

Abstract
cAMP-stimulated anion conductance is defective in cystic fibrosis (CF). The regulatory domain of CFTR, the anion channel protein encoded by the CF gene, possesses an unusually high density of consensus sequences for phosphorylation by protein kinase A (14 in a stretch of <200 amino acids). Thus it is not surprising that CFTR is viewed primarily as a cAMP-stimulated anion channel, and most studies have focused on this mode of activation. However, there is growing evidence that CFTR also responds to Ca2+-mobilizing secretagogues and contributes substantially to cholinergic and purinergic responses in native tissues. G protein-coupled receptors that signal through Gαq can stimulate CFTR channels by activating Ca2+-dependent adenylyl cyclase and tyrosine kinases, and also by inhibiting protein phosphatase type 2A. Here we review evidence for these novel mechanisms of CFTR activation and discuss how they may help explain previous observations.
 |
Arnaud Billet (left) received his Doctorate degree in cellular and molecular biology from the Université de Poitiers (France) for studies of CFTR structure–function. He currently works as a Postdoctoral Fellow in the laboratory of John Hanrahan within the Cystic Fibrosis Translational Research centre (CFTRc) at McGill University (Canada). John W. Hanrahan (right) became interested in epithelial Cl− transport during his Doctoral work at the University of British Columbia. He has studied CFTR channels in epithelial cells since 1989 and in various expression systems since 1990.
Introduction
Fluid secretion into the airway lumen is driven by Cl− transport, which is mediated by at least two types of anion channels that respond to different stimuli. Secretagogues that elevate cAMP such as vasoactive intestinal peptide, adrenaline (epinephrine), adenosine and serine proteases stimulate secretion by causing protein kinase A (PKA)-mediated phosphorylation of the anion channel CFTR (cystic fibrosis transmembrane conductance regulator). By contrast, secretagogues that mobilize Ca2+ from endoplasmic reticulum (ER) stores such as acetylcholine and ATP induce secretion by stimulating Ca2+-activated Cl− channels (CaCCs) such as the recently identified TMEM16A (reviewed by Frizzell & Hanrahan, 2012).
This paradigm of separate anion conductances activated by cAMP and Ca2+ has provided a framework for understanding epithelial transport since early studies suggested that cAMP-dependent secretion is defective in CF whereas Ca2+-activated secretion is preserved (Sato & Sato, 1984; Anderson & Welsh, 1991). It also fits with the classical scheme for signalling by G protein-coupled receptors (GPCRs), which utilize cAMP and Ca2+ as second messengers for Gs- and Gq/11-coupled receptors, respectively. The importance of cAMP/PKA is further suggested by the exceptionally large number of consensus sequences for PKA phosphorylation on the regulatory (R) domain of CFTR. Early functional studies of CFTR using inside-out membrane patches and planar lipid bilayers revealed that the catalytic subunit of PKA + MgATP can activate CFTR channels whereas exposure to Ca2+ either alone, or in combination with calmodulin, has no effect, in contrast to the CaCCs characterized previously in exocrine glands (Evans & Marty, 1986).
Convergent cAMP and Ca2+ signalling by muscarinic agonists
Despite all the evidence for two separate anion conductances that respond specifically to cAMP or Ca2+, some studies indicate overlap between these pathways. For example in pancreatic duct cells, Ca2+ signals were shown to stimulate CFTR-dependent HCO3− secretion (Namkung et al. 2003). The results were interpreted in the context of apical anion exchange, but Ca2+ may also have stimulated CFTR conductance, which is thought to mediate substantial HCO3− efflux in guinea pig and mouse pancreatic ducts (Ishiguro et al. 2009). Moreover, Ca2+-stimulated anion secretion may not be as well preserved in CF as has been widely assumed. Carbachol-stimulated HCO3− secretion is reduced by 75–100% in the intestine of cftr null mice (Hogan et al. 1997; Seidler et al. 1997). In submucosal glands from human airways, the Ca2+-mobilizing muscarinic agonist pilocarpine stimulates robust fluid secretion, and this response is reduced by ∼60% in CF glands. The apparent decrease of 60% is even more striking when one considers that it is probably an underestimate, and that the rates would have been reduced more if normalized per unit area of epithelium to correct for 2- to 3-fold hypertrophy of CF glands. It is not immediately obvious why pilocarpine-induced secretion should be compromised in CF submucosal glands if it occurs via CaCCs, yet it is a consistent observation in glands from CF piglet trachea (Joo et al. 2010) and nasal turbinate (Cho et al. 2011), and tracheal xenografts (Sun et al. 2010). Pig glands exposed to the inhibitor CFTRinh-172 (Thiagarajah et al. 2004) also have diminished responses to cholinergic agonists. Thus in some tissues, the premise that muscarinic (i.e. Ca2+-activated) secretion is preserved in CF does not hold up well under scrutiny.
