of pERK1 and pERK2 bands; phosphorylation of both ERK1 and ERK2 was increased by BDNF. Representative Western blots are shown (Molecular weights: BDNF, 14kDa; TrkB, 140kDa; ERK1 and ERK2, 44kDa and 42kDa, respectively). All data (mean ± SEM) are expressed as percent of mean values in the non-injected (N) hemisphere.
Next, we evaluated AMPAR surface expression 30 min after BDNF injection into the NAc core (0.75μg/0.5μl) (Fig. 2). For rats that received a unilateral BDNF injection (B), BDNF levels were significantly increased in the injected hemisphere (I) compared to the non-injected hemisphere (N) (Fig. 2, BDNF), while no differences was detected in rats that received vehicle injections (V). At 30 min time-point, we also detected elevated ERK phosphorylation in the BDNF-injected side. Most importantly, we found a significant increase in surface (S) expression of GluA1 [Fig. 2, GluA1(S)] in the BDNF-injected hemisphere compared to the non-injected hemisphere, whereas no changes were detected when different aliquots of the same tissue were probed with GluA2 or GluA2/3 antibodies [Fig. 2, GluA2(S) and GluA2/3(S)]. Total GluA1 levels, derived by summing surface and intracellular bands, showed a trend towards an increase after BDNF [Fig. 2, GluA1(T), p=0.08], whereas no changes or trends were observed for GluA2 or GluA2/3 [Fig. 2, GluA2(T) and GluA2/3(T)]. Vehicle injection did not alter surface or total levels of any AMPAR subunit (Fig. 2).
Figure 2.

An increase in GluA1 surface expression was observed 30 min after BDNF injection into the NAc core. Each rat received a unilateral injection of vehicle (V; sterile PBS-saline, 1:1, v/v) or BDNF (B; 0.75 μg/0.5 μL), and the injected hemisphere (I) was compared to the non-injected hemisphere (N). BDNF injection increased levels of BDNF protein (t10=8.01, ***p<0.0005) and phosphorylated ERK (pERK; expressed as the sum of pERK1 and pERK2 bands) (t10=2.60, *p<0.05). BDNF also increased surface (S) GluA1 (t10=3.19, *p<0.05) and produced a trend towards increased total (T) GluA1 (t10=2.00, p=0.08), whereas GluA2 and GluA2/3 immunoreactivity were unaffected. Representative surface bands are shown above bars. Total AMPAR subunit levels were determined by summing surface and intracellular bands. n=6 rats/group. All data (mean ± SEM) are expressed as percent of mean values in the non-injected (N) hemisphere.
In other systems, BDNF has been shown to increase AMPAR synaptic targeting through ERK and protein synthesis dependent mechanisms (see Discussion). In fact, the trend towards an increase in total GluA1 30 min after BDNF injection (Fig. 2) suggested a possible effect of BDNF on GluA1 protein synthesis. To test involvement of protein synthesis and ERK, rats received an i.p. injection of anisomycin (an inhibitor of protein synthesis) or SL327 (an inhibitor of ERK activation) ~30 min prior to intracranial injection of BDNF (0.75 μg/0.5μL). Results from these rats are shown in Fig. 3 by the middle and right pairs of bars in each graph, labeled “A+B” to indicate anisomycin + BDNF treatment and “S+B” to indicate SL327 + BDNF treatment, respectively. Controls received i.p. saline followed by vehicle microinjection into the NAc core (V+V; left pair of bars). BDNF injection produced an elevation of BDNF levels in anisomycin and SL327 pretreated rats (Fig. 3A) similar to that observed in rats that were not pretreated (Fig. 2). However, anisomycin and SL327 completely blocked the effect of BDNF on surface and total GluA1 [Fig. 3C, GluA1(S) and GluA1(T)]. To verify that SL327 was blocking ERK activation, we measured pERK in all treatment groups. As expected, SL327 pretreatment blocked BDNF-induced ERK activation (Fig. 3B left) and reduced basal pERK levels by ~70% (Fig. 3B right), whereas ERK activation was not affected by anisomycin (Fig. 3B left and right). Together, these results suggest that BDNF produces a rapid ERK-dependent increase in GluA1 protein synthesis that leads to increased GluA1 surface expression. To determine if anisomycin or SL327 given alone had effects on AMPAR surface expression, we compared the non-injected hemispheres of the anisomycin or SL327 pretreated rats to the non-injected hemisphere of the saline pretreated group. No differences were found, indicating that 30 min of protein synthesis inhibition or ERK inhibition do not significantly alter AMPAR subunit surface expression in the NAc (data not shown).
