

Abstract
Pathological left ventricle (LV) hypertrophy (LVH) results in reactive and replacement fibrosis. Volume overload LVH (VOH) is less profibrotic than pressure overload LVH (POH). Studies attribute subendocardial fibrosis in POH to ischaemia, and reduced fibrosis in VOH to collagen degradation favouring dilatation. However, the mechanical origin of the relative lack of fibrosis in VOH is incompletely understood. We hypothesized that reduced ischaemia propensity in VOH compared to POH accounted for the reduced replacement fibrosis, along with reduced reactive fibrosis. Rats with POH (ascending aortic banding) evolved into either compensated-concentric POH (POH-CLVH) or dilated cardiomyopathy (POH-DCM); they were compared to VOH (aorta–caval fistula). We quantified LV fibrosis, structural and haemodynamic factors of ischaemia propensity, and the activation of profibrotic pathways. Fibrosis in POH-DCM was severe, subendocardial and subepicardial, in contrast with subendocardial fibrosis in POH-CLVH and nearly no fibrosis in VOH. The propensity for ischaemia was more important in POH versus VOH, explaining different patterns of replacement fibrosis. LV collagen synthesis and maturation, and matrix metalloproteinase-2 expression, were more important in POH. The angiotensin II-transforming growth-factor β axis was enhanced in POH, and connective tissue growth factor (CTGF) was overexpressed in all types of LVH. LV resistin expression was markedly elevated in POH, mildly elevated in VOH and independently reflected chronic ischaemic injury after myocardial infarction. In vitro, resistin is induced by angiotensin II and induces CTGF in cardiomyocytes. Based on these findings, we conclude that a reduced ischaemia propensity and attenuated upstream reactive fibrotic pathways account for the attenuated fibrosis in VOH versus POH.
Key points
Pressure overload hypertrophy is profibrotic while volume overload hypertrophy is not profibrotic.
Fibrosis occurs in the form of replacement or reactive fibrosis.
Replacement fibrosis in pressure overload is considered ischaemic in origin.
There is less propensity for ischaemia in volume overload, explaining the relative lack of fibrosis.
Reactive fibrosis pathways are more active in pressure than in volume overload.
Local resistin expression reflects replacement fibrosis in chronic ischaemia.
Introduction
Left ventricular (LV) fibrosis complicates pathological LV hypertrophy (LVH) (Kehat & Molkentin, 2010) with complex functional effects (Brower et al. 2006). Experimental models of ‘pure’ pressure overload LV hypertrophy (POH) and volume overload LV hypertrophy (VOH) consistently reveal severe fibrosis in POH, with little or no fibrosis in VOH (Borer et al. 2006; Kehat & Molkentin, 2010; Toischer et al. 2010), with the exception of some rodent studies consistently reporting fibrosis in aortic regurgitation (AR) (Plante et al. 2004, 2008; Nakanishi et al. 2007; Zendaoui et al. 2012). The reasons for these differences have been explored but are incompletely understood. In contrast, clinical studies are heterogeneous in terms of fibrosis in VOH (Fuster et al. 1977; Piper et al. 2003; Borer et al. 2006; Sparrow et al. 2006; Lopes et al. 2007). Myocardial fibrosis is reported in a clinical study of mitral insufficiency on biopsy specimens (Lopes et al. 2007). In patients with aortic insufficiency, Sparrow et al. (2006) measured myocardial fibrosis by magnetic resonance imaging T1 mapping and found mildly increased T1 relaxation time in myocardial segments with abnormal motion in some patients, highly suggestive of fibrosis, with focal areas of delayed gadolinium enhancement, but there was no evidence of diffuse fibrosis. In both studies, patients were enrolled before valve surgery and care was taken to exclude coronary artery disease (Sparrow et al. 2006; Lopes et al. 2007). Nevertheless, the myocardial fibrosis in human mitral or aortic regurgitation is difficult to attribute to volume overload alone, since the degenerative or inflammatory (rheumatic) cause of the valvular disease (Sparrow et al. 2006; Lopes et al. 2007) can affect the myocardial extracellular matrix.
Collagen digestion in VOH and the complex role of MMPs
Numerous studies on VOH explain the lack of fibrosis in VOH by an imbalance between matrix metalloproteases (MMPs) and tissue inhibitors of metalloproteases (TIMP) favouring collagen degradation in AR, as opposed to aortic stenosis (AS) (Borer et al. 2006), and allowing ventricular dilatation in VOH (Borer et al. 2006; Hutchinson et al. 2010). Studies in VOH by mitral regurgitation (MR) have documented extracellular matrix loss and collagen degradation without an increase in collagen I synthesis and with a decrease in the expression and activity of profibrotic cytokines (Zheng et al. 2009). In rat VOH by aorta–caval fistula, recent studies point to inflammation-induced collagen loss as a mechanism for LV dilatation and dysfunction (Wei et al. 2012). This paradigm appears to be challenged by Beeri et al. (2008) who studied sheep with myocardial infarction (MI) and a LV-to-left atrial shunt implanted to superimpose MR on the MI. A time course comparison between MR + MI and MI alone revealed a biphasic increase–decrease in MMPs and a progressive rise in TIMP; interestingly, in that study, significant LV dilatation and dysfunction associated with the MR was observed at a later time, when MMPs had decreased and TIMP increased (Beeri et al. 2008). Thus, this latter study challenges the paradigm of collagen digestion leading to ventricular dilatation in VOH, suggesting a context-dependent and a more complex role for MMPs in LV remodelling. More recently, Zendaoui et al. (2012) studied a rat model of AR with fibrosis and found no change in the mRNA of MMP2 but an increase in the mRNA of TIMP-1 in AR.
Increased MMP activity in LVH and failure is not specific of primary VOH (Peterson et al. 2001). In rat heart failure from POH, MMP activity was increased along with increased fibrosis, with reduced LV remodelling by MMP inhibition (Peterson et al. 2001), indicating that increased MMPs alone does not reduce collagen content, and that MMPs affect ventricular remodelling beyond their collagenase activity (Peterson et al. 2001; Eckhouse & Spinale, 2012). Furthermore, in a study of mouse POH, TIMP-2 was the dominantly increased TIMP, and transverse aortic constriction on TIMP-2 knock-out increased both collagenase activity and myocardial fibrosis (Kandalam et al. 2011). This latter study and others emphasize the rather profibrotic effect of MMPs (Spinale, 2011). Together with the increased fibrosis in dilated ventricles from VOH (Nakanishi et al. 2007; Plante et al. 2008; Zendaoui et al. 2012) and other causes (Beltrami et al. 1994; van den Borne et al. 2010), these data appear to contradict a mere antifibrotic effect of volume overload leading to collagen digestion and further LV dilatation as a complete explanation for the differential fibrotic response between VOH and POH. Importantly, the remote (non-infarcted) area in myocardial infarction undergoes fibrosis, despite a volume overload physiology with lengthening of cardiac myocytes (Beltrami et al. 1994; Hutchinson et al. 2010; van den Borne et al. 2010). At least theoretically, a vicious circle of dilatation–digestion would promote LV rupture (Heymans et al. 1999), not typically observed in primary VOH. Moreover, the lack of activation or downregulation of profibrotic cytokines is documented in VOH but the origin of this pattern is not well understood (Zheng et al. 2009).
