

Abstract
Plasma membrane microdomains are features based on the physical properties of the lipid and sterol environment and have particular roles in signaling processes. Extracting sterol-enriched membrane microdomains from plant cells for proteomic analysis is a difficult task mainly due to multiple preparation steps and sources for contaminations from other cellular compartments. The plasma membrane constitutes only about 5-20% of all the membranes in a plant cell, and therefore isolation of highly purified plasma membrane fraction is challenging. A frequently used method involves aqueous two-phase partitioning in polyethylene glycol and dextran, which yields plasma membrane vesicles with a purity of 95% 1. Sterol-rich membrane microdomains within the plasma membrane are insoluble upon treatment with cold nonionic detergents at alkaline pH. This detergent-resistant membrane fraction can be separated from the bulk plasma membrane by ultracentrifugation in a sucrose gradient 2. Subsequently, proteins can be extracted from the low density band of the sucrose gradient by methanol/chloroform precipitation. Extracted protein will then be trypsin digested, desalted and finally analyzed by LC-MS/MS. Our extraction protocol for sterol-rich microdomains is optimized for the preparation of clean detergent-resistant membrane fractions from Arabidopsis thaliana cell cultures.
We use full metabolic labeling of Arabidopsis thaliana suspension cell cultures with K15NO3 as the only nitrogen source for quantitative comparative proteomic studies following biological treatment of interest 3. By mixing equal ratios of labeled and unlabeled cell cultures for joint protein extraction the influence of preparation steps on final quantitative result is kept at a minimum. Also loss of material during extraction will affect both control and treatment samples in the same way, and therefore the ratio of light and heave peptide will remain constant. In the proposed method either labeled or unlabeled cell culture undergoes a biological treatment, while the other serves as control 4.
Keywords: Issue 79, Cellular Structures, Plants, Genetically Modified, Arabidopsis, Membrane Lipids, Intracellular Signaling Peptides and Proteins, Membrane Proteins, Isotope Labeling, Proteomics, plants, Arabidopsis thaliana, metabolic labeling, stable isotope labeling, suspension cell cultures, plasma membrane fractionation, two phase system, detergent resistant membranes (DRM), mass spectrometry, membrane microdomains, quantitative proteomics
Introduction
In 1972, Jonathan Singer and Garth Nicolson proposed the fluid mosaic model a structure model of cellular membranes, replacing the protein-lipid-protein sandwich model that was generally accepted in the early 1960s. Singer and Nicolson postulated that the biological membrane can be considered as a two-dimensional liquid where all lipid and protein molecules diffuse freely and easily 5. Since that time, structure model of the plasma membrane and knowledge of the membrane composition became even more complex. Particularly, within the plasma membrane, structures such as protein complexes and lipid/sterol based structurally disordered microdomains can be observed. In artificial model membranes 6,7, sterols and sphingolipids can laterally segregate from other lipid species to form regions with altered physical features. This segregation within the cellular membrane is mainly caused by the self-associating properties between sterols and highly saturated hydrocarbon chains of phopsho- and sphingolipids 8. Particularly, the rigid sterol rings favor interactions with stiffer and straighter saturated lipid species and these interactions force neighboring hydrocarbon chains into more extended conformations, increasing membrane thickness and hardness.
One of the commonly observed features of sterol enriched membrane microdomains was their insolubility upon treatment with non-ionic detergents such a Triton X-100 or Brij 35. These fractions were thought to be identical with membrane microdomains and were called detergent resistant membranes (DRM) based on their biochemical preparation method 2. The use of nonionic detergents during DRM extraction received some criticism as the biochemical DRM preparation may not directly correspond to any specific membrane compartment within the living cell 9. Particularly, the detergent to protein ratio seems crucial in such preparations, as different detergents, as well as different detergent amounts can yield different composition of the detergent resistant membrane fraction 10. However, there is evidence that particular protein species specifically associate with these cellular sterol-rich membrane domains, and that these proteins are well enriched in biochemical preparations of detergent-resistant membrane fractions 11. The core of proteins that were found in plant DRM fraction, and for which the presence in DRMs was sterol dependent, were particularly GPI-anchored proteins, such as fasciclin-like arabinogalactan proteins (FLAs) and members of the SKU protein family. Also some signaling proteins, such as receptor-like kinases or phospholipases were found 11. These results are consistent with many proteomic studies on mammalian membrane microdomains 12,13. Also in plants there is increasing evidence for the role of membrane microdomains in context of stress response 14-16.
The protocol described here provides a robust method for fractionation of plasma membrane microdomains and particularly uses a protein to detergent concentration that allows us to depict stress induced alterations of the sterol-rich membrane compartment 4,11,14.
Protocol
PROCEDURE
Common reagents and buffers used in the extraction protocol:
- JPL medium for Arabidopsis thaliana suspension cell cultures
- 3 μM H3BO3
- 3 μM MnSO4 x H2O
- 1.1 μM ZnSO4 x 7 H2O
- 0.15 μM KJ
- 0.03 μM Na2MoO4 x 2 H2O
- 3 nM CoCl2 x 6 H2O
- 3 nM CuSO4 x 5 H2O
- 0.9 mM CaCl2 x 2 H2O
- 0.5 mM MgSO4 x 7 H2O
- 0.5 μM FeSO4 x 7 H2O
- 0.5 μM Na2EDTA x 2 H2O
- 0.12 μM thiamine HCl
- 0.16 μM nicotinic acid
- 0.097 μM pyridoxine HCl
- 0.107 μM NAA
- 0.375 mM KH2PO4
- 0.061 mM NaH2PO4 x 2 H2O
- 0.039 mM Na2HPO4
- 0.027 mM glycine
- 0.56 mM myo-inositol
- 1.5% sucrose
- 10 mM K14NO3 or 10 mM K15NO3
NOTE: pH of the JPL medium should be adjusted to 5.7 with KOH. The medium must be sterilized by filtration or autoclaving prior to use.