Figure 3.
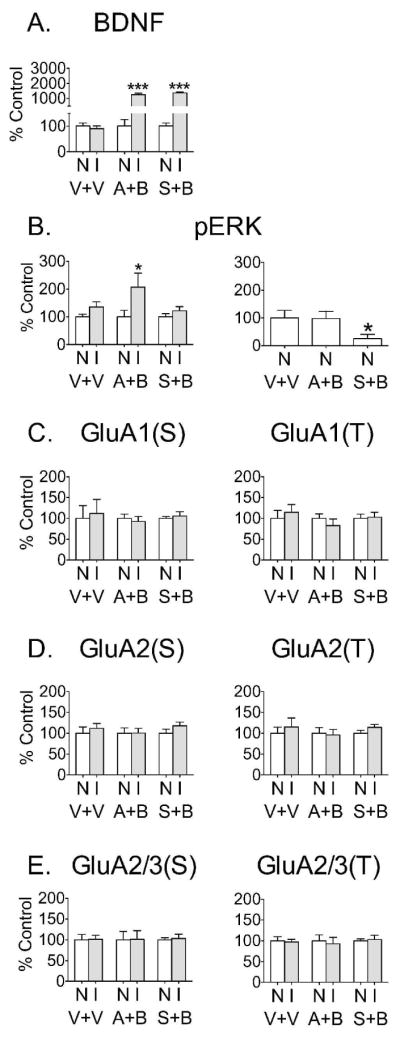
The increased GluA1 surface expression observed 30 min after BDNF injection into the NAc core was dependent on both protein synthesis and ERK activation. Three groups of rats were pretreated with vehicle (V; saline, i.p.), anisomycin (A; 150mg/kg, i.p.) or SL327 (S; 30mg/kg, i.p.). Thirty minutes later, vehicle rats (V+V, n=6) received unilateral vehicle injection (V; sterile PBS-saline, 1:1, v/v) into the NAc core, while the other two groups (A+B and S+B, n=6 and n=10, respectively) received unilateral BDNF injection (0.75 μg/0.5 μL). (A) BDNF levels were significantly elevated on the injected side (A+B, t10=11.06; S+B, t18=20.13; both ***p<0.05 compared to non-injected hemisphere). The elevated BDNF levels observed in anisomycin and SL327 pretreated rats did not differ from those observed in rats that were not pretreated (compare to Fig. 2). (B, left): The BDNF-induced elevation in pERK (t10=2.36, *p<0.05) was intact in anisomycin pretreated rats (compare to Fig. 2), while SL327 completely blocked the effect of BDNF on pERK. Data in this panel are normalized to the non-injected side of each group. (B, right): Comparison of pERK in the non-injected sides of vehicle, anisomycin and SL327 pretreated rats revealed that SL327 reduced basal pERK levels by ~70% [ANOVA, F(2,19)=4.41; *p<0.05, Dunnett’s test]. Data in this panel are normalized to the vehicle + vehicle group. (C) Both anisomycin and SL327 pretreatment abolished the effect of BDNF on GluA1 surface (S) and total (T) protein levels. (D–E) GluA2 and GluA2/3 surface (S) and total (T) levels were unaffected by BDNF injection regardless of pretreatment. All data (mean ± SEM) are expressed as percent of mean values in the non-injected (N) hemisphere, except for the right-hand graph in panel B (see above for explanation of this panel).
To determine if the effect of BDNF on GluA1 persisted longer than 30 min, rats were injected unilaterally into NAc core with either BDNF or vehicle and killed 3 h, 24 h or 3 days later for comparison of AMPAR levels in non-injected and injected hemispheres (Fig. 4). No changes in GluA1 or GluA2/3 surface levels [Fig. 4, GluA1(S), GluA2/3(S)] or total levels (data not shown) were detected at these time-points. These experiments used the same BDNF dose as the 30 min time-point (0.75 μg/0.5 μL) except for the 3-day experiment, which used 0.25μg/0.5μL BDNF. Both doses of BDNF had a similar effect in a prior study demonstrating BDNF-mediated enhancement of the incubation of cocaine craving (Lu et al., 2004), a phenomenon relevant to the goals of the present study because incubation is associated with increased levels of CP-AMPARs in the NAc (Conrad et al., 2008). Furthermore, since our goal is to understand the consequences of cocaine-induced elevation of NAc BDNF, both BDNF concentrations used here are relevant since both produce a greater elevation of NAc BDNF levels than those observed after cocaine treatment (e.g., Grimm et al., 2003; Huang et al., 2011). We also examined the BDNF-induced activation of signaling pathways at the later time-points. While pERK remained elevated 3 h after BDNF injection (Fig. 4A), no significant differences were detected between the BDNF-injected and non-injected hemispheres after 24 h and 3 days (data not shown). Taken together, these results indicated a transient, protein synthesis- and ERK-dependent effect of BDNF on GluA1 surface expression in the NAc core.