Serous cells express both CFTR and CaCCs, and respond to VIP and muscarinic stimuli. Consequently they can generate responses that are synergistic at several levels. One synergy occurs when cAMP enhances Ca2+ release from intracellular stores by stimulating PKA phosphorylation of the inositol trisphosphate (IP3) receptor. The IP3 receptor normally interacts with a protein called IRBIT (IP3 receptor-binding protein released with IP3), which inhibits IP3 receptor channel function and is released when the receptor binds IP3 (Ando et al. 2003). After dissociating from the endoplasmic reticulum, IRBIT can interact with several acid–base transporters and regulate their activity at the plasma membrane, including the sodium bicarbonate cotransporter NBCe1-B and CFTR (Yang et al. 2011). IRBIT can mediate synergism between cAMP and Ca2+ because its release from the endoplasmic reticulum is enhanced by PKA phosphorylation of the IP3 receptor (Park et al. 2013). Conversely, carbachol can amplify the response to VIP by stimulating Ca2+-activated high conductance and intermediate conductance K+ channels, thereby hyperpolarizing the cell membrane and increasing the driving force for apical anion efflux. cAMP and Ca2+ signalling during transepithelial transport has been discussed in a recent review article (Kunzelmann & Mehta, 2013), which emphasizes the upregulation of Ca2+ signalling in CF cells (Antigny et al. 2007; Balghi et al. 2011; Martins et al. 2011) and functional interactions between CFTR and the Ca2+-activated Cl− channel TMEM16A (Kunzelmann et al. 2012).
Cross-talk and synergy between VIP and cholinergic signalling have been appreciated since 1982 (Schultzberg et al. 1982) and are clearly important, but they do not explain why cholinergic responses are 60% smaller in CF glands than in non-CF glands. In this topical review we argue that CFTR itself mediates a substantial fraction of the ‘CaCC conductance’ during muscarinic stimulation of airway submucosal glands and perhaps some other tissues, and that CFTR activation results from Ca2+-dependent phosphorylation by both PKA and tyrosine kinases. A contribution of CFTR to the cholinergic response of airway submucosal glands has already been suggested (Cho et al. 2011), and carbachol regulation of CFTR has recently been demonstrated directly by co-expressing CFTR together with the type 3 muscarinic receptor (M3R) in baby hamster kidney (BHK) cells (Billet et al. 2013). Remarkably, CFTR currents stimulated by carbachol in this heterologous expression system were about 40% larger than those induced by forskolin.
Cholinergic stimulation of airway epithelial cells is mediated by the M3R and, to a lesser extent, the M1 receptor. The M3R signals through Gαq/11 to phospholipase C, which generates IP3 and mobilizes Ca2+ from intracellular stores (Berridge, 2012) (see http://www.cellsignallingbiology.org). Although CFTR is not stimulated directly by Ca2+, several adenylyl cyclase isoforms are Ca2+ activated (types 1, 8 and perhaps 3; Halls & Cooper, 2011) and thus could couple the phosphorylation of CFTR by PKA to muscarinic Ca2+ responses. This regulation may involve a localized pool of cAMP near the plasma membrane (Monterisi et al. 2012), which is isolated from the bulk cytoplasm by phosphodiesterases 4D (Barnes et al. 2005) and/or 3A (Penmatsu et al. 2010).