- Buffer H
- 100 mM HEPES-KOH (pH 7.5)
- 250 mM sucrose
- 10% (w/v) glycerol
- 5 mM EDTA
- 5 mM ascorbic acid
- 0.6% (w/v) PVP K-25 or K-30
- 5 mM DTT (add fresh)
- 1 mM PMSF (add fresh)
- protease and phosphatase inhibitors (add fresh)
- 50 mM NaF
- 1 mM Na3VO4
- 1 mM benzamidin
- 0.3 μM mikrocystin
- 4 μM leupeptin
- protease inhibitor cocktail
- Buffer R
- 5 mM potassium phosphate
- 0.33 M sucrose
- 3 mM KCl
- 0.1 mM EDTA
- 1 mM DTT (add fresh)
- Buffer TNE
- 25 mM Tris-HCl, pH 7.5
- 150 mM NaCl
- 5 mM EDTA
- Two-phase system (6 g-system is suitable for up to 15 g of sample fresh weight) preparation method In 15 ml Falcon tube mix ingredients listed below and mix thoroughly:
- 20% (w/w) dextran T500 - 2.6 g
- 40% (w/w) poly(ethylene glycol) (PEG 3350) - 1.3 g
- sucrose - 0.678 g
- 0.2 M Potassium phosphate, pH 7.8 - 0.15 ml
- 2 M KCl - 0.014 ml
- add water until total mass of the two-phase system is 6 g.
- Sucrose solutions in TNE buffer
- 2.4 M, 1.6 M, 1.4 M, 0.15 M
- Reagents for trypsin digestion
- UTU: 6 M urea, 2 M thiourea (pH 8.0 with 10 mM Tris-HCl)
- Reduction buffer (1 μg/μl DTT in water; 6.5 mM)
- Alkylation buffer (5 μg/μl iodoacetamide in water; 27 mM)
- LysC Endopeptidase (0.5 μg/μl)
- Trypsin, modified sequencing grade (0.4 μg/μl)
- Reagents for peptide desalting over C18
- resuspension solution: 2% trifluoroacetic acid (TFA), 5% acetonitrile in water
- solution A: 0.5% acetic acid
- solution B: 80% acetonitrile, 0.5% acetic acid
- Reagents for phosphopeptide enrichment
- Solution A: 0.1% TFA, 5% acetonitrile
- Solution B: 0.1% TFA, 80% acetonitrile
- TiO2 10 μm
- Ammonia (stock 25% solution)
- Piperidine (stock 100%)
PROTOCOLS
1. Metabolic Labeling of Arabidopsis thaliana Cell Suspension Cultures
Grow Arabidopsis Col-0 cell suspension cultures derived from leaves 17 in full 14N-JPL medium (18; see "Common reagents and buffers" section) and one set of cultures in 15N-JPL medium in sterile flask at constant light condition at 80 to 100 μmol/m2s, 23 °C, under constant shaking at 120 rpm.
To maintain the cell cultures, inoculate 400 ml of fresh JPL medium with 40 ml of seven day old cell culture in 1 L flasks.
Harvesting of cell cultures occurs via vacuum suction through a wide glass funnel with a stainless steel mesh. Cells accumulate on the mesh plate and in the funnel and can easily be collected from there. Cells are recommended to be frozen at -80 °C or in liquid nitrogen before grinding.
NOTE: For 15N metabolically labeled cell cultures use the K15NO3 as the only source of nitrogen for at least two passages over two weeks 19. In experiments for comparative proteomics, use a 15N labeled cell culture and also maintain an unlabeled culture in normal medium. Biological treatment will then be applied to either labeled or unlabeled culture, while the other serves as control (Figure 1). For protein preparation, both cultures will be combined 20. When planning the experimental setup, we recommend considering a reciprocal labeling setup 4 in which the same treatment is applied once to the 15N-labeled cells and once to the unlabeled cells and the respective unlabeled or 15N-labeled cells serve as controls. In this case, the double amounts of cell cultures are needed.
2. Plasma Membrane Purification
NOTE: All further steps are carried out in the cold room and/or on ice unless it is noted otherwise.
Homogenize cells from about 1 L of 7 days old cell suspension cultures in liquid nitrogen. The material can be stored at -80 °C until further use.
Mix the same amounts of labeled and unlabeled frozen cell cultures based on fresh weight in a beaker and add 2 volumes of buffer H immediately. In case of membrane microdomain preparation it is suggested to use at least 7 g fresh weight of both labeled and unlabeled cells.
Place the beaker on a shaker and shake until the material is melted and the solution is liquid and without ice crystals.
Filter the homogenate through one layer of Miracloth into 50 ml centrifuge tubes.
Balance samples with buffer H and centrifuge at 10,000 x g for 8 min.
Load the supernatant into pre-cooled ultra-centrifuge tubes (e.g. for rotor Beckman Coulter SW31Ti), with volume of about 32 ml
Balance samples with buffer H and pellet the microsomes by centrifuging at 100,000 x g at 4 °C for 30 min using Beckman SW32Ti rotor.
Remove the supernatant after centrifugation.
NOTE: The Supernatant contains soluble proteins. A small amount of this fraction can be saved for further protein precipitation and subsequent analysis also of soluble proteins.
Resuspend the membrane pellet of each sample in respective amount of buffer R and load onto the top of U1 two-phase system (Figure 2). If the 6 g two-phase system is used, load exactly 2 g of resuspended membrane pellet.
NOTE: The final volume of buffer R depends on the size of the two phase system that is going to be used (~1.6 ml of buffer R is needed when using 6 g system). It is recommended to use less buffer for resuspension so pure buffer can be added onto two-phase system to obtain the demanded weight, rather than using too much buffer for solubilization and not be able to load the whole sample onto the two phase system.
Load the same volume of pure buffer R as used for the sample resuspension onto U2.
Slowly mix U1 and U2 tubes by inverting them 30x (approximately 1 inversion/sec).
NOTE: Do not vigorously shake the two-phase system. It can cause a lack of separation of the phases in the ultracentrifugation step.
Centrifuge the samples at 1,500 x g at 4 °C for 10 min.
Remove the supernatant after centrifugation.