Figure 4.

No effect on AMPAR expression was observed at longer times following unilateral BDNF injection into the NAc core. (A) Three hours after NAc core injection of vehicle (V) or BDNF (B; 0.75 μg/0.5 μL), no differences in surface (S) GluA1 or GluA2/3 were observed between N and I hemispheres, whereas elevated levels of BDNF (t12=22.14, ***p<0.0005) and pERK (t12=3.30, *p<0.05) were still detected in the BDNF-injected hemisphere. n=7 rats/group. (B) No changes in surface (S) GluA1 or GluA2/3 were found 24 h after BDNF injection into the NAc core (0.75 μg/0.5 μL). n=10 rats/group. (C) No changes in surface (S) GluA1 or GluA2/3 were found 3 days after BDNF injection into the core (0.25 μg/0.5 μL). n=6 rats/group. Total AMPAR subunit levels were also unchanged (data not shown). BDNF levels in the injected hemisphere remained significantly elevated at the 24 h and 3 day time-points (data not shown). All data (mean ± SEM) are expressed as percent of mean values in the non-injected (N) hemisphere.
Effects of BDNF in NAc shell
We checked the biological activity of injected BDNF by measuring levels of phosphorylated TrkB and ERK. As shown in Fig. 5A, levels of BDNF and pTrkB were significantly elevated 10 min after BDNF injection (0.75μg/0.5μl) into the NAc shell, and there was a trend towards increased pERK. The magnitude of BDNF elevation in shell was somewhat less than occurred when the same amount of BDNF was injected into core (compare to Fig. 1), possibly due to more diffusion into the ventricle following shell injections. However, the level of TrkB activation was at least as great in the shell, and the magnitude of the trend towards increased pERK was similar to the significant increase observed in the core (see Fig. 1).
Figure 5.

No effect on AMPAR expression was observed after unilateral BDNF injection into the NAc shell. (A) Ten minutes after BDNF injection into NAc shell (0.75 μg/0.5 μL), BDNF protein levels (t6=4.91, *p<0.05) and phosphorylated TrkB (pTrkB; t6=3.17, *p<0.05) were significantly elevated in the injected hemisphere (I) compared to the non-injected hemisphere (N). n=4 rats/group. (B) Tissue was analyzed 30 min after injection of BDNF (B; 0.75 μg/0.5 μL) or vehicle (V) into the NAc shell. While BDNF was still significantly elevated 30 min after BDNF injection (t14=6.38, ***p<0.0005), no change in pERK levels or AMPAR surface (S) expression was found in the injected hemisphere compared to the non-injected hemisphere. Total AMPAR subunit levels (T) were also unchanged. n=8 rats/group. (C) No changes in AMPAR subunit surface expression were found 3 h or 3 days after BDNF injection into the NAc shell (0.25 μg/0.5 μL). Total AMPAR subunit levels were also unchanged (data not shown). BDNF levels in the injected hemisphere remained significantly elevated 3 h but not 3 days after BDNF injection (data not shown; n=8 rats/group for 3 h time-point and 5 rats/group for 3 day time-point). All data (mean ± SEM) are expressed as percent of mean values in the non-injected hemisphere.
BDNF levels remained elevated 30 min after BDNF injection (0.75μg/0.5μl) into the NAc shell (Fig. 5B). However, in contrast to results obtained in the core, pERK levels were no longer elevated and no changes in cell surface (S) or total (T) AMPAR subunit levels were observed at this time-point (Fig. 5B). There were also no changes in AMPAR surface expression at 3 h and 3 day time-points (Fig. 5C and 5D, respectively; these experiments used the lower BDNF dose of 0.25μg/0.5μl). Total AMPAR subunit levels were also unaltered after 3 h or 3 days (data not shown).