Ischaemic replacement fibrosis characterizes POH and concentric LVH
Subendocardial fibrosis in POH (Rudolph et al. 2009) and primary hypertrophic cardiomyopathies (HCM) is considered to be, at least in part, a replacement fibrosis secondary to ischaemic cardiomyocyte loss (Maron et al. 2009), with oxygen supply/demand mismatch, according to the ‘necrotic core’ hypothesis (Rudolph et al. 2009). Coronary microvascular dysfunction is reported in POH, involving vascular rarefaction, vascular compression and perivascular fibrosis (Camici & Crea, 2007). In HCM, wall thickening was associated with reduced myocardial blood flow and replacement subendocardial fibrosis (Maron et al. 2009), similar to primarily ischaemic cardiomyopathies (Beltrami et al. 1994). To our knowledge, this ischaemia–fibrosis paradigm is not studied in VOH and in direct POH–VOH comparisons; furthermore, dogs with mitral regurgitation (MR) and LV dysfunction had normal coronary flow reserve in a study by Carabello et al. (1991).
Hypotheses of the present study
In this study, we hypothesized that the relative lack of replacement fibrosis in VOH was due to less propensity for ischaemia in VOH than POH, at equal levels of LVH and LV dilatation from the two mechanisms. We systematically studied structural and mechanical factors determining myocardial oxygen supply and demand in rat models of POH with compensated and concentric LVH (CLVH), POH with LV dilatation (DCM) and VOH by aorta–caval fistula (Chemaly et al. 2012). Our study design allows us to investigate the effect of LV dilatation on fibrosis in the presence and absence of concomitant POH. We also sought to integrate the expression pattern and the actions of resistin, a novel profibrotic cytokine (Chemaly et al. 2011), with the different patterns of replacement and reactive fibrosis. The adipocytokine resistin was shown to induce myocardial hypertrophy, dysfunction and fibrosis when overexpressed in vectors (Kim et al. 2008; Chemaly et al. 2011; Kang et al. 2011). Resistin serum levels predicted clinical heart failure (Frankel et al. 2009), and resistin expression increased in diabetic hearts (Kim et al. 2008) and in POH (Kang et al. 2011).
Methods
Ethical approval and animal surgical models of POH and VOH
Animals were obtained and handled as approved by the Icahn School of Medicine at Mount Sinai's Institutional Animal Care and Use Committee in accordance with the Principles of Laboratory Animal Care by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 86-23, revised 1996).
Male Sprague–Dawley rats (body weight 70–100 g) underwent ascending aortic constriction under general anesthesia with ketamine up to 85 mg kg−1 and xylazine up to 10 mg kg−1 intraperitoneal as previously described (Sakata et al. 2007; Chemaly et al. 2012) to induce POH. Male rats (body weight 250–300 g) underwent aorta–caval fistula creation under ketamine up to 85 mg kg−1 and xylazine up to 10 mg kg−1, intraperitoneally, as previously described (Takewa et al. 2009; Chemaly et al. 2012). Rats underwent left anterior descending artery ligation, which was either permanent or followed by reperfusion, under general anesthesia with pentobarbital 60 mg kg−1 intraperitoneal as previously described (Chen et al. 2011). Sham-operated animals were used as controls.
Echocardiography
Echocardiography was performed under sedation with ketamine up to 80 mg kg−1 intraperitoneal as previously described (Chemaly et al. 2011, 2012; Arias et al. 2013) using a Vivid echocardiography apparatus with a 13–14 MHz linear array probe (General Electric, New York, NY, USA).
Animal disease model assessment and group assignment
Echocardiography 2 months after aortic constriction distinguished compensated-concentric LV hypertrophy (POH-CLVH) from dilated cardiomyopathy (POH-DCM) based on LV end-diastolic volume (EDV) >750 μl, end-systolic volume (ESV) >200 μl and ejection fraction (EF) <70% defining DCM as previously described (Chemaly et al. 2012). POH animals underwent terminal echocardiography and haemodynamics 4 months after surgery. VOH animals were studied 3 months after aorta–caval fistula creation (Fistula group). Rat models of MI were used as a source of tissue and we have previously published the characterization of these animals (Chen et al. 2011).
Invasive haemodynamics
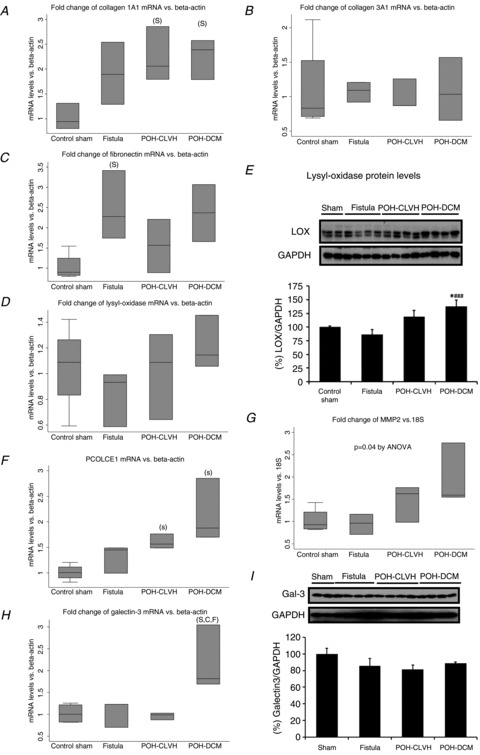
Haemodynamics were recorded subsequently through a Scisense Advantage Pressure–Volume (P–V) Control Unit (FY897B) under general anesthesia with inhaled isoflurane 0.75–1% as previously described in detail (Chemaly et al. 2012). Stroke work (SW) in mmHg ×μl was obtained from the area of the P–V loop representing LV ejection. Meridional LV wall stress σ was calculated as reported by Grossman et al. (1975) as 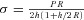 where P is LV pressure, R is LV cavity radius and h is LV wall thickness, measured at corresponding parts of the cardiac cycle (end-systole or end-diastole).
where P is LV pressure, R is LV cavity radius and h is LV wall thickness, measured at corresponding parts of the cardiac cycle (end-systole or end-diastole).
Animal humane killing and cardiac tissue sampling
Rats were killed at the end of the invasive hemodynamic measurements. Briefly, at the end of the open chest LV pressure-volume measurements, isoflurane was increased to a maximal dose of 5%, the animal was exsanguinated by sectioning the inferior vena cava and the heart removed. Right ventricle and LV were separated and weighed; then, a transverse ring of LV tissue at the mid-papillary level was embedded in OCT for histological analysis, and the rest of the LV tissue was frozen for protein and RNA extraction.