Transfer the upper phase from U1 onto U2 and repeat centrifugation at 1,500 x g at 4 °C for 10 min.
Move the upper phase from U2 into the SW41Ti ultracentrifuge tubes and dilute it with five volumes of buffer R and mix thoroughly.
NOTE: The final upper phase may have to be divided into separate ultracentrifuge tubes before it can be diluted five times.
Centrifuge the samples at 200,000 x g at 4 °C for one hour using the SW41Ti rotor.
Resuspend membrane pellet in small amount of TNE buffer (typically 200 μl of buffer is used) for isolation of detergent resistant membranes.
NOTE: A small amount of this fraction can be saved for the analysis of unfractionated plasma membrane.
NOTE: The plasma membrane fraction can be stored at 4 °C overnight before fractionation into detergent resistant membranes and detergent soluble fractions.
3. Detergent Resistant Membrane Preparation
Check the concentration of the plasma membrane fraction by Bradford assay.
Add Triton X-100 to the plasma membrane fraction. The final concentration of detergent should stay between 0.5-1%. The detergent to protein weight to weight ratio should stay between 13-15.
Shake samples at around 100 rpm at 4 °C for 30 min.
Add three volumes of 2.4 M sucrose to one volume of the treated plasma membrane to obtain 1.8 M final concentration of sucrose.
Prepare the sucrose gradient in SW41Ti ultracentrifuge tubes by loading the respective sucrose solutions on top of the 1.8 M fraction as illustrated in Figure 3.
Centrifuge the samples at 250,000 x g at 4 °C for 18 hr using the Beckman SW41Ti rotor.
Carefully remove the ultracentrifuge tubes from the rotor to avoid disrupting of the low-density band which may be visible as a milky ring at the interface of 0.15 M/1.4 M sucrose. Sometimes nothing can be seen but the fraction still contains the detergent resistant protein fraction.
Remove fraction of 0.75 ml volume from the top of the gradient. Fractions 2 and 3 which cover volume of around 1.5 ml above the interphase ring and 0.5 ml below represent the detergent resistant membrane fraction. Collect these fractions into 15 ml Falcon tubes. Fractions 9 and 10 contain the detergent soluble membrane fraction and may also be collected for comparison. For further analysis, pool fractions 2 and 3 as well as fractions 9 and 10.
4. Extraction of Proteins from the Detergent Resistant Fraction by Methanol/Chloroform
NOTE: All steps are carried out in the room temperature.
Add 4 volumes of methanol to the collected fractions and vortex thoroughly.
Add 1 volume of chloroform, vortex thoroughly.
Add 3 volumes of water, vortex thoroughly.
Centrifuge the samples at 2,000 x g for 5 min using a benchtop centrifuge (e.g. Eppendorf 5417R).
Remove the aqueous phase above the protein layer in the interphase.
Add 3 volumes of methanol, vortex thoroughly.
Centrifuge the samples at 4,000 x g for 10 min.
Remove supernatant and dry the pellet. The dried pellet is ready for in-solution digestion.
5. In-solution Trypsin Digestion
NOTE: In this procedure all steps are done at room temperature to reduce unwanted derivatization of amino acid side-chains by the denaturants.
Dissolve sample in small volume of 6 M urea, 2 M thiurea, pH 8 (UTU). Use as low volume as is compatible with the sample (usually approximately 40 μl).
Spin samples solubilized in UTU at 5,000 x g in a benchtop centrifuge (e.g. Eppendorf 5417R) for 10 min to pellet any insoluble material.
The pH of the final solution should be near 8.0 for optimal trypsin digestion. Check with pH strips.
Add 1 μl of reduction buffer for every 50 μg of sample protein and incubate 30 min at room temperature.
NOTE: Only a rough estimate of protein content is necessary. When sample amount is limited it is better to sacrifice accuracy rather than wasting sample on a protein assay.
Add 1 μl of alkylation buffer for every 50 μg sample protein and incubate 20 min at room temperature in dark.
Add 0.5 μl of LysC per 50 μg sample protein and incubate for three hours at room temperature with constant shaking at 700 rpm. If necessary, the digest can also be carried out overnight. However, extended time at warm temperatures is not recommended as it can increase peptide loss due to adsorption to plastic tube material under aqueous pH 8 conditions.
Dilute sample with four volumes 10 mM Tris-HCl, pH 8.
NOTE: This step is absolutely necessary to dilute the urea concentration as trypsin is very sensitive to high salt.
Add 1 μl of trypsin per 50 μg sample protein and incubate overnight at room temperature with constant shaking at 700 rpm.
Acidify the samples to 0.2% TFA final concentration to reach pH 2 (add approximately 1/10 volume of 2% trifluoracetic acid).
NOTE: Samples can be stored at - 20 °C until used further, but it is better to store them on StageTips at 4 °C if it is for a short time (one week) or store them after StageTips desalting.
6. Manufacturing of C18-StageTips
Place an Empore Disk C18 on a flat, clean surface such as a disposable plastic Petri dish.
Punch out a small disk using a blunt-tipped hypodermic needle with diameter of 1.5 mm. The disk sticks in the needle and can be transferred into a pipette tip.
Push the disk out of the needle and fix it in the tapering of a pipette tip by a piece of fused silica or tubing fitting in the inside of the needle.
NOTE: StageTips can be stored dry at room temperature 21.
7. Use of C18-StageTips for Peptide Desalting and Concentration
Condition a C18-StageTip by placing 50 μl of solution B onto the prepared StageTip. Spin the tip in the centrifuge at 2,000 rpm for 2 min in a benchtop centrifuge (e.g. Eppendorf 5417R).
NOTE: Use adapters to spin StageTips in an Eppendorf centrifuge, liquid will be collected in a 2 ml reaction tube. For larger-scale preparations, stage tips can also be placed into a tip rack with 96 of the 200 μl tips and liquid can be collected in a microtiter plate.
NOTE: Never use higher speeds than 3,000 x g in a benchtop centrifuge (e.g. Eppendorf 5417R) due to the risk of spinning out the C18 disk from the tip.