Discussion
We undertook these studies to bridge three prior areas of investigation: 1) studies demonstrating that cocaine can increase BDNF levels in the NAc and that this can affect cocaine-related behaviors, 2) studies implicating NAc AMPAR transmission in cocaine seeking, and 3) studies showing that BDNF modulates AMPAR expression and trafficking in vitro (see Introduction). Our results demonstrate that the acute injection of exogenous BDNF into the NAc core of adult rats rapidly (30 min) up-regulated GluA1 surface expression in the core via a protein synthesis and ERK dependent mechanism. This effect of BDNF dissipated quickly and had no delayed consequences for AMPAR expression, as no changes in surface GluA1 were found 3 h, 24 h or 3 days after BDNF injection. When injected into the shell, BDNF had no effect on shell GluA1 levels. BDNF did not alter GluA2 or GluA2/3 levels in either subregion. To our knowledge, this is the first demonstration that BDNF alters AMPAR trafficking in vivo.
It is important to acknowledge that our studies were restricted to drug-naïve and anesthetized rats, that intracranial injection of BDNF does not reproduce temporal features of cocaine-induced increases in endogenous BDNF levels, and that the BDNF concentrations achieved after intracranial injection are much higher than those reported after cocaine exposure (e.g., Grimm et al., 2003; Huang et al., 2011). Nevertheless, our results raise the possibility that cocaine-induced elevations in endogenous BDNF levels lead to enhanced AMPAR transmission in the NAc and therefore have potentially important implications for understanding the mechanisms by which BDNF influences drug seeking and other behaviors in cocaine experienced rats.
Potential explanations for different BDNF effects in the core and shell of drug naïve rats
Although core and shell differ in a number of properties, it is unclear which of these may explain our observation that BDNF increased AMPAR surface expression in core but not shell. Different roles for core and shell in particular behaviors have been attributed both to differences in connectivity (Groenewegen et al., 1999; Voorn et al., 2004) and to different intrinsic properties of core and shell neurons, including differences in neuron size, dendritic organization, spine density, and subcellular localization of dopamine and glutamate receptors (Meredith & Totterdell, 1999; Meredith et al., 2008). Differences in intrinsic properties seem more likely to explain our results, since we are applying BDNF directly into the NAc and measuring AMPAR distribution at the same location. For example, perhaps TrkB receptors in the core are located closer to spines where AMPAR trafficking or local protein synthesis may be occurring. Although no studies have assessed such possibilities, a difference in BDNF signaling in the two subregions may be indicated by our observation that pERK in the core was significantly elevated 10 min, 30 min and 3 h after BDNF injection, whereas pERK in the shell was completely normalized by 30 min. In fact, even at an earlier time-point (10 min after BDNF injection), pERK showed only a trend towards elevation in the shell, despite very robust increases in BDNF levels and TrkB phosphorylation. More studies are needed to determine the molecular basis for core-shell differences in the effects of BDNF on ERK and AMPAR trafficking.
Core and shell play distinct functional roles in cocaine experienced rats (e.g., Ito et al., 2004) and some adaptations produced by repeated cocaine exposure differ between the subregions, including adaptations potentially related to local protein synthesis (e.g., Wang et al., 2010). Therefore, it is possible that the core-shell selectivity we have observed in drug naïve rats is lost or altered due to neuroadaptations produced by repeated cocaine exposure. For example, a relationship between elevated BDNF and AMPAR trafficking may emerge in the shell of cocaine-experienced rats, or be altered in the core. Exploring these possibilities is an important goal for future studies because it may help explain some results discussed in the next section pertaining to core-shell differences in the role of BDNF in cocaine seeking behavior.
Possible core-shell differences in the role of BDNF after cocaine exposure
There is growing evidence for distinct BDNF-cocaine interactions in core versus shell. For example, BDNF mRNA levels were increased in the shell but not the core after acute or repeated cocaine injections, while TrkB mRNA levels declined more robustly in core than shell (Filip et al., 2006). Furthermore, a transient increase in BDNF protein levels was found in the shell but not in the core immediately after cocaine self-administration, but not 1 day later; this increase was linked to maintaining higher cocaine intake and facilitating relapse to cocaine seeking after abstinence (Graham et al., 2007). TrkB levels were also elevated in shell but not core immediately after cocaine self-administration, and knocking down TrkB in the NAc reduced cocaine reward in cocaine place conditioning experiments and produced a downward shift in the cocaine self-administration dose-response curve (Graham et al., 2009). These data provide evidence for facilitation of cocaine reward by BDNF-TrkB activation in the NAc shell.