Tyrosine phosphorylation contributes to muscarinic stimulation of CFTR
Activation of CFTR by carbachol-induced elevation of cAMP was recently demonstrated in BHK cells that coexpress CFTR and M3R (Billet et al. 2013). However, to our surprise, only about half of the cholinergic response could be attributed to Ca2+ activation of adenylyl cyclase; ∼50% of CFTR's response to carbachol persisted when PKA was inhibited by Rp-cAMPS. Similar results were obtained using a CFTR mutant that is unresponsive to forskolin due to removal of 15 PKA consensus sequences (Seibert et al. 1999). Thus about half of CFTR's response to carbachol does not involve activation of adenylyl cyclase by Ca2+, yet the CFTR response to carbachol was clearly Ca2+ dependent since it was strongly inhibited by buffering intracellular Ca2+ with BAPTA-AM (Fig. 1). This raises the question, ‘What Ca2+-dependent pathway mediates CFTR activation by carbachol when the response of CFTR to PKA is blocked?’
Figure 1. Time course and current–voltage relations of CFTR activation by carbachol and their dependence on intracellular Ca2+.
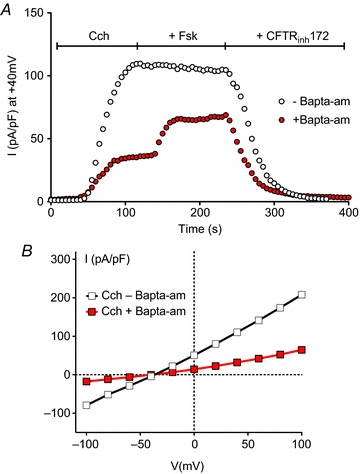
A, representative whole cell currents measured in BHK cells made to co-express the muscarinic type 3 receptor and CFTR. Cells were exposed sequentially to carbachol (Cch), forskolin (Fsk) and CFTRinh172, each at 10 μmol l−1. Experiments were performed with or without 60 min preincubation with the membrane-permeant Ca2+ buffer BAPTA-AM (500 μmol l−1) in the bath and pipette solutions. Note that stimulation was more robust with carbachol than with forskolin, that BAPTA-AM inhibited the Cch response >60%, and the CFTR inhibitor CFTRinh172 virtually abolished the response to Cch + Fsk. B, current–voltage relationship for currents measured in A during the plateau phase of stimulation.
Exposing excised patches to the tyrosine kinase Src increases fast CFTR channel gating and open probability (Fischer & Machen, 1996). Although the upstream stimulus for Src was not identified, many GPCRs are now known to recruit and activate Src. Therefore we examined the possible role of Src during muscarinic stimulation of CFTR and found that Src Inhibitor-1 (Inh-1) abolished the PKA-independent component of muscarinic stimulation (Billet et al. 2013). To explain how a brief Ca2+ transient during carbachol exposure might activate Src, we also investigated Pyk2, a Ca2+-activated, proline-rich tyrosine kinase related to focal adhesion kinase (FAK). Pyk2 forms a complex with Src thereby facilitating their reciprocal phosphorylation and activation. Pyk2 has already been implicated in the regulation of CFTR (Liang et al. 2011) as has FAK itself (Marshall et al. 2009). Interestingly, like Src Inh-1, the specific Pyk2 inhibitor tyrphostin A9 abolished the PKA-independent component of CFTR stimulation by carbachol. These and other results led us to propose that Ca2+ activates CFTR, at least in part, through activation of the Pyk2/Src complex and tyrosine phosphorylation (Fig. 2).
Figure 2. Simplified scheme for Ca2+ activation of CFTR via PKA and tyrosine kinases.
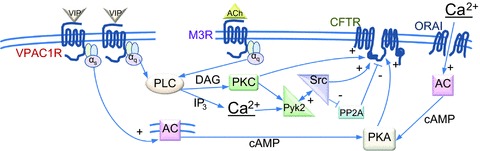
VIP receptors couple strongly to Gαs and weakly to Gαq, which stimulate adenylyl cyclase (AC) and phospholipase C (PLC), respectively. The muscarinic type 3 receptor (M3R) couples strongly to Gαq. cAMP activates CFTR channels by means of the canonical PKA pathway. Diacylglycerol and inositol trisphosphate (IP3) activate protein kinase C (PKC) and mobilize Ca2+, which in turn activate the Pyk2/Src complex. Src stimulates CFTR activity by phosphorylating it directly and also by inhibiting its dephosphorylation through inactivation of protein phosphatase type 2A (PP2A). Under basal conditions, constitutive Ca2+ entry through store-operated Ca2+ channels partially activates adenylyl cyclase and induces tonic CFTR activity.