Equilibrate the StageTip using 100 μl solution A. Spin the tip in the centrifuge at 5,000 x g in a benchtop centrifuge (e.g. Eppendorf 5417R).
Repeat step 2.
Load sample onto disk by carefully pipetting the sample into the pipette tip with the disc inside.
NOTE: One disc can bind approx. 100 μg of protein.
Spin the tips in the centrifuge until the whole volume of sample pass through the C18 disk.
Wash the StageTips two times with 100 μl of solution A. Spin the tips in the centrifuge.
NOTE: The washed and loaded StageTips can be stored at 4 °C for up to a week.
Elute sample with 40 μl of solution B. Collect the eluate in a fresh 1.5 ml reaction tube.
Concentrate the sample in the speed vac. Under ideal conditions, stop the dehydration process as there is just about 1 or 2 μl liquid left.
NOTE: Dried samples can be stored in the -80 °C for years.
Add a final volume of 9 μl of resuspension solution to the sample and transfer it to the microtiterplate for mass spec analysis.
NOTE: The final volume of the used resuspension buffer is dependent on the needs of experimenter and the sensitivity of the mass spectrometer used.
8. Alternative Protocol for Phosphopeptide Enrichment
NOTE: For phosphopeptide enrichment, protein extraction in step 2 must be done in the presence of phosphatase inhibitors.
NOTE: Exact protein concentration does not need to be determined for the following steps. It is enough to have a rough estimate of protein content to avoid unnecessary sample loss
Prepare the C8-StageTips (follow the same protocol for preparation of C18-StageTips at step 6).
Transfer 1 mg of TiO2-beads to the top of prepared C8-StageTips (use 1 mg TiO2 per 100 μg of protein).
Equilibrate the TiO2-tips with 100 μl of solution C, spin at 2,000 x g for 5 min in a benchtop centrifuge (e.g. Eppendorf 5417R).
Mix 100 μg of digested and desalted peptides from step 7.7 (it should be in 100 μl of solution B) with 100 μl of solution A.
Load mixed sample onto the equilibrated TiO2 columns; spin at 1,000 x g, 5 min.
Collect the flow-through and load it onto the same tip again (keep flow through after second pass).
Wash tip with 100 μl of solution A; spin at 2,000 x g, 5 min in a benchtop centrifuge (e.g. Eppendorf 5417R).
Elute sample with 50 μl of 5% ammonia.
Elute sample with 50 μl of 5% piperidine (both eluates can be combined).
Immediately acidify the sample using 50 μl of 20% phosphoric acid. pH should be at around 2.
NOTE: Save the TiO2 tips and retrieve the powder. It can be washed with solution B and used again for one or two more rounds.
Again desalt acidified samples over C18 tips (see step 8).
9. LC-MS/MS Analysis of Peptide Mixtures
Resuspend desalted phosphopeptides in resuspension buffer.
Load resuspended sample onto LC-MS/MS system of choice and acquire spectra in data dependent mode at suggested resolution of 60,000 full widths at half maximum.
NOTE: For optimal fragmentation, either neutral-loss scanning should be applied 22, or if available the multistage activation 23. If ETD is available it will allow peptide backbone fragmentation without loss of phosphoric acid 24.
NOTE: Peak lists from raw data need to be extracted and submitted to database identification as well as for quantitation. Here, we describe settings for using Mascot and MSQuant, which works for raw data files from LTQ, LTQ-Orbitrap and LTQ-FT instruments (Thermo Scientific).
Extract peak list using DTAsupercharge within the "Helper" Menu in MSQuant. Use default settings.
Submit resulting peak list file to Mascot search engine. Critical settings: precursor ion mass tolerance 10 ppm, MS/MS mass tolerance 0.5 Da. Fixed modifications: carbamidomethylation of cysteines, variable modifications: oxidation of methionine, phosphorylation of serine, threonine, tyrosine. Quantitation method: 15N metabolic labeling.
Save Mascot result as web page file.
Load Mascot result file and raw file into MSQuant. Select all proteins of interest and run "Automatic quantitation". The program will read full scan spectra from the raw file and do peak integration for labeled and unlabeled partners of each peptide.
Export quantitation results to a spreadsheet program or to a tab-delimited text file. The data can then be submitted to further statistical analysis in Excel or using further software packages such as Statquant 25 or cRacker 26.
Representative Results
With the presented protocol using metabolically labeled Arabidopsis cell cultures it is possible to isolate plasma membranes from plant tissue (step2, Figures 2 and 4), and enrich for detergent resistant membrane fractions within the plasma membrane (step 3, Figures 3 and 5). Subsequently, the protocol allows extraction of proteins from these detergent resistant membrane fractions (step 4) and digestion of the protein for comparative proteomic analysis (step 5). Finally, the optional enrichment of phosphopeptides (step 8) is particularly interesting in studying cellular signaling processes. The last step then is the quantitative data analysis (step 9, Figure 6) of protein abundance ratios in membrane fractions from labeled and unlabeled cell culture.
Typical results after mixing of the 2-phase system (step 2) and subsequent centrifugation are a clear colored upper phase and a green colored lower phase (Figure 2). Plasma membrane vesicles preferentially associate with the upper PEG phase, while membrane vesicles from green chloroplasts and other interior membranes are incorporated into the lower dextran phase. If the color separation cannot be observed, step 2 was not carried out correctly and plasma membranes will be contaminated with significant amounts of internal membranes. Enrichment of plasma membrane proteins can be shown by proteomic analysis of small aliquots of plasma membrane fractions and proteins in the lower phase of the two-phase system. The lower phase contains the inner membranes of the cell. For example, particularly typical known plasma membrane proteins, such as receptor kinase proteins showed high abundance in the plasma membrane fraction (PM) and low abundance in the lower phase fraction (Figure 4A) containing the internal membranes (IM). In turn, known proteins of the endoplasmatic reticulum show high abundance in the inner membrane fraction and were not observed in the plasma membrane fraction (Figure 4B).