Fibrosis staining using Trichrome and Sirius Red
Frozen 8 μm tissue sections of rat LV transverse sections were stained by Masson Trichrome and Sirius Red. Areas of fibrosis (blue colour) on digital images of Trichrome-stained ×20 microscopic fields (5–12 images per animal randomly selected from tissue sections) were quantified using the software Image J (National Institutes of Health). The distinction between blue (fibres) and red (cells) on the Masson Trichrome micrographs was made possible by the decomposition of the images into red–blue–green layers. Fibrosis area quantification accounted for interstitial fibrosis and did not account for perivascular fibrosis, presented separately.
Protein extraction and Western blot
Proteins were prepared from rat LV tissue using a lysis buffer containing protease inhibitors (Roche) and phosphatase inhibitor cocktails (Sigma). Cell lysates were separated by SDS-PAGE and transferred onto nitrocellulose membrane. The membranes were blocked with 5% non-fat milk and incubated overnight at 4°C with anti-resistin (Biovision, Milpitas, CA, USA), anti-galectin-3 (GeneTex, Irvine, CA, USA) and anti-LOX (Abcam, Cambridge, MA, USA) antibodies. The membranes were incubated with appropriate secondary antibodies and signal intensities were visualized by enhanced chemiluminescence (EMD Millipore, Billerica, MA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control.
RNA extraction and quantitative real-time polymerase chain reaction
After RNA extraction and cDNA synthesis, quantitative real-time PCR was performed with SYBR-green-based detection of double-stranded DNA (Applied Biosystems 7500 cycler, Life technologies, Carlsbad, CA). Results were expressed as fold change in mRNA expression compared to the sham (reference group) using reference genes (ΔΔCt approach, fold change = 2ΔΔCt).
Capillary density measurement
Frozen 7 μm tissue sections of rat LV were stained by immunofluorescence for capillary membranes using an anti-caveolin anti-caveolin-1α antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and cardiomyocyte membranes using an anti-vinculin antibody (Sigma, St Louis, MO, USA). Appropriate secondary antibodies were used to detect the proteins of interest (Invitrogen, Eugene, OR, USA). Capillaries and cardiomyocytes were counted using Image J on digital images of ×10 fluorescence microscopic fields (one image per animal). Capillary density was quantified in capillaries per square micrometre and capillaries per cardiomyocyte.
Isolation of rat cardiomyocytes and cardiac fibroblasts, and adenoviral infection
Adult rat cardiomyocytes were isolated as previously described (Kim et al. 2008). Adult rat cardiac fibroblasts were collected from the supernatant after cardiomyocyte centrifugation, suspended in DMEM with 10% fetal bovine serum, penicillin and streptomycin (100 U ml−1) and grown in plastic culture dishes. Fibroblasts were passaged at 80% confluence and cells from passages 2–3 were used for experiments. Cardiomyocytes and fibroblasts were infected with recombinant adenoviruses expressing resistin or β-galactosidase at a concentration of 100 multiplicities of infection (MOI) as previously described (Kim et al. 2008), for 36 h. Cells of both types were also treated with angiotensin II (10 nm), endothelin 1 (10 nm) and phenylephrine (10 μm), for 36 h. At the end of the 36 h incubations, proteins were harvested from the cells. Additional analysis of the effect of adenovirus-resistin on transcription was performed by incubating the same cell types with 5 MOI of adenovirus.
Statistical analysis
Groups were compared using analysis of variance (ANOVA). Post hoc two-by-two comparisons used a Tukey–Kramer correction for multiple testing. To avoid type 2 error due to the correction for multiple testing, uncorrected P values were reported separately when pertinent (Chemaly et al. 2012). Graphic representation of data in box plots show the median value flanked by the 25th and 75th percentiles as edges of the box, with bars representing the adjacent values to the 25th and 75th percentiles, and dots representing additional values. In the particular case of quantitative PCR data, fold changes of gene expression (fold change = 2ΔΔCt) were reported on the graphs; however, due to the potentially skewed distribution of the exponential fold change, statistical testing was performed on the corresponding values of ΔΔCt for each sample. Statistical significance was set at P < 0.05 and we used Stata version 10.1 software (Stata, College Station, TX, USA).
Results
Models of LV hypertrophy and failure by POH and VOH
We have recently published a comprehensive physiological characterization of the chronic ventricular loading models used (Chemaly et al. 2012) which is summarized in Fig. 1. Briefly, similar levels of LVH were reached in all models, with similar right ventricle (RV) hypertrophy for POH-DCM and VOH (Fig. 1A). POH groups had similar pressure overload. The POH-DCM group had markedly elevated LV end-diastolic pressure (LVEDP; Fig. 1B). POH-DCM and VOH animals showed similar LV dilatation, although more severe in VOH. LVEF was reduced in POH-DCM and VOH (Fig. 1C). Wall thickness was increased in POH, particularly POH-CLVH (Fig. 1C). Tissue from MI rats was only used to measure resistin protein levels; we have published the physiological characteristics of MI models and their cardiac fibrosis assessment (Chen et al. 2011).
Figure 1. Physiological characteristics of animal models of POH and VOH based on Tables 2 and 3 of Chemaly et al. (2012).
Groups were compared by ANOVA followed by Tukey–Kramer correction: ‘C’ significant vs. POH-CLVH, ‘D’ significant vs. POH-DCM, ‘F’ significant vs. Fistula, ‘S’ significant vs. Sham. A, morphometric analysis of LV and RV hypertrophy. n= 14–21 animals per group; P < 0.0001 for ANOVA. LVW/BW, LV weight/body weight; RVW/BW, RV weight/body weight. B, pressure overload measured by LV maximal pressure, and congestive heart failure measured by LVEDP; n= 7–17 animals per group. LV maximal pressure and LVEDP: P < 0.0001 for ANOVA; P= 0.003 uncorrected for POH-CLVH vs. Sham. C, LV dimensions and function by echocardiography, demonstrating LV dilatation with low LVEF in Fistula and POH-DCM, and wall thickening in POH. n= 14–26 animals per group; end-diastolic volume (EDV), end-systolic volume (ESV), ejection fraction (EF), anteroseptal diastolic wall thickness: P < 0.0001 by ANOVA.