In contrast to these results, there is evidence that elevation of BDNF in the prefrontal cortex (PFC) is a homeostatic response that opposes drug seeking (Berglind et al., 2007; Berglind et al., 2009; Sadri-Vakili et al., 2010; Whitfield et al., 2011; but see Lu et al., 2010). Interestingly, a single injection of BDNF into the dorsal PFC, which is sufficient to suppress subsequent cocaine seeking, increased BDNF levels not only in the PFC but also in the NAc (presumably via anterograde transport in PFC-NAc projections) (Berglind et al., 2007). Intra-PFC injection of BDNF also normalized alterations in NAc glutamate transmission linked to reinstatement of cocaine seeking (Berglind et al., 2009). Microdialysis probes in the latter study were primarily located in the core, and the region of PFC injected with BDNF in both studies projects primarily to the NAc core (see Pierce et al., 1998). Thus, one way to reconcile these results with those of Graham and coworkers is to propose that elevation of BDNF-TrkB signaling in the core suppresses cocaine seeking, whereas the opposite relationship holds in the shell (Graham et al., 2007; Graham et al., 2009). However, it is also possible that presynaptic and postsynaptic effects of BDNF in the NAc promote different behaviors, and that PFC infusion mimics a predominantly presynaptic effect of BDNF. Furthermore, the temporal pattern of BDNF release likely differed between studies, and it has been shown that the temporal aspects of BDNF application can significantly alter the duration of TrkB activation and its downstream consequences (Ji et al., 2010).
In addition to core-shell differences, evidence for cell-type specificity of BDNF action in the NAc of cocaine-experienced rodents is provided by a recent study showing that TrkB deletion had opposite effects on cocaine reward in D1 receptor-positive versus D2 receptor-positive NAc neurons (Lobo et al., 2010). Studies that did not distinguish between NAc subregions or cell types generally suggest that elevating NAc BDNF signaling promotes cocaine-related behaviors (Grimm et al., 2003; Horger et al., 1999; Bahi et al., 2008).
BDNF and CP-AMPARs
Our observation that BDNF increases surface expression of GluA1 but not GluA2 or GluA2/3 implicates homomeric GluA1 receptors, which are CP-AMPARs. Some previous results also suggest a selective effect of BDNF on CP-AMPARs: 1) In hippocampal cultures, incubation with BDNF for 30 min produced a protein synthesis-dependent increase in cell surface GluA1 but not GluA2 that normalized by 3 h, suggesting a transient increase in CP-AMPARs very similar to what we have observed in vivo (Caldeira et al., 2007). 2) In pond turtle brainstem, bath application of BDNF for 80 min increased synaptic delivery of GluA1 and GluA4, reproducing cellular changes associated with classical conditioning in this in vitro model (Li & Keifer, 2008; Li & Keifer, 2009). 3) Mutant mice with lower BDNF levels showed reduced hippocampal expression of GluA1 but not GluA2 or GluA3 (Giralt et al., 2009). However, other results suggest that BDNF can regulate protein levels of GluA2 and GluA3 as well as GluA1 (Caldeira et al., 2007; Narisawa-Saito et al., 1999) and induce a rapid translocation of GluA2-containing AMPARs to the cell surface (Narisawa-Saito et al., 2002). Furthermore, our preliminary studies in cultured NAc neurons have found that 30 min of incubation with BDNF increased cell surface GluA1 and GluA2 levels, as well as their co-localization (Reimers et al., 2010). Together, these results indicate that BDNF can selectively modulate CP-AMPARs, but also influences GluA2-containing AMPARs under some conditions.
TrkB signaling after BDNF infusion
Our results at the 30 min time-point in the core showed that the transient effect of BDNF on GluA1 surface expression is ERK-dependent, which is consistent with results obtained in the turtle brainstem (Li & Keifer, 2009). ERK activation is also implicated in upregulation of GluA1A2-containing AMPARs in the NAc of cocaine-sensitized rats (Boudreau et al., 2007; Schumann & Yaka, 2009). However, some of the present results indicate dissociation between ERK activation and GluA1 surface expression. Thus, pERK remained elevated at the 3 h time-point, whereas surface GluA1 had normalized. Furthermore, while anisomysin prevented the BDNF-induced increase in surface GluA1 at the 30 min time-point, ERK activation was not altered. Similarly, a reduction in BDNF levels can be associated with decreased GluA1 expression in the absence of altered ERK phosphorylation (Giralt et al., 2009). It should be noted that our study measured pERK in tissue homogenates, leaving open the possibility of compartmentalized changes in ERK activation that parallel the observed changes in GluA1 surface expression.
We also monitored TrkB phosphorylation at Tyr706/707 to verify BDNF’s biological activity after in vivo injection. It is well established that positionally equivalent residues in TrkA, B and C correspond to autophosphorylation sites in the catalytic domain; phosphorylation of these sites is necessary for TrK activation and subsequent activation of downstream pathways including the MAPK pathway (Segal et al., 1996; Cunningham et al., 1997; Huang & Reichardt, 2003). In our studies, we found a significant elevation in pTrkB 10 min after BDNF injection into either the core or shell, indicating that infused BDNF triggered TrkB activation. Unfortunately, we were unable to measure pTrkB at later time-points, since tissue collected at these time-points was crosslinked with BS3 to enable measurement of AMPAR surface expression, and the pTrkB antibody was not able to detect crosslinked TrkB receptors.