The subsequent enrichment of detergent resistant membrane fractions (step 3) will in ideal cases result in formation of a milky ring of low-density membrane vesicles at approximately 1/5th of the tube height (Figure 3). If the adjustment of Triton X-100 concentrations or the detergent to protein ratio (step 3.2) is optimal for the separation into detergent resistant and detergent soluble fractions, additional rings can be observed in lower parts of the gradient and there will also be membrane clumps in the low density band. In these cases, detergent to protein ratio or total detergent amount is different from the critical micelle concentration necessary for reproducible separation of the two membrane domain fractions. Using the final detergent concentration and detergent to protein ratio described here, Enrichment of typical microdomain proteins, such as remorin (AT3G61260) 27 could be confirmed by a comparative mass spectrometric analysis of DRM fraction, detergent soluble fraction (step 3.8) as well as unfractionated plasma membrane and internal membranes. Highest abundance of remorin protein was found in DRM fractions as expected (Figure 6A). Proteins not present in the membrane microdomains, such as the ABC-transporter AT1G59870 (Figure 6B), do not show an increased abundance in the DRM fraction. Also typical contaminants, such as a mitochondrial protein (AT2G07707) from the ATPase F0 complex (Figure 6C) and ribosomal protein (At1G001100) from the 60S ribosome (Figure 6D) do not show an enriched abundance in the DRM fraction, although minimal amounts can still be identified.
After mass spectrometric analysis, protein abundances ratios of protein phosphorylation status in plasma membrane fractions of labeled and unlabeled cell cultures can be quantitatively distinguished (Figure 6). Typical results from the MSQuant (msquant.alwaysdata.net) quantitation window should show an ion intensity ratio of one to one for most of the identified peptides (Figure 6A). Those peptides with ion intensity ratio significantly deviating from 1:1 are considered as differential regulated between the labeled and unlabeled cell culture, and are candidates for matching to proteins affected by the treatment applied (Figure 6C) . A good quality control of successful metabolic labeling is the co-elution of both labeled and unlabeled peptide versions as a single peak in the nano-HPLC separation (step 9) as shown in Figures 6B and D.
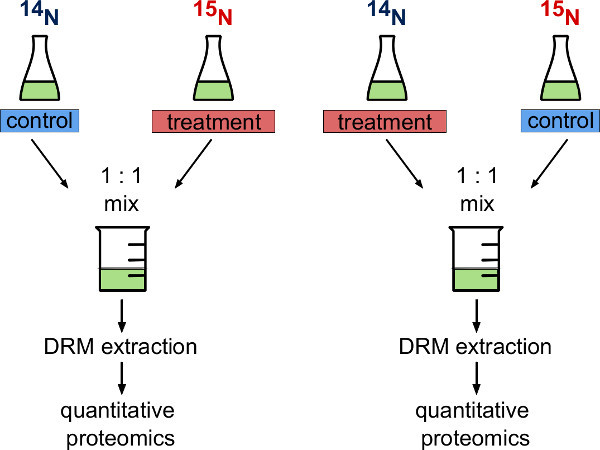 Figure 1. Metabolic labeling experimental strategy. Schematic representation of two variants of experimental design when using fully metabolic labeled and unlabeled A. thaliana cell cultures. The experimenter can either decide to use 14N cells (unlabeled) as a control and 15N cells (metabolically labeled) that undergo biological treatment or vice versa. In a reciprocal experimental design both variants are carried out in parallel.
Figure 1. Metabolic labeling experimental strategy. Schematic representation of two variants of experimental design when using fully metabolic labeled and unlabeled A. thaliana cell cultures. The experimenter can either decide to use 14N cells (unlabeled) as a control and 15N cells (metabolically labeled) that undergo biological treatment or vice versa. In a reciprocal experimental design both variants are carried out in parallel.
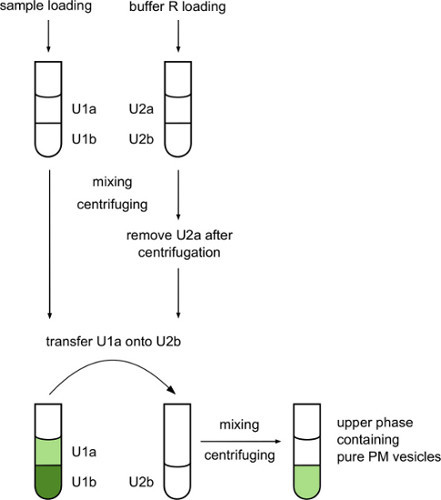 Figure 2. Workflow for separation of plant membrane vesicles on an aqueous two-phase system based on polyethylene glycol (PEG) and dextran. Plasma membrane vesicles separated into upper PEG phase while chloroplast and other interior membranes associated with the bottom dextran phase after centrifugation. With additional washing of upper phase better purification can be achieved, but also material loss is increased.
Figure 2. Workflow for separation of plant membrane vesicles on an aqueous two-phase system based on polyethylene glycol (PEG) and dextran. Plasma membrane vesicles separated into upper PEG phase while chloroplast and other interior membranes associated with the bottom dextran phase after centrifugation. With additional washing of upper phase better purification can be achieved, but also material loss is increased.
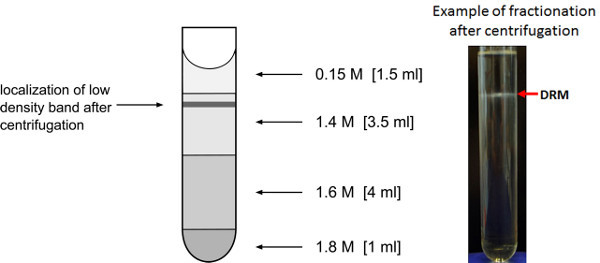 Figure 3. Enrichment of low-density membrane fractions. Representation of sucrose gradient for the enrichment of low-density detergent resistant membrane fractions. The expected location after the overnight centrifugation of the low density membrane vesicle band is indicated.
Figure 3. Enrichment of low-density membrane fractions. Representation of sucrose gradient for the enrichment of low-density detergent resistant membrane fractions. The expected location after the overnight centrifugation of the low density membrane vesicle band is indicated.