Fibrosis is nearly absent in VOH compared to POH at similar levels of LV hypertrophy and dilatation
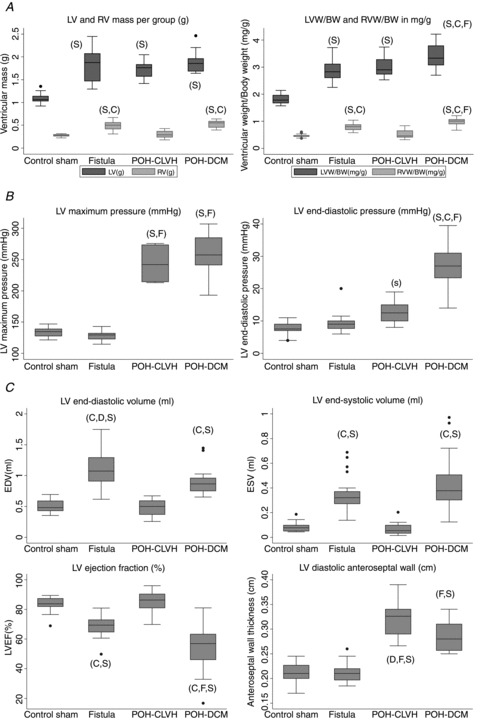
Fibrosis area quantification excluded perivascular fibrosis (probably the vascular impact of pressure overload on the coronary vasculature) and only quantified interstitial fibrosis, more directly related to myocardial processes. This distinction is based on the study by Schwartzkopff et al. (1992). Illustrative micrographs of Trichrome staining are shown in Fig. 2A. POH-DCM showed the highest interstitial fibrosis, with no significant fibrosis in VOH and intermediate levels in POH-CLVH (Fig. 2A). Fibrosis was demonstrated by Sirius Red staining, revealing a subendocardial–subepicardial wavefront of fibrosis, starting in the subendocardium in the POH-CLVH group and gaining the subepicardium in the more diseased POH-DCM group (Fig. 2B). In addition to interstitial fibrosis, all POH animals showed periarterial fibrosis (Fig. 2C), and POH-DCM (heart failure) animals perivenous fibrosis, a likely consequence of RV failure (Fig. S1 in Supplemental material, available online).
Figure 2. Fibrosis by histology in POH and VOH.
Groups were compared by ANOVA followed by Tukey–Kramer test: ‘C’ significant vs. POH-CLVH, ‘D’ significant vs. POH-DCM, ‘F’ significant vs. Fistula, ‘S’ significant vs. Sham. A, representative ×20 micrographs of Trichrome-stained LV sections. Quantitative analysis of LV interstitial fibrosis (percentage of area) showing no fibrosis in VOH vs. Sham and gradual fibrosis in POH with or without dilatation; P= 0.0018 by ANOVA; (s) P < 0.05 by uncorrected t test in POH-CLVH vs. Sham. B, fibrosis (Sirius Red) in POH reveals an endocardial–epicardial fibrosing wavefront starting in the subendocardium (POH-CLVH) and progressing to the subepicardium (POH-DCM). C, periarterial fibrosis in all POH animals by Trichrome and Sirius Red.
Replacement fibrosis in POH occurs in the form of microscopic scarring
Importantly, myocardial fibrosis in POH is perimyocytic, with clear evidence of cardiomyocyte loss and replacement of cardiomyocytes by collagen in POH animals, representing microscopic scarring. This is shown in both interstitial and perivascular spaces, with Masson Trichrome and Sirius Red staining, in the representative images in Fig. 2, and in more detail in the Supplemental Figs S1 and S2. Furthermore, Fig. 2B and Supplemental Fig. S2C illustrate the endocardial–epicardial wavefront of fibrosis in POH.
Distinct expression profile of fibrosis-related genes in VOH vs. POH reveals reduced collagen maturation in VOH
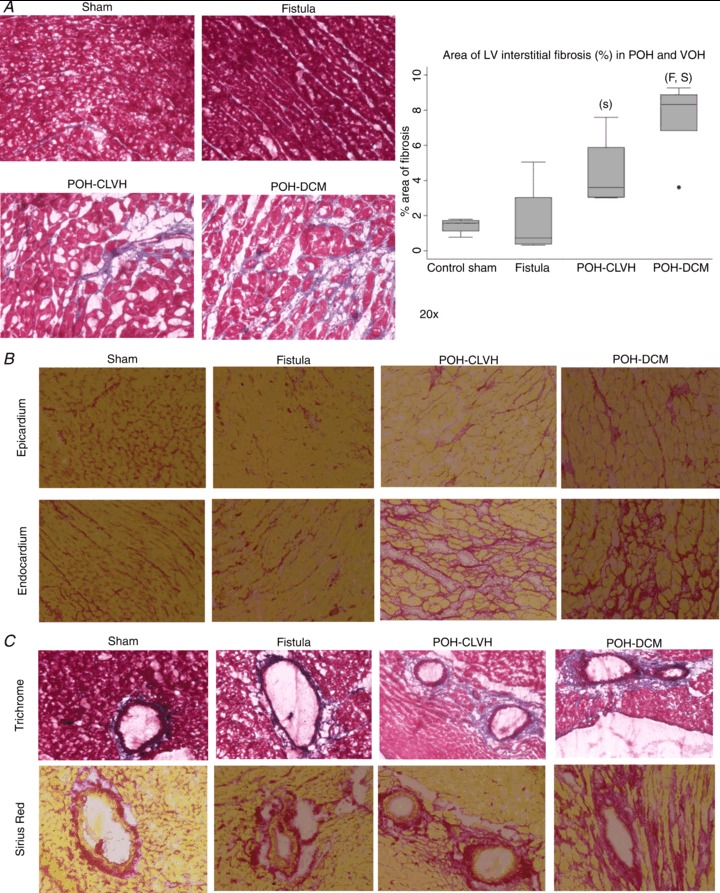
Collagen type I gene 1A1 (COL1A1) mRNA levels were significantly elevated in POH vs. sham and intermediate in VOH (Fig. 3A). Collagen type III gene 3A1 (COL3A1) mRNA levels were unchanged (Fig. 3B). Fibronectin, a non-collagen component of the extracellular matrix, was highest in dilated ventricles, specifically VOH (Fig. 3C). To evaluate collagen maturation, we measured mRNA levels of lysyl-oxidase (LOX), which only trended to be highest in POH-DCM and lowest in VOH (Fig. 3D); however, protein levels of LOX revealed a similar pattern and were significantly higher in POH-DCM vs. sham and VOH (Fig. 3E). Procollagen C-endopeptidase enhancer 1 (PCOLCE1) mRNA levels were highest in POH models and intermediate in VOH (Fig. 3F). MMP2 mRNA was highest in POH (Fig. 3G) with MMP9 mRNA unchanged between groups (data not shown). mRNA (Fig. 3H) and protein (Fig. 3I) levels of galectin-3, a novel marker of myocardial fibrosis (Sharma et al. 2004; Ho et al. 2012) were also measured, with an increase in galectin-3 mRNA seen only in the POH-DCM group.
Figure 3. Expression profile of fibrosis-related genes reveals distinct patterns in VOH and POH.
Statistical testing used ANOVA followed by Tukey test on ΔΔCt for real-time PCR data. ‘C’ significant vs. POH-CLVH, ‘D’ significant vs. POH-DCM, ‘F’ significant vs. Fistula, ‘S’ significant vs. Sham. n= 3–4 animals per group. A, collagen COL1A1 mRNA expression; P= 0.02 by ANOVA. B, collagen COL3A1 mRNA expression; P= n.s. by ANOVA. C, fibronectin mRNA expression; P < 0.05 by ANOVA. D, lysyl oxidase (LOX) mRNA expression; P= n.s. by ANOVA. E, LOX protein expression; *P < 0.05 vs. Sham and ###P < 0.001 vs. Fistula by t test. Graphics represent mean ± SEM, n= 2–4 animals per group. F, PCOLCE1 mRNA expression; P= 0.005 by ANOVA. G, MMP2 mRNA expression; P= 0.04 by ANOVA. H, galectin-3 mRNA expression is only increased in POH-DCM; P= 0.011 for ANOVA on ΔΔCt; n= 3–4 animals per group. I, galectin-3 protein expression is not altered by LVH; n= 2–4 animals per group.