Due to the rapid protein synthesis dependent nature of the BDNF-induced increase in GluA1 in the NAc core, we speculate that the increased GluA1 protein was translated from local mRNA. Indeed, some results in other systems are consistent with an ability of BDNF to modulate local translation of GluA1 (e.g., Narisawa-Saito et al., 1999; Slipczuk et al., 2009). However, we failed to detect changes in levels or phosphorylation of some candidate signaling molecules that could contribute to local protein synthesis regulation after BDNF, including AKT (protein kinase B) and mTOR (mammalian target of rapamycin) (data not shown). Our results could be explained by the involvement of diverse mechanisms in BDNF-induced local protein synthesis (Santos et al., 2010).
In conclusion, our in vivo results extend prior in vitro findings suggesting that BDNF can selectively modulate CP-AMPARs (Caldeira et al., 2007; Li & Keifer, 2008; 2009). Thus, future studies are warranted to determine if there is a relationship between chronic cocaine-induced changes in BDNF levels (Ghitza et al., 2010) and AMPAR transmission (Wolf & Ferrario, 2010), particularly in animal models of cocaine addiction in which CP-AMPARs are implicated (Conrad et al., 2008; Mameli et al., 2009; Ferrario et al., 2011; McCutcheon et al., 2011).
Acknowledgments
We thank Mike Milovanovic and Kerstin Ford for assistance with the experiments. These studies were supported by DA015835, DA000453 and DA029099 to M.E.W. Some of these results were presented previously in poster form (Li et al., 2009).
Abbreviations
- AKT
protein kinase B
- BDNF
brain-derived neurotrophic factor
- BS3
bis(sulfosuccinimidyl) suberate
- CP-AMPAR
Ca2+ permeable AMPA receptor
- ERK
extracellular signal-regulated kinase
- LTP
long-term potentiation
- mTOR
mammalian target of rapamycin
- MAPK
mitogen-activated protein kinase
- NAc
nucleus accumbens
- pERK
phosphorylated extracellular signal-regulated kinase
- PI3-K
phosphoinositide 3-kinase
- pTrkB
phosphorylated tropomyosin receptor kinase B
- TrkA
tropomyosin receptor kinase A
- TrkB
tropomyosin receptor kinase B
Footnotes
Financial Disclosures
Marina E. Wolf has no biomedical financial interests but has a patent on A Possible Therapy For Cue-Induced Cocaine Craving Leading to Relapse in Abstinent Cocaine Abusers Based on Blockade of GluR2-lacking AMPA Receptors in the Nucleus Accumbens. Xuan Li reports no biomedical financial interests or potential conflicts of interest.
References
- Bahi A, Boyer F, Dreyer JL. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology (Berl) 2008;199:169–182. doi: 10.1007/s00213-008-1164-1. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitefield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi RE, Lattal KM, Berger SP. Anisomycin disrupts a contextual memory following reactivation in a cocaine-induced locomotor activity paradigm. Behav Neurosci. 2007;121:156–163. doi: 10.1037/0735-7044.121.1.156. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy- 5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine raving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks KR, Kleven DT, Rodriguiz RM, Wetsel WC, McNamara JO. TrkB signaling is required for behavioral sensitization and conditioned place preference induced by a single injection of cocaine. Neuropharmacology. 2010;58:1067–1077. doi: 10.1016/j.neuropharm.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Cunningham ME, Stephens RM, Kaplan DR, Greene LA. Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. J Biol Chem. 1997;272:10957–10967. doi: 10.1074/jbc.272.16.10957. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Robinson TE. Amphetamine-evoked gene expression in stratopallidal neurons: regulation by corticostriatal afferents and the ERK/MAPK signaling cascade. J Neurochem. 2004;91:337–348. doi: 10.1111/j.1471-4159.2004.02712.x. [DOI] [PubMed] [Google Scholar]
- Ferrario CR, Li X, Wang X, Reimers JM, Uejima JL, Wolf ME. The role of glutamate receptor redistribution in locomotor sensitization to cocaine. Neuropsychopharmacology. 2010;35:818–833. doi: 10.1038/npp.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Loweth JA, Milovanovic M, Ford KA, Galiñanes GL, Heng L-J, Tseng KY, Wolf ME. Alterations in AMPA receptor subunits and TARPs in the rat nucleus accumbens related to the formation of Ca2+-permeable AMPA receptors during the incubation of cocaine craving. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.01.021. Epub Jan 27 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filip M, Faron-Górecka A, Ku mider M, Go3da A, Frankowska M, Dziedzicka-Wasylewska M. Alterations in BDNF and trkB mRNAs following acute or sensiizing cocaine treatments and withdrawal. Brain Res. 2006;1071:218–225. doi: 10.1016/j.brainres.2005.11.099. [DOI] [PubMed] [Google Scholar]