This tyrosine kinase pathway suggests yet another mode of cross-talk between the Ca2+ and cAMP signalling pathways through the activity of a protein phosphatase. Protein phosphatase type 2A (PP2A) is known to interact with, and dephosphorylate, CFTR (Hwang et al. 1994; Luo et al. 1998; Thelin et al. 2005) and is strongly inhibited when phosphorylated by Src at residue Y307 (Chen et al. 1992). Thus Ca2+-dependent activation of Src (presumably through its association with Pyk2) can enhance channel activity by inhibiting the dephosphorylation of PKA sites on CFTR. In support of this idea, Src Inh-1 causes a ∼2-fold greater inhibition of carbachol-stimulated current when tested alone than when tested with calyculin A (a specific inhibitor of PP2A) present to mitigate its action on the phosphatase. These results indicate that Src normally suppresses PP2A activity, and when this inhibition is partially relieved by Src Inh-1 the dephosphorylation of PKA sites is enhanced. Calyculin A would prevent the upregulation of PP2A activity by Src Inh-1, thereby reducing the relative inhibition of the carbachol response by Src Inh-1. Exposing CFTR to protein kinase C (PKC) enhances subsequent activation of the channel by PKA, and since some conventional PKC isoforms are Ca2+ dependent, this suggests yet another mechanism by which Ca2+ could regulate CFTR. A CFTR mutant lacking nine predicted PKC sites was partially activated by M3 muscarinic receptors, apparently through tyrosine phosphorylation since the response was abolished by tyrosine kinase inhibitors (Billet et al. 2013). Although the ‘priming’ effect of PKC is not required for activation of CFTR by tyrosine phosphorylation, further studies are needed to establish whether Ca2+-dependent PKC activity enhances the component of the muscarinic response that is mediated by PKA.
In summary, CFTR can operate as a Ca2+-activated Cl− channel and respond to cholinergic stimulation. There is evidence for at least three Ca2+-dependent mechanisms of CFTR activation: (1) upregulation of adenylyl cyclase by Ca2+ leading to conventional PKA-mediated phosphorylation of CFTR, (2) stimulation of Pyk2/Src, which inhibits PP2A and thus increases serine phosphorylation on CFTR, and (3) direct tyrosine phosphorylation of CFTR by the Pyk2/Src complex.
Basal CFTR activity in epithelial cells: possible role of Ca2+ entry
Calcium mobilization is necessary for CFTR activation during muscarinic stimulation, but does Ca2+ also influence basal CFTR activity? This was explored using unstimulated Calu-3 monolayers, which have substantial anion secretion (∼1 μequiv cm−2 h−1) that is abolished by the CFTR inhibitor GlyH101. Basal anion secretion was inhibited 68–100% by the adenylyl cyclase inhibitor MDL-12330A and 68–88% by the PKA inhibitor Rp-cAMPS (Shan et al. 2012). Those pharmacological experiments suggested that low-level phosphorylation by PKA maintains a basal rate of anion secretion. Since basal anion secretion was strongly inhibited (65–90%) by 2-APB, an antagonist of store-operated Ca2+ entry, we proposed that slow leakage into resting cells stimulates Ca2+-dependent adenylyl cyclase, raising [cAMP] locally and leading to partial PKA activation of CFTR (Fig. 2). Thus CFTR may contribute directly to Ca2+-activated Cl− conductance even in the absence of secretagogue. Moreover Ca2+ signalling is regulated by CFTR, which suppresses Ca2+ release from intracellular stores (Antigny et al. 2007) and reduces Ca2+ influx through its modulation of store-operated Ca2+ channels (ORAI1; Balghi et al. 2011). CFTR normally inhibits the insertion of ORAI1 channels and suppresses Ca2+ entry by ∼50%; however, this downregulation of Ca2+ entry is lost in CF cells, resulting in exaggerated Ca2+ influx during stimulation by secretagogues that cause depletion of Ca2+ stores. In summary, although there is growing evidence that Ca2+ signalling and CFTR function are interdependent, the precise mechanisms remain to be determined. Ca2+ clearly mediates most of the CFTR response to muscarinic stimulation and probably contributes to tonic CFTR activity in some cell types.