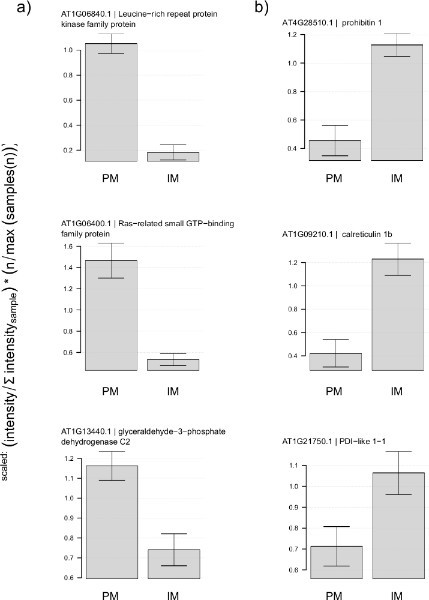 Figure 4. Enrichment of plasma membrane proteins after purification in two-phase system. Normalized protein ion intensities for proteins measured in plasma membrane (PM) and internal membranes (IM) fraction. (A) Typical known constituents of plasma membrane show much higher abundance in PM fraction than in IM fraction. (B) Natural known components of internal membranes shows the opposite behavior. For normalization, ion intensity sums for each protein were expressed as a fraction of total ion intensity per sample and then averaged between replicates. Fraction of total ion intensities were then expressed as relative to the average between the two treatments. Error bars represent the standard deviation of three preparations from independently grown cell cultures. Click here to view larger figure.
Figure 4. Enrichment of plasma membrane proteins after purification in two-phase system. Normalized protein ion intensities for proteins measured in plasma membrane (PM) and internal membranes (IM) fraction. (A) Typical known constituents of plasma membrane show much higher abundance in PM fraction than in IM fraction. (B) Natural known components of internal membranes shows the opposite behavior. For normalization, ion intensity sums for each protein were expressed as a fraction of total ion intensity per sample and then averaged between replicates. Fraction of total ion intensities were then expressed as relative to the average between the two treatments. Error bars represent the standard deviation of three preparations from independently grown cell cultures. Click here to view larger figure.
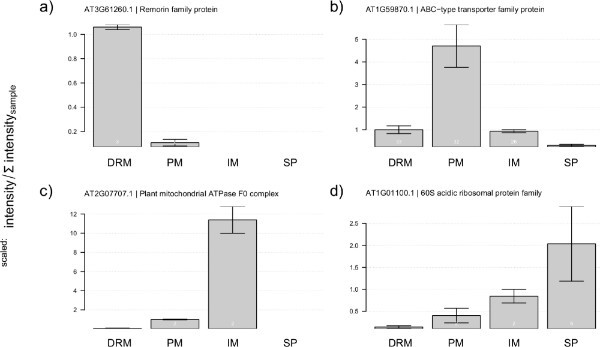 Figure 5. Abundance of standard protein markers within measured fractions. Plots represent the distribution of normalized protein ion intensities of chosen protein markers to particular subcellular compartments (DRM - sterol-rich microdomains, PM - plasma membrane, IM - internal membranes, SP - soluble proteins). Abundance patterns of (A) remorin as a marker for sterol-rich membrane microdomains, (B) ABC-type transporter family protein as a representative of a not sterol-dependent protein, (C) subunit of mitochondrial F0 complex, (D) 60S subunit of ribosomes as a typical co-purifying contaminant. For normalization, ion intensity sums for each protein were expressed as a fraction of total ion intensity per sample and then averaged between replicates. Fraction of total ion intensities were then expressed as relative to the average between the two treatments. Error bars represent the standard deviation of three preparations from independently grown cell cultures. Click here to view larger figure.
Figure 5. Abundance of standard protein markers within measured fractions. Plots represent the distribution of normalized protein ion intensities of chosen protein markers to particular subcellular compartments (DRM - sterol-rich microdomains, PM - plasma membrane, IM - internal membranes, SP - soluble proteins). Abundance patterns of (A) remorin as a marker for sterol-rich membrane microdomains, (B) ABC-type transporter family protein as a representative of a not sterol-dependent protein, (C) subunit of mitochondrial F0 complex, (D) 60S subunit of ribosomes as a typical co-purifying contaminant. For normalization, ion intensity sums for each protein were expressed as a fraction of total ion intensity per sample and then averaged between replicates. Fraction of total ion intensities were then expressed as relative to the average between the two treatments. Error bars represent the standard deviation of three preparations from independently grown cell cultures. Click here to view larger figure.
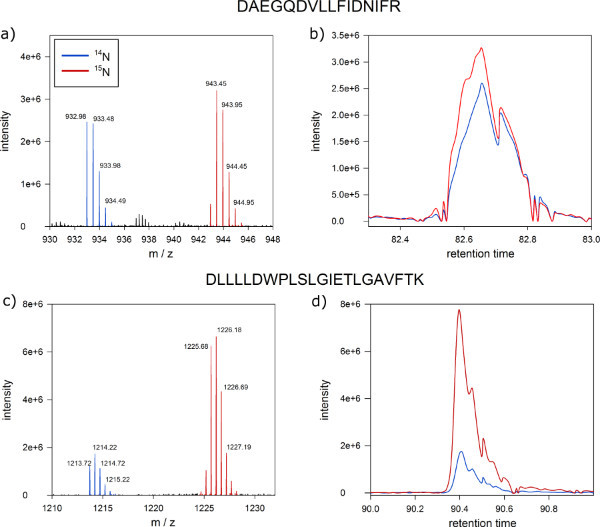 Figure 6. Expected results of quantitative protein mass spectrometry. Screen shot from MSQuant showing the expected 1:1 ion intensity ratio of a peptide from a 1:1 mixture of protein extracts from labeled and unlabeled Arabidopsis cells. (A) Full scan spectrum of peptide DNNLLGK clearly separating labeled (right peak) and unlabeled (left peak) version of the peptides on the m/z axis. The monoisotopic mass and the first isotope are indicated by blue marks. (B) Labeled (blue) and unlabeled (red) versions of the peptide co-elute as a single peak during nano-HPLC chromatography. (C) Full scan spectrum of a peptide with ion intensity ratio of 1:5 as a candidate for a differentially regulated protein. (D) Also proteins with ratios different from 1:1 co-elute in reversed-phase chromatography. Click here to view larger figure.