- Flood JF, Rosenzweig MR, Bennett EL, Orme AE. The influence of duration of protein synthesis inhibition on memory. Physiol Behav. 1973;10:552–562. doi: 10.1016/0031-9384(73)90221-7. [DOI] [PubMed] [Google Scholar]
- Freeman AY, Soghomonian JJ, Pierce RC. Tyrosine kinase B and C receptors in the neostriatum and nucleus accumbens are co-localized in enkephalin-positive and enkephalin-negative neuronal profiles and their expression is influenced by cocaine. Neuroscience. 2003;117:147–156. doi: 10.1016/s0306-4522(02)00802-3. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Di Pasquale L, Caffino L, Racagni G, Riva MA. Repeated exposure to cocaine differently modulates BDNF mRNA and protein levels in rat striatum and prefrontal cortex. Eur J Neurosci. 2007;26:2756–2763. doi: 10.1111/j.1460-9568.2007.05918.x. [DOI] [PubMed] [Google Scholar]
- Ghitza UE, Zhai H, Wu P, Airavaara M, Shaham Y, Lu L. Role of BDNF and GDNF in drug reward and relapse: a review. Neurosci Biobehav Rev. 2010;35:157–171. doi: 10.1016/j.neubiorev.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Rodrigo T, Martín ED, Gonzalez JR, Milà M, Ceña V, Dierssen M, Canals JM, Alberch J. Brain-derived neurotrophic factor modulates the severity of cognitive alterations induced by mutant huntingtin: involvement of phospholipaseCgamma activity and glutamate receptor expression. Neuroscience. 2009;158:1234–1250. doi: 10.1016/j.neuroscience.2008.11.024. [DOI] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Graham DL, Krishnan V, Larson EB, Graham A, Edwards S, Bachtell RK, Simmons D, Gent LM, Berton O, Bolanos CA, DiLeone RJ, Parada LF, Nestler EJ, Self DW. Tropomyosin-related kinase B in the mesolimbic dopamine system: region-specific effects on cocaine reward. Biol Psychiatry. 2009;65:696–701. doi: 10.1016/j.biopsych.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewegen HJ, Wright CI, Beijer AV, Voorn P. Convergence and segregation of ventral striatial inputs and outputs. Ann NY Acad Sci. 1999;877:49–63. doi: 10.1111/j.1749-6632.1999.tb09260.x. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yeh CM, Wu MY, Chang AY, Chan JY, Chan SH, Hsu KS. Cocaine withdrawal impairs metabotropic glutamate receptor-dependent long-term depression in the nucleus accumbens. J Neurosci. 2011;31:4194–4203. doi: 10.1523/JNEUROSCI.5239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120–1127. doi: 10.1038/nn.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Ito R, Robbins TW, Everitt BJ. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci. 2004;7:389–397. doi: 10.1038/nn1217. [DOI] [PubMed] [Google Scholar]
- Ji Y, Lu Y, Yang F, Shen W, Tang TT, Feng L, Duan S, Lu B. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci. 2010;13:302–310. doi: 10.1038/nn.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer J, Zheng Z. AMPA receptor trafficking and learning. Eur J Neurosci. 2010;32:269–277. doi: 10.1111/j.1460-9568.2010.07339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Abel T. Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci. 2001;21:5773–5780. doi: 10.1523/JNEUROSCI.21-15-05773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Diaz J, Sokoloff P. A single cocaine exposure increases BDNF and D3 receptor expression: implications for drug-conditioning. NeuroReport. 2005;16:175–178. doi: 10.1097/00001756-200502080-00022. [DOI] [PubMed] [Google Scholar]
- Li W, Keifer J. Coordinate action of pre- and postsynaptic brain-derived neurotrophic factor is required for AMPAR trafficking and acquisition of in vitro classical conditioning. Neuroscience. 2008;155:686–697. doi: 10.1016/j.neuroscience.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Keifer J. BDNF-induced synaptic delivery of AMPAR subunits is differentially dependent on NMDA receptors and requires ERK. Neurobiol Learn Mem. 2009;91:243–249. doi: 10.1016/j.nlm.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Bahi A, DeJoseph MR, Urban JH, Dreyer JL, Wolf ME. Brain-derived neurotrophic factor (BDNF) and AMPA receptor trafficking in the nucleus accumbens core. Soc Neurosci Abstr. 