Conclusions
Further studies of the CaCC activity of CFTR should clarify CFTR's role in anion secretion and other cellular processes. It has been a longstanding puzzle that loss of CFTR function in airway submucosal glands should have such dire consequences when the more robust CaCC response to carbachol is preserved. This becomes more understandable if CFTR contributes significantly to both Ca2+- and cAMP-mediated secretagogues, although we note that the CaCC function of CFTR depends on cell type and may not occur in all tissues. For example, the response of salivary secretion to cholinergic stimulation is not affected noticeably in cftr null mice (Best & Quinton, 2005), suggesting it is mediated by bona fide CaCCs. Further studies should examine whether these differences in CFTR regulation arise at the level of receptors or downstream signalling. If other CaCCs are present in large numbers, they may dominate responses to moderate stimuli. Also, the CaCC activity of CFTR may be obscured during maximal stimulation if apical anion conductance is not the rate-limiting step for secretion.
The identification of TMEM16A (also known as ANO1, DOG1, ORAOV2 and TAOS2) as a CaCC has been an exciting development in the field. Its role in airway and intestinal epithelia has been questioned based on studies with the new TMEM16A inhibitor T16Ainh-A01, which is a potent blocker of TMEM16A but has little effect on CaCC in those tissues (Namkung et al. 2011). This discrepancy may reflect the expression of alternatively spliced forms of TMEM16A, which differ in their functional properties and are expressed in a tissue-specific manner (Ferrera et al. 2009), or perhaps the presence of other members of the anoctamin family, which may differ in their sensitivity to pharmacological inhibitors. It will be interesting to see if residual CFTR activity also contributes to the T16Ainh-A01-insensitive component in airway and intestinal epithelia. This would be important because pharmacological stimulation of Ca2+-activated Cl− conductance has been proposed as a therapeutic strategy for bypassing defective CFTR, but would not succeed if CFTR itself contributed a substantial fraction of the CaCC activity. Finally, elevated cytokine release by CF epithelial cells has been reported by many groups and can be suppressed by expressing an anion conductance even in the absence of secretagogue (Veit et al. 2012). The paradox that CFTR must be functional to suppress cytokine release, yet does not need to be activated by a secretagogue, might be explained if Ca2+ entry caused tonic CFTR activation as observed in Calu-3 (Shan et al. 2012). While there is little doubt that the CFTR channel functions (albeit indirectly) as a CaCC, we are just beginning to appreciate the complex relationship between intracellular Ca2+ signalling and CFTR-mediated secretion.
Acknowledgments
None.
Additional information
Competing interests
None declared.
Funding
A.B. was supported by fellowships from the Groupe d’étude des protéines membranaires (GÉPROM), the Groupe de Recherche Axé sur la Structure des Protéines (GRASP) and by the CIHR Program in Chemical Biology at McGill. The research was supported by the Canada Foundation for Innovation and Canadian Institutes for Health Research.
References
- Anderson MP, Welsh MJ. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc Natl Acad Sci U S A. 1991;88:6003–6007. doi: 10.1073/pnas.88.14.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H, Mizutani A, Matsu-ura T, Mikoshiba K. IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J Biol Chem. 2003;278:10602–10612. doi: 10.1074/jbc.M210119200. [DOI] [PubMed] [Google Scholar]
- Antigny F, Norez C, Becq F, Vandebrouck C. Calcium homeostasis is abnormal in cystic fibrosis airway epithelial cells but is normalized after rescue of F508del-CFTR. Cell Calcium. 2007;43:175–183. doi: 10.1016/j.ceca.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Balghi H, Robert R, Rappaz B, Zhang X, Wohlhuter-Haddad A, Evagelidis A, Luo Y, Goepp J, Ferraro P, Roméo P, Trebak M, Wiseman PW, Thomas DY, Hanrahan JW. Cystic fibrosis enhances store-operated Ca2+ entry by increasing plasma membrane insertion of Orai1. FASEB J. 2011;25:4274–4291. doi: 10.1096/fj.11-187682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Livera G, Huang P, Sun C, O’Neal WK, Conti M, Stutts MJ, Milgram SL. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem. 2005;280:7997–8003. doi: 10.1074/jbc.M407521200. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Cell Signalling Biology. 2012. doi: 10.1042/csb0001002. [Google Scholar]