The angiotensin II–TGFβ1–CTGF reactive fibrosis pathway interacts with resistin and is attenuated in VOH compared to POH
Angiotensin II is known to promote cardiac fibrosis, at least in part, through the induction of the transforming growth factor (TGF) β1 which in turn induces the connective tissue growth factor (CTGF; Leask, 2010). PCR primers for the common coding sequence of the AT1AR and AT1BR subtypes of the rat AT1 receptor of angiotensin II were used to quantify mRNA expression of angiotensin II receptor type 1 (AT1R) in POH and VOH. AT1R mRNA levels were significantly downregulated in POH but not in VOH, indicating higher myocardial angiotensin II activity in POH (Fig. 4A). Moreover, POH hearts displayed the highest mRNA levels of TGFβ1 (Fig. 4B).
Figure 4. Interrelationship of reactive fibrosis, resistin expression and resistin action in cardiac haemodynamic and neurohormonal stress.
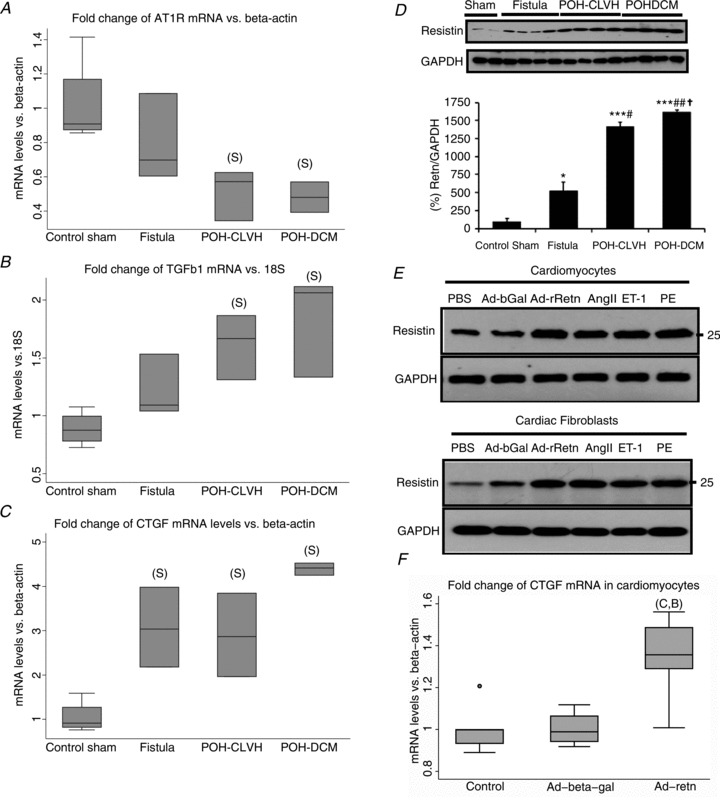
A, AT1R mRNA is significantly downregulated in POH; P= 0.01 for ANOVA on ΔΔCt; ‘S’, significant vs. Sham by Tukey test; n= 3–4 animals per group. B, TGFβ1 mRNA expression; P= 0.005 by ANOVA. C, CTGF mRNA expression; P= 0.0004 by ANOVA. D, resistin protein levels are markedly elevated in POH and mildly elevated in VOH. Mean ± SEM, by t test: *P < 0.05 vs. Sham, ***P < 0.001 vs. Sham, #P < 0.05 vs. Fistula, ##P < 0.01 vs. Fistula, †P < 0.05 vs. POH-CLVH, n= 2–4 per group. E, resistin expression in adult rat cardiomyocytes and cardiac fibroblasts is induced by neurohormonal stress. Ad-bGal, adenovirus overexpressing β-galactosidase; Ad-rRetn, adenovirus overexpressing rat resistin; AngII, angiotensin II; ET-1, endothelin 1; PE, phenylephrine; PBS, phosphate-buffered saline. F, CTGF mRNA expression in adult rat cardiomyocytes infected with adenovirus (Ad) expressing resistin. P= 0.0094 for ANOVA on ΔΔCt; Ad-resistin is significantly higher than Ad-β-galactosidase ‘B’ and control ‘C’ by Tukey test; n= 4–5 samples per group.
Interestingly, CTGF mRNA was elevated in POH and VOH alike (Fig. 4C). Resistin protein expression was severely elevated in POH and mildly elevated in VOH (Fig. 4D). To better situate resistin in the profibrotic cascade initiated by angiotensin II, we exposed cardiac myocytes and cardiac fibroblasts to angiotensin II, phenylephrine and endothelin 1 and found all stimuli to induce resistin expression (Fig. 4E). We investigated the downstream effects of resistin on TGFβ1 and CTGF in adult rat cardiomyocytes and cardiac fibroblasts overexpressing resistin. In cardiomyocytes, resistin increased the expression of CTGF (Fig. 4F) but not TGFβ1 (data not shown). The expression of TGFβ1 and CTGF were unaffected by resistin in adult cardiac fibroblasts (data not shown).
Local resistin expression reflects replacement fibrosis in chronic ischaemia
Differential resistin protein expression in POH and VOH reflected myocardial fibrosis (Fig. 4D). In Fig. 4, we connect resistin expression and action to key factors of reactive fibrosis and the dynamic relationship is summarized in Fig. 5A. Our main hypothesis is that the absent or reduced replacement fibrosis in VOH compared to POH is related at least in part to less propensity for ischaemia and ischaemic injury in VOH. To assess whether increased myocardial resistin expression was a local phenomenon in response to replacement fibrosis, we measured resistin expression in infracted LV tissue, which was elevated compared to sham (Fig. 5B); this elevation in resistin expression was not seen in the remote area of the LV (Fig. 5C) indicating its local (paracrine and autocrine) origin. Details of infarct size and the lack of fibrosis in the remote area (Fig. 5C) of the MI models have been previously published by our group (Chen et al. 2011).
Figure 5. Local resistin expression interconnects with reactive fibrosis and replacement fibrosis after ischaemic injury.