2009;35:550.12. [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after long withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci. 2011;31:5737–5743. doi: 10.1523/JNEUROSCI.0350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Baldo BA, Andrezjewski ME, Kelley AE. The structural basis for mapping behavior onto the ventral striatum and its subdivisions. Brain Struct Funct. 2008;213:17–27. doi: 10.1007/s00429-008-0175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S. Microcircuits in nucleus accumbens’ shell and core involved in cognition and reward. Psychobiology. 1999;27:165–186. [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1999;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Iwakura Y, Kawamura M, Araki K, Kozaki S, Takei N, Nawa H. Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. J Biol Chem. 2002;277:40901–40910. doi: 10.1074/jbc.M202158200. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; San Diego, CA, USA: 1998. [Google Scholar]
- Pierce RC, Reeder DC, Hicks J, Morgan ZR, Kalivas PW. Ibotenic acid lesions of the dorsal prefrontal cortex disrupt the expression of behavioral sensitization to cocaine. Neuroscience. 1998;82:1103–1114. doi: 10.1016/s0306-4522(97)00366-7. [DOI] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers JM, Loweth JA, Milovanovic M, Wolf ME. Brain-derived neurotrophic factor (BDNF) alters AMPA receptor trafficking in primary cultures of rat nucleus accumbens neurons. Soc Neurosci Abstr. 2010;36:366.15. [Google Scholar]
- Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56:73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Kumaresan V, Schmidt HD, Famous KR, Chawla P, Vassoler FM, Overland RP, Xia E, Bass CE, Terwilliger EF, Pierce RC, Cha JH. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AR, Comprido D, Duarte CB. Regulation of local translation at the synapse by BDNF. Prog Neurobiol. 2010;92:505–516. doi: 10.1016/j.pneurobio.2010.08.004. [DOI] [PubMed] [Google Scholar]
- Schumann J, Yaka R. Prolonged withdrawal from repeated noncontingent cocaine exposure increases NMDA receptor expression and ERK activity in the nucleus accumbens. J Neurosci. 2009;29:6955–6963. doi: 10.1523/JNEUROSCI.1329-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Bhattacharyya A, Rua LA, Alberta JA, Stephen RM, Kaplan DR, Stiles DR. Differential utilization of Trk autophosphorylation sites. J Biol Chem. 1996;271:20175–20181. doi: 10.1074/jbc.271.33.20175. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;4(6):e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronel S, Milekic M, Alberini CM. Linking new information to a reactivated memory requires consolidation and not reconsolidation mechanisms. PLoS Biol. 2005;3(9):e293. doi: 10.1371/journal.pbio.0030293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corbillé AG, Bertran-Gonzalez J, Hervé D, Girault JA. Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci U S A. 2006a;103:2932–2937. doi: 10.1073/pnas.0511030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Trzaskos JM, Girault JA, Hervé D. Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006b;7:20. doi: 10.1186/1471-2202-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM. Putting a spin on the dorsal–ventral divide of the striatum. Trends Neurosci. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wang X, Luo YX, He YY, Li FQ, Shi HS, Xue LF, Xue YX, Lu L. Nucleus accumbens core mammalian target of rapamycin signaling pathway is critical for cue-induced reinstatement of cocaine seeking in rats. J Neurosci. 2010;30:12632–12641. doi: 10.1523/JNEUROSCI.1264-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther. 2003;304:172–178. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield TW, Jr, Shi X, Sun WL, McGinty JF. The suppressive effect of an intra-prefrontal cortical infusion of BDNF on cocaine-seeking is Trk receptor and extracellular signal-regulated protein kinase mitogen-activated protein kinase dependent. J Neurosci. 2010;31:834–842. doi: 10.1523/JNEUROSCI.4986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Ferrario CR. AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neurosci Biobehav Rev. 2010;35:185–211. doi: 10.1016/j.neubiorev.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Zhang L, Lou DW, Nakabeppu Y, Zhang J, Xu M. The dopamine D1 receptor is a critical mediator for cocaine-induced gene expression. J Neurochem. 2002;82:1453–1464. doi: 10.1046/j.1471-4159.2002.01089.x. [DOI] [PubMed] [Google Scholar]