Figure 6. Expected results of quantitative protein mass spectrometry. Screen shot from MSQuant showing the expected 1:1 ion intensity ratio of a peptide from a 1:1 mixture of protein extracts from labeled and unlabeled Arabidopsis cells. (A) Full scan spectrum of peptide DNNLLGK clearly separating labeled (right peak) and unlabeled (left peak) version of the peptides on the m/z axis. The monoisotopic mass and the first isotope are indicated by blue marks. (B) Labeled (blue) and unlabeled (red) versions of the peptide co-elute as a single peak during nano-HPLC chromatography. (C) Full scan spectrum of a peptide with ion intensity ratio of 1:5 as a candidate for a differentially regulated protein. (D) Also proteins with ratios different from 1:1 co-elute in reversed-phase chromatography. Click here to view larger figure.
Discussion
The protocol presented in this paper contains many steps and all of them are crucial to obtain pure and representative fractionation of the plant plasma membrane into detergent resistant membranes and detergent soluble fractions. Therefore, it is important to follow each step as instructed.
Treatment of the plasma membrane fraction with non-ionic detergent (step 3.2) has the strongest influence on the quality of membrane microdomain fractionation. To obtain reproducible results between different preparations, and to be able to compare different samples from the same experiment, it is very important to precisely evaluate the concentration of proteins in plasma membrane and to apply the detergent always in the same ratio with respect to protein content and always to the same final concentration. Practically, it means that sometimes it is necessary to dilute samples with high protein concentration or to use different concentration stocks of Triton X-100.
In biochemical studies, recently there has been a tendency among researchers to use a detergent-free method for isolation of membrane microdomains 28-30. These methods are based on a mechanical disruption of the plasma membrane by shearing through a very small needle in analogy to the French pressure cell press. These procedures were successfully applied to mammalian cell cultures rich in membrane microdomains, but application to cell wall containing organisms (yeast, plant cells) has not been reported. Using quantitative proteomics together with sterol-disrupting agents, such as methyl-beta-cyclodextrin, it was shown that the DRM fractions prepared from plant plasma membrane do contain proteins that are markers for membrane microdomains 11.
Regarding the final results of mass spectrometric quantitation, the typical results shown in Figure 6 represent the ideal situation in which the ion intensity for the majority of labeled and unlabeled peptides are close to identical. However, in some cases a certain degree of biological variation between the sets of labeled and unlabeled cell cultures can be observed. Thus, before biological treatment of either labeled or unlabeled cell culture is carried out, analysis of a mixture of labeled and unlabeled control cells, both without any treatment, allows identification of peptides with a ratios divergent from the ideal one to one situations. These proteins with divergent ratios are indications for biological variation. To overcome the challenge of distinguishing treatment effects from biological variation, we therefore propose to use the reciprocal labeling in paired experiments 20. In the reciprocal experimental setup, both variants of the experiment are performed as proposed in Figure 1 and the steps of the protocol are followed as described. Then, comparing the ion intensity ratios of proteins from both of the reciprocal experiments, one can distinguish if the differences between ion intensity ratios are due to biological variation or as an effect of the applied treatment.
Another method for addressing the problem differentiating between treatment effects and biological variation is the use of an internal labeled standard as a reference for all the treatments 31. In that approach, unlabeled control and unlabeled cells that undergo biological treatment are mixed with the same amount of heavy labeled control cells, coming from the same batch. It allows eliminating the influence of biological variation on peptide ratio, because proteins are considered as significant, if ion intensity ratios of 14N-control/15N-control and 14N-treatment/15N-control are significantly different. This is possible, as the labeled standard is the exact same in all cases.
One of the drawbacks of the DRM enrichment procedure described here are the co-purifying proteins that can be identified in the detergent resistant membrane fraction. In the list of proteins identified from the low-density detergent-resistant membrane fraction, we can regularly observe a large number of ribosomal proteins. Due to the relatively low density of the ribosomal proteins, they migrate in the same fraction as sterol dependent membrane proteins in the sucrose gradient. It was shown without any doubts that the ribosomal proteins are clearly not constituents of membrane microdomains 11. To exclude these and other co-purifying proteins not actually associated with the detergent resistant low-density membrane fractions, we propose to analyze also the proteomic composition of the intracellular membranes (IM) which can be extracted from the lower dextran phase from the two-phase system and to compare the abundance ratios of putative contaminants between the inner membrane and the DRM fraction (see examples in Figure 5). DRM-co-purifying proteins should have similar abundances in both fractions (IM and DRM).
Fractionation of cell extracts and membrane microdomains is a common strategy to reduce complexity in a sample for proteomic analysis 32. Therefore, the protocol described here is useful for all studies of signaling processes at the plasma membrane of plant cells. Biological applications are in studying stress responses as well as pathogen infections which all to a large degree induce alterations in plasma membrane composition and modification of membrane proteins 14,15. Besides studying stress responses, quantitation of protein abundances in different membrane fractions can greatly aid annotation of yet unknown proteins 33 34,35.
Disclosures
The authors declare that they have no competing financial interests.
The author, Witold Szymanski, is an employee of the Max Planck Institute of Molecular Plant Physiology. The author, Waltraud Schulze is an employee at the University of Hohenheim, Germany.