- Best JA, Quinton PM. Salivary secretion assay for drug efficacy for cystic fibrosis in mice. Exp Physiol. 2005;90:189–193. doi: 10.1113/expphysiol.2004.028720. [DOI] [PubMed] [Google Scholar]
- Billet A, Luo Y, Balghi H, Hanrahan JW. Role of tyrosine phosphorylation in the muscarinic activation of the cystic fibrosis transmembrane conductance regulator (CFTR) J Biol Chem. 2013;288:21815–21823. doi: 10.1074/jbc.M113.479360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- Cho H-J, Joo NS, Wine JJ. Defective fluid secretion from submucosal glands of nasal turbinates from CFTR−/– and CFTRΔF508/ΔF508 pigs. PloS One. 2011;6:e24424. doi: 10.1371/journal.pone.0024424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MG, Marty A. Calcium-dependent chloride currents in isolated cells from rat lacrimal glands. J Physiol. 1986;378:437–460. doi: 10.1113/jphysiol.1986.sp016229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, Pagani F, Galietta LJV. Regulation of TMEM16A chloride channel properties by alternative splicing. J Biol Chem. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Machen TE. The tyrosine kinase p60c-src regulates the fast gate of the cystic fibrosis transmembrane conductance regulator chloride channel. Biophys J. 1996;71:3073–3082. doi: 10.1016/S0006-3495(96)79501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzell RA, Hanrahan JW. Physiology of chloride and fluid secretion. In: Riordan JR, Boucher RC, Quinton PM, editors. Cystic Fibrosis: Molecular Basis, Physiological Changes, and Therapeutic Strategies. Cold Spring, NY, USA: Cold Spring Harbor Press; 2012. [Google Scholar]
- Halls ML, Cooper DMF. Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb Perspect Biol. 2011;3:a004143. doi: 10.1101/cshperspect.a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DL, Crombie DL, Isenberg JI, Svendsen P, Schaffalitzky de Muckadell OB, Ainsworth MA. CFTR mediates cAMP- and Ca2+-activated duodenal epithelial HCO3− secretion. Gastroenterology. 1997;113:533–541. doi: 10.1152/ajpgi.1997.272.4.G872. [DOI] [PubMed] [Google Scholar]
- Hwang T-C, Baukrowitz T, Nagel G, Horie M, Nairn AC, Gadsby DC. Regulation of the gating of cardiac CFTR Cl channels by phosphorylation and ATP hydrolysis. J Gen Physiol. 1994;104:34a. [Google Scholar]
- Ishiguro H, Steward MC, Naruse S, Ko SB, Goto H, Case RM, Kondo T, Yamamoto A. CFTR functions as a bicarbonate channel in pancreatic duct cells. J Gen Physiol. 2009;133:315–326. doi: 10.1085/jgp.200810122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo NS, Cho H-J, Khansaheb M, Wine JJ. Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J Clin Invest. 2010;120:3161–3166. doi: 10.1172/JCI43466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann K, Mehta A. CFTR: a hub for kinases and cross-talk of cAMP and Ca2+ FEBS J. 2013;280:4417–4429. doi: 10.1111/febs.12457. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Tian Y, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, Wolf L, Schreiber R. Airway epithelial cells – functional links between CFTR and anoctamin dependent Cl− secretion. Int J Biochem Cell Biol. 2012;44:1897–1900. doi: 10.1016/j.biocel.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Liang L, Woodward OM, Chen Z, Cotter R, Guggino WB. A novel role of protein tyrosine kinase2 in mediating chloride secretion in human airway epithelial cells. PloS One. 2011;6:e21991. doi: 10.1371/journal.pone.0021991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Pato MD, Riordan JR, Hanrahan JW. Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am J Physiol Cell Physiol. 1998;274:C1397–C1410. doi: 10.1152/ajpcell.1998.274.5.C1397. [DOI] [PubMed] [Google Scholar]