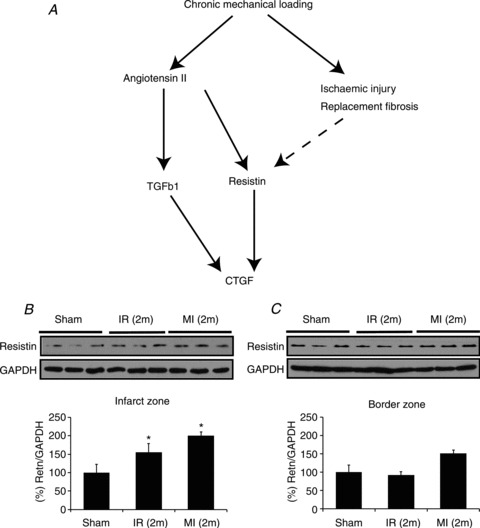
A, recapitulative pathway based on the mRNA and protein expression data in Fig. 4. B, local resistin expression is associated with chronic ischaemic injury in rats with myocardial infarction 2 months after permanent left anterior descending artery ligation (MI (2m)) or after a 30 min ischaemia–reperfusion sequence (IR (2m)): LV resistin was elevated compared to Sham rats. C, no significant increase in resistin expression was seen in the remote area (border zone). By t test: *P < 0.05 vs. sham, n= 3 animals per group.
Capillary density is reduced in advanced POH (POH-DCM) but preserved in VOH
To differentiate the propensity for ischaemia in POH and VOH, capillary density per area of LV tissue was measured using caveolin-1α and vinculin immunofluorescence staining (Fig. 6A). Capillary density (in capillaries μm−2) was preserved in VOH, significantly reduced in POH-DCM compared to sham and VOH animals, and intermediate in POH-CLVH (Fig. 6B). The number of capillaries per cardiomyocyte, however, was elevated in VOH and POH-CLVH reflecting hypertrophy, but reduced and close to control levels in POH-DCM indicating capillary loss in advanced POH (Fig. 6C). Thus, capillary density is markedly reduced in POH-DCM compared to VOH at similar LV hypertrophy and dilatation.
Figure 6. Capillary density is reduced in advanced POH.
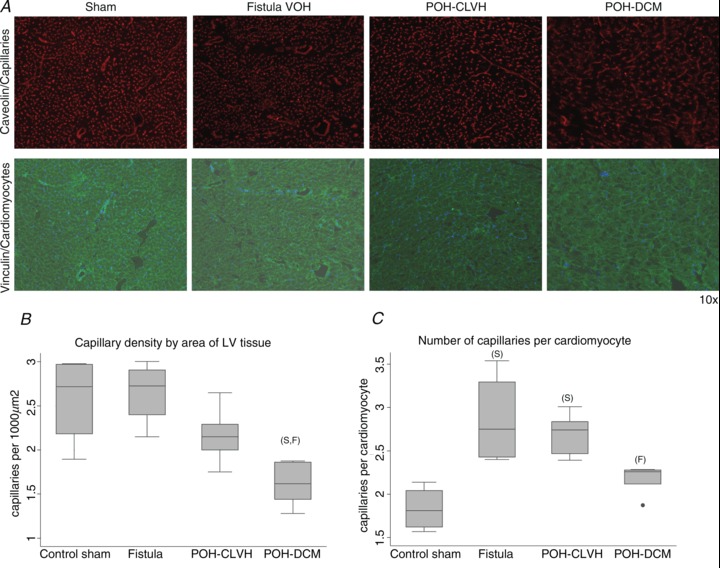
ANOVA followed by Tukey test, ‘F’ significant vs. Fistula, ‘S’ significant vs. Sham, n= 4–5 animals per group. A, representative ×10 micrographs of capillaries and cardiomyocyte membranes in POH and VOH. B, capillary density per area is reduced in advanced POH (POH-DCM) while it is preserved in compensated POH and intact in VOH; P= 0.003 for ANOVA. C, the number of capillaries per cardiac myocyte increases in both VOH and compensated POH (POH-CLVH) and regresses to control levels in the advanced POH (POH-DCM); P= 0.0014 for ANOVA.
Additional haemodynamic determinants of the LV oxygen supply–demand ratio and of the propensity for ischaemia in chronic loading
We hypothesized that the differential fibrotic response in POH and VOH (Figs 2 and 3) was related to a lesser propensity for ischaemia in VOH, based on more favourable energetics (oxygen supply–demand ratios) in VOH. The mechanical and morphometric parameters in Fig. 1 affect the LV oxygen supply–demand balance; furthermore, LV perfusion is impaired by perivascular fibrosis (Fig. 2) and reduced capillary density (Fig. 6) in POH. Heart rate, which affects myocardial oxygen demand and supply, was mildly but significantly reduced in POH-DCM (Fig. 7A). Stroke work increased in all models, and further increased in POH-DCM compared to VOH (Fig. 7B). Wall stress was higher in dilated ventricles, and highest in POH-DCM (Fig. 7C; Chemaly et al. 2012). Although stroke work was similarly elevated in POH-CLVH and in VOH, we distinguished stroke work increase by pressure development (POH) from stroke work increase by increased stroke volume (VOH). Figure 7D shows that LV developed pressure (difference between LV maximal pressure and LVEDP; Fig. 1) is highest in POH. Distinguishing pressure-driven stroke work from volume-driven stroke work is critical in predicting oxygen demand, since the same myocardial work performed in tension development was shown to consume more oxygen than external work (Coleman et al. 1969).
Figure 7. Additional haemodynamic determinants of myocardial oxygen supply–demand balance in POH and VOH.
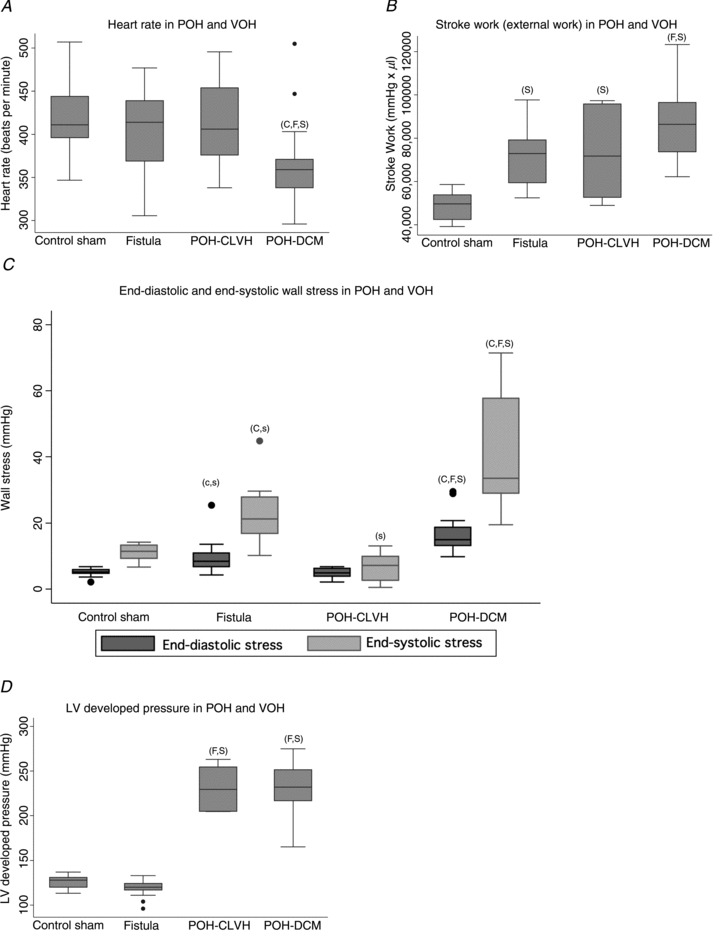
ANOVA with Tukey-Kramer test, ‘C’ significant vs. POH-CLVH, ‘F’ significant vs. Fistula, ‘S’ significant vs. Sham. A, heart rate (echocardiography) was mildly decreased in POH-DCM (Table 1 of Chemaly et al. 2012), P= 0.0034 by ANOVA, n= 14–26 animals per group. B, all models produced larger LV stroke work than control rat LV, with larger stroke work in POH-DCM than in VOH; P < 0.0001 by ANOVA; n= 7–17 animals per group. C, wall stress was increased in dilated ventricles, highest in POH-DCM (Table 9 of Chemaly et al. 2012). End-diastolic wall stress: P < 0.0001 by ANOVA, POH-DCM higher than other groups by Tukey test, Fistula higher than Sham and CLVH by uncorrected t test (indicated by ‘c,s’). End-systolic wall stress: P < 0.0001 for ANOVA, POH-DCM higher than other groups and Fistula higher than POH-CLVH by Tukey test; Fistula higher than Sham; also POH-CLVH lower than Sham by uncorrected t test (s); n= 7–17 animals per group. D, pressure development is higher in POH models, P < 0.0001 by ANOVA, n= 7–17 animals per group.