References
- Alexandersson E, Saalbach G, Larsson C, Kjellbom P. Arabidopsis plasma membrane proteomics identifies components of transport, signal transduction and membrane trafficking. Plant Cell Physiol. 2004;45:1543–1556. doi: 10.1093/pcp/pch209. [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Lanquar V, et al. 15N-metabolic labeling for comparative plasma membrane proteomics in Arabidopsis cells. Proteomics. 2007;7:750–754. doi: 10.1002/pmic.200600791. [DOI] [PubMed] [Google Scholar]
- Kierszniowska S, Walther D, Schulze WX. Ratio-dependent significance thresholds in reciprocal 15N-labeling experiments as a robust tool in detection candidate proteins responding to biological treatment. Proteomics. 2009;9:1916–1924. doi: 10.1002/pmic.200800443. [DOI] [PubMed] [Google Scholar]
- Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–5672. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Baumgart T, et al. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvius JR. Partitioning of membrane molecules between raft-and non raft-domains: insights from model membrane studies. Biochim. Biophys. Acta. 2005;1746:193–202. doi: 10.1016/j.bbamcr.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Tanner W, Malinsky J, Opekarova M. In plant and animal cells, detergent-resistant membranes do not define functional membrane rafts. Plant Cell. 2011. [DOI] [PMC free article] [PubMed]
- Lauwers E, Andre B. Association of yeast transporters with detergent-resistant membranes correlates with their cell-surface location. Traffic. 2006;7:1–15. doi: 10.1111/j.1600-0854.2006.00445.x. [DOI] [PubMed] [Google Scholar]
- Kierszniowska S, Seiwert B, Schulze WX. Definition of Arabidopsis sterol-rich membrane microdomains by differential treatment with methyl-ß-cyclodextrin and quantitative proteomics. Mol. Cell. Proteomics. 2009;8:612–623. doi: 10.1074/mcp.M800346-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Kinoshita T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 2011;50 doi: 10.1016/j.plipres.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Keinath NF, et al. PAMP-induced changes in plasma membrane compartmentalization reveal novel components of plant immunity. J. Biol. Chem. 2010;285:39140–39149. doi: 10.1074/jbc.M110.160531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanislas T, et al. Quantitative proteomics reveals a dynamic association of proteins to detergent resistant membranes upon elicitor signaling in tobacco. Mol. Cell. Proteomics. 2009;8:2186–2198. doi: 10.1074/mcp.M900090-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami A, et al. Alterations in detergent-resistant plasma membrane microdomains in Arabidopsis thaliana during cold acclimation. Plant Cell Physiol. 2008;50:341–359. doi: 10.1093/pcp/pcn202. [DOI] [PubMed] [Google Scholar]
- Pauly M, Eberhard S, Albersheim P, Darvill A, York WS. Effects of the mur1 mutation on xyloglucans produced by suspension-cultured Arabidopsis thaliana cells. Planta. 2001;214:67–74. doi: 10.1007/s004250100585. [DOI] [PubMed] [Google Scholar]
- Jouanneau JP, Peaud-Lenoel C. Growth and synthesis of proteins in cell suspensions of a kinetin dependent tobacco. Physiol. Plant. 1967;20:834–850. [Google Scholar]
- Engelsberger WR, Erban A, Kopka J, Schulze WX. Metabolic labeling of plant cell cultures with K15NO3 as a tool for quantitative analysis of proteins and metabolites. Plant Methods. 2006;2:1–11. doi: 10.1186/1746-4811-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsova B, Kierszniowska S, Schulze WX. The use of heavy nitrogen in quantitative proteomics experiments in plants. Trends Plant Sci. 2012;17:102–112. doi: 10.1016/j.tplants.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. Stop And Go Extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Macek B. High accuracy mass spectrometry in large-scale analysis of protein phosphorylation. Methods Mol. Biol. 2009;492:131–142. doi: 10.1007/978-1-59745-493-3_7. [DOI] [PubMed] [Google Scholar]
- Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 2004;76:3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- Frese CK, et al. Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ-Orbitrap Velos. Journal of Protrome Research. 2011;10:2377–2388. doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- van Breukelen B, vanden Toorn HW, Drugan MM, Heck AJ. StatQuant: a post-quantification analysis toolbox for improving quantitative mass spectrometry. Bioinformatics. 2009;25:1472–1473. doi: 10.1093/bioinformatics/btp181. [DOI] [PubMed] [Google Scholar]
- Zauber H, Schulze WX. Proteomics wants cRacker: Automated standardized data analysis of LC/MS derived proteomic data. J. Proteome Res. 2012;11:5548–5555. doi: 10.1021/pr300413v. [DOI] [PubMed] [Google Scholar]
- Raffaele S, Mongrand S, Gamas P, Niebel A, Ott T. Genome-wide annotation of remorins, a plant-specific protein family: Evolutionary and functional perspectives. Plant Physiol. 2007;145:593–600. doi: 10.1104/pp.107.108639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MB, Sehgal PB. Nondetergent isolation of rafts. Methods Mol. Biol. 2007;398:21–28. doi: 10.1007/978-1-59745-513-8_3. [DOI] [PubMed] [Google Scholar]
- Persaud-Sawin D-A, Lightcap S, Harry GJ. Isolation of rafts from mouse brain tissue by a detergent-free method. Journal of Lipid Research. 2009;50:759–767. doi: 10.1194/jlr.D800037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald LJ, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. Journal of Lipid Research. 2005;46:1061–1067. doi: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- Mühlhaus T, Weiss J, Hemme D, Sommer F, Schroda M. Quantitative shotgun proteomics using a uniform 15N-labeled standard to monitor proteome dynamics in time course experiments reveals new insights into the heat stress response of Chlamydomonas reinhardtii. Mol. Cell. Proteomics. 2011;10:M110.004739. doi: 10.1074/mcp.M110.004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze W, Usadel B. Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 2010;61:491–516. doi: 10.1146/annurev-arplant-042809-112132. [DOI] [PubMed] [Google Scholar]
- Foster LJ, de Hoog C, Mann M. Unbiased quantiative proteomics of lipid rafts reveales high specificity for signaling factors. Proc. Natl. Acad. Sci. USA. 2003;100:5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley KS, Dupree P. Plant organelle proteomics. Curr. Opin. Plant Biol. 2007;10:594–599. doi: 10.1016/j.pbi.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Sadowski PG, Groen AJ, Dupree P, Lilley KS. Sub-cellular localization of membrane proteins. Proteomics. 2008;8:3991–4011. doi: 10.1002/pmic.200800217. [DOI] [PubMed] [Google Scholar]