- Marshall WS, Watters KD, Hovdestad LR, Cozzi RR, Katoh F. CFTR Cl− channel functional regulation by phosphorylation of focal adhesion kinase at tyrosine 407 in osmosensitive ion transporting mitochondria rich cells of euryhaline killifish. J Exp Biol. 2009;212:2365–2377. doi: 10.1242/jeb.030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins JR, Kongsuphol P, Sammels E, Dahimene S, Aldehni F, Clarke LA, Schreiber R, de Smedt H, Amaral MD, Kunzelmann K. F508del-CFTR increases intracellular Ca2+ signaling that causes enhanced calcium-dependent Cl− conductance in cystic fibrosis. Biochim Biophys Acta. 2011;1812:1385–1392. doi: 10.1016/j.bbadis.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Monterisi S, Favia M, Guerra L, Cardone RA, Marzulli D, Reshkin SJ, Casavola V, Zaccolo M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J Cell Sci. 2012;125:1106–1117. doi: 10.1242/jcs.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Lee JA, Ahn W, Han W, Kwan SW, Ahn DS, Kim KH, Lee MG. Ca2+ activates cystic fibrosis transmembrane conductance regulator- and Cl−-dependent HCO3− transport in pancreatic duct cells. J BiolChem. 2003;278:200–207. doi: 10.1074/jbc.M207199200. [DOI] [PubMed] [Google Scholar]
- Namkung W, Phuan P-W, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Shcheynikov N, Hong JH, Zheng C, Suh SH, Kawaai K, Ando H, Mizutani A, Abe T, Kiyanari H, Seki G, Yule D, Mikoshiba K, Muallem S. Irbit mediates synergy between Ca2+ and cAMP signaling pathways during epithelial transport in mice. Gastroenterology. 2013;145:232–241. doi: 10.1053/j.gastro.2013.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsu H, Zhang W, Yarlagadda S, Li C, Conoley VG, Yue J, Bahouth SW, Guddington RK, Zhang G, Nelson DJ, Sonecha MD, Manganiello V, Wine JJ, Naren AP. Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol Biol Cell. 2010;21:1097–1110. doi: 10.1091/mbc.E09-08-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Sato F. Defective beta adrenergic response of cystic fibrosis sweat glands in vivo and in vitro. J Clin Invest. 1984;73:1763–1771. doi: 10.1172/JCI111385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultzberg M, Hökfelt T, Lundberg JM. Coexistence of classical transmitters and peptides in the central and peripheral nervous systems. Br Med Bull. 1982;38:309–313. doi: 10.1093/oxfordjournals.bmb.a071778. [DOI] [PubMed] [Google Scholar]
- Seibert FS, Chang X-B, Aleksandrov AA, Clarke DM, Hanrahan JW, Riordan JR. Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochim Biophys Acta. 1999;1461:275–283. doi: 10.1016/s0005-2736(99)00163-7. [DOI] [PubMed] [Google Scholar]
- Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent HCO3− secretion. J Physiol. 1997;505:411–423. doi: 10.1111/j.1469-7793.1997.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Liao J, Huang J, Robert R, Palmer ML, Fahrendrug SC, O’Grady SM, Hanrahan JW. Bicarbonate-dependent chloride transport drives fluid secretion by the human airway epithelial cell line Calu-3. J Physiol. 2012;590:5273–5297. doi: 10.1113/jphysiol.2012.236893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ, Joo NS, Zhang Y, Zhou W, Yi Y, Kinyon JM, Lei-Butters DC, Griffin MA, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine JJ, Meyerholz DK, Engelhardt JF. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelin WR, Kesimer M, Tarran R, Kreda SM, Grubb BR, Sheehan JK, Stutts MJ, Milgram SL. The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J Biol Chem. 2005;280:41512–41520. doi: 10.1074/jbc.M507308200. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Song Y, Haggie PM, Verkman AS. A small-molecule CFTR inhibitor produces cystic fibrosis-like submucosal gland fluid secretions in normal airways. FASEB J. 2004;18:875–877. doi: 10.1096/fj.03-1248fje. [DOI] [PubMed] [Google Scholar]
- Veit G, Bossard F, Goepp J, Verkman AS, Galietta LJ, Hanrahan JW, Lukacs GL. Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol Biol Cell. 2012;23:4188–4202. doi: 10.1091/mbc.E12-06-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Shcheynikov N, Muallem S. IRBIT: It is everywhere. Neurochem Res. 2011;36:1166–1174. doi: 10.1007/s11064-010-0353-6. [DOI] [PMC free article] [PubMed] [Google Scholar]