The multifactorial propensity for ischaemia is higher in POH than VOH
To comprehensively assess the different propensity for ischaemia in POH and VOH, the association matrix in Fig. 8 represents the different indicators of increased oxygen demand and/or reduced supply in a heat map where red favours ischaemia and blue prevents it. All parameters shown in Fig. 8 were quantified in the present study, except for the ex vivo LV oxygen consumption as a function of the pressure–volume area (PVA) which were measured by our group in POH and VOH blood-perfused non-working isolated rat hearts (Sakata et al. 2007; Takewa et al. 2009). At equal levels of PVA, oxygen consumption was higher in POH than sham rat LV (Sakata et al. 2007), while oxygen consumption of VOH LV was lower than sham (Takewa et al. 2009). Endocardial oxygen supply and coronary flow are impaired in all POH by wall thickening and periarterial fibrosis, and, in advanced POH, by reduced capillary density and increased LVEDP (Figs 1, 2 and 4). Wall stress, representing potential energy, is elevated in dilated ventricles from VOH and POH-DCM, and is the only feature of increased oxygen consumption in VOH compared to POH-CLVH (Figs 7 and 8). The results presented in Fig. 8 are aligned with the amount and distribution of interstitial fibrosis shown in Fig. 2.
Figure 8. Heat map.
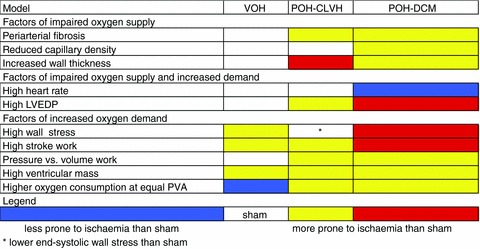
Heat map (association matrix) showing higher multifactorial ischaemia propensity in POH than VOH.
Discussion
We propose to explain the nearly absent fibrosis in VOH by aorta–caval fistula compared to the progressive fibrosis in POH by (1) a different pattern of activation of key profibrotic pathways, leading to less reactive fibrosis in VOH, and (2) a lower propensity for ischaemia in VOH than POH, at similar LVH and dilatation, explaining why the replacement fibrosis found in POH is not found in VOH.
Differences in reactive fibrosis and resistin expression in POH and VOH relate to differential neurohormonal activation in VOH and POH
The renin–angiotensin system is known to be overactive in both POH (Rockman et al. 1994) and VOH (Ruzicka et al. 1993); however, compared to POH, the therapeutic benefits of renin–angiotensin system inhibition in VOH are controversial at best (Zheng et al. 2009). Thus, there remains the possibility that, at similar levels of LVH, myocardial angiotensin II activity may be lower in VOH than POH, leading to less therapeutic efficacy and explaining less fibrosis in VOH. To further evaluate the neurohormonal origin of the differential fibrosis in POH and VOH, we measured mRNA expression of AT1R in LV tissue as an indicator of prolonged angiotensin II activity (Schultz et al. 2002). AT1R mRNA was markedly downregulated in POH, in contrast with VOH (Fig. 4A). Thus, differences in fibrosis and resistin expression between POH and VOH may indeed reflect differences in the cellular actions of neurohormonal stressors between models (Fig. 4). This is shown in the expression of TGFβ1, a downstream effector of angiotensin II (Leask, 2010). Interestingly, CTGF, which has a less critical role in fibrosis (Leask, 2010), was elevated in POH and VOH alike in our present study (Fig. 4). CTGF, but not TGFβ1, was inducible by resistin overexpression in cardiac myocytes but not in fibroblasts (Fig. 4), in accordance with our previous in vivo study (Chemaly et al. 2011). Interestingly, Zendaoui et al. report a decrease in myocardial fibrosis upon spironolactone administration in a rat model of AR, suggesting that the profibrotic effects of the renin–angiotensin–aldosterone system may also be at work in VOH (Zendaoui et al. 2012).
Different ischaemia propensity and replacement fibrosis drives the different fibrotic response between POH and VOH along a continuum
Fibrosis in either POH or HCM is considered ischaemic in origin (Maron et al. 2009; Rudolph et al. 2009), and the predominantly subendocardial fibrosis reflects the greater vulnerability of the subendocardium to ischaemia (Duncker & Bache, 2008), particularly in LVH (Bache & Dai, 1990). The basis of our work is that both chronic haemodynamic loading and LVH result in an imbalance of oxygen demand and supply on the cardiomyocyte leading to progressive ischaemic damage. Taken together with our other studies (Sakata et al. 2007; Takewa et al. 2009), our results indicate more imbalance towards ischaemia in POH than VOH (Fig. 8) resulting in more fibrosis, and further increase in fibrosis in POH-DCM, driven by higher LVEDP and higher wall stress (Figs 1–8).
Previous studies attributed the lack of fibrosis in VOH to extracellular matrix degradation by MMPs allowing LV dilatation and dysfunction (Zheng et al. 2009; Hutchinson et al. 2010; Wei et al. 2012); however, some studies on VOH contradict this paradigm (Beeri et al. 2008; Zendaoui et al. 2012) and the profibrotic effect of MMPs is clearly demonstrated elsewhere (see Introduction). Our study concentrates on reactive and replacement fibrosis as positive responses, and shows that their primary stimuli are attenuated in VOH versus POH. As shown in Fig. 8, VOH animals do have some propensity for subendocardial ischaemia, driven by increased stroke work and wall stress, and features of fibrosis in VOH can be detected by molecular analysis (Fig. 3). This is consistent with a recent study of aorta–caval fistula in rats showing less or similar fibrosis by Sirius Red than sham animals at 8 and 15 weeks, with increased mRNA of profibrotic molecules (Hutchinson et al. 2011). A previous study had shown fibronectin-based, non-collagen fibrosis in AR (Borer et al. 2002); interestingly, our results with fibronectin mRNA being highest in VOH and POH-DCM suggest a distinct response of fibronectin to LV dilatation and myocyte lengthening of various causes. Since fibronectin is part of the basement membrane of cardiomyocytes, its expression is likely to respond to longitudinal cardiomyocyte growth occurring in eccentric LV hypertrophy.
Furthermore, since reactive and replacement fibrosis are attenuated, and not inverted, in VOH, our conclusion is compatible with fibrosis development in VOH models with further increases in stroke work and wall stress. We studied a rat model of aorta–caval fistula, known for its (relative) lack of fibrosis; and fibrosis is reported in rodent models of AR (Plante et al. 2004; Nakanishi et al. 2007; Zendaoui et al. 2012) but not in a recent study of rat MR (Kim et al. 2011). This series of studies, along with ours, reflects a continuum between three models of VOH with gradually increased afterload and wall stress: aorta–caval fistula, MR and AR, as recently proposed (Carabello, 2012). In their report on fibrosis in rat AR, Zendaoui et al. found an increase in LVEDP (which probably results from fibrosis, but also impairs subendocardial perfusion) along with reduced capillary density in AR (Zendaoui et al. 2012).
In our study, galectin-3 mRNA was only increased in the POH-DCM group, the most severe in terms of LV dysfunction (Chemaly et al. 2012) and fibrosis (Figs 2 and 3). In a recent study, Kortekaas et al. evaluated heart failure patients with functional MR, in whom volume overload is superimposed on advanced heart failure, and found low preoperative levels of galectin-3 to predict a therapeutic response (reverse remodelling) after mitral valve repair (Kortekaas et al. 2013). These results are consistent with our findings and highlight the specificity of enhanced galectin-3 expression for advanced heart failure.
Our study placing POH and VOH along a continuum of ischaemia propensity parallels a recent classification of LVH as a continuum between physiological and pathological hypertrophy, VOH being closer to physiological hypertrophy than POH (Dorn, 2007). The physiological–pathological hypertrophy continuum may therefore reflect the propensity for ischaemia, maximal in POH with high afterload and concentric LVH, and minimal in aerobic exercise and pregnancy. Distinct processes are thought to lead to physiological or pathological LVH (Dorn, 2007); however, we suggest that any process can lead to pathological LVH and replacement fibrosis depending on the oxygen supply–demand ratio and ischaemia propensity. Endurance exercise training LVH is the archetype of physiological hypertrophy (Dorn, 2007) without fibrosis (Dorn, 2007) and without arrhythmias (Kehat & Molkentin, 2010); nonetheless, intense endurance exercise may lead to these adverse outcomes (O’Keefe et al. 2012).
Resistin interacts with profibrotic pathways
Another novel aspect of our work is the pattern of resistin expression in LVH: highest in fibrosing models (i.e. POH, Fig. 4D) and in local chronic ischaemic injury (Fig. 5). We show the resistin expression and action pattern to interact with profibrotic pathways. Resistin expression reflects neurohormonal activation (Fig. 4) and mirrors the expression of AT1R mRNA, which was downregulated in POH but not VOH (Fig. 4A), confirming a previous report of POH in dogs (Schultz et al. 2002). A recent study using inhibitors of the circulating and tissue angiotensin-converting enzyme (ACE) in patients with clinical coronary artery disease and elevated plasma resistin levels has indeed documented a stronger resistin lowering by tissue-type ACE inhibition (Krysiak et al. 2010). Furthermore, resistin drives the expression of CTGF in vitro in cardiac myocytes (Fig. 4E).
Functional implications
Fibrosis is accompanied by an increase in passive myocardial stiffness and diastolic dysfunction (Brower et al. 2006), which we did not observe in our study of the rat model of aorta–caval fistula (Chemaly et al. 2012). As for systolic dysfunction, it is difficult to view it as a direct consequence of fibrosis. Moreover, collagen degradation may indeed impair myocardial force transmission and cause systolic dysfunction (Brower et al. 2006). However, and in contrast with the conclusions of the study by Ryan et al. (2007), our data on the aorta–caval fistula model reveal normal baseline systolic performance when measured by the stroke-volume-to-wall-stress-ratio, and normal contractile reserve on dobutamine (Chemaly et al. 2012), along with a relative lack of fibrosis. In contrast, the pressure-overload hypertrophy (POH) groups studied in the present work show impaired contractile reserve (Chemaly et al. 2012). In rats with compensated-concentric POH (POH-CLVH), stroke-volume-to-wall-stress-ratio is normal while it is markedly reduced in dilated cardiomyopathy following POH (POH-DCM; Chemaly et al. 2012). A prudent conclusion regarding the link between fibrosis and ventricular systolic dysfunction is that they both result from common processes leading to cardiomyocyte dysfunction and loss and may therefore follow a parallel course.
Clinical implications
Our study has clinical implications, notably in explaining the different tolerability and evolution of chronic valvular heart disease. In two parallel studies, chronic asymptomatic severe MR was well tolerated (Rosenhek et al. 2006) compared to asymptomatic severe AS, in which myocardial fibrosis may not be reversible after valve replacement (Rosenhek et al. 2000). Another study revealed the impact of MR severity on outcomes in asymptomatic patients, worse in larger regurgitant volumes (Enriquez-Sarano et al. 2005). The latter paradigm can also be interpreted in the light of our data, from which we suggest that at equal levels of LV hypertrophy, POH shows more fibrosis than VOH due to a higher propensity for ischaemia. Since we show the main determinants of increased oxygen demand in VOH to be wall stress and increased stroke work, VOH of increased severity probably leads to a subendocardial supply–demand imbalance with ischaemic myocardial damage and fibrosis similar to POH. Also, since chronic increase in afterload (POH) is more detrimental than chronic increase in preload (VOH) based on ischaemia propensity, this may explain why pharmacological afterload reduction has a clear benefit on heart failure disease progression (Mullens et al. 2009), whereas the benefit of preload reduction is less evident but possible (Faris et al. 2012). Based on our study, we suggest that a better reduction in subendocardial ischaemia propensity is achieved with afterload reduction, which reduces developed pressure and wall stress, and improves forward flow, as opposed to preload reduction, which mainly reduces diastolic wall stress and filling pressures.
Limitations
We did not assess coronary flow reserve and oxygen consumption and supply in our animal models, although we previously demonstrated preserved contractile reserve upon dobutamine infusion in VOH, but not in severe POH (Chemaly et al. 2012). Besides, we considered ischaemia to be an ongoing phenomenon, as in HCM (Maron et al. 2009). Although, in HCM, resting myocardial blood flow was normal with impaired reserve, normal myocardial blood flow in the face of high demand may explain ongoing ischaemic damage, along with ‘bouts of ischaemia’ leading to progressive myocyte loss (Maron et al. 2009) according to the ‘necrotic core’ hypothesis (Rudolph et al. 2009). In that regard, our current study and our previous work on myocardial energetics in POH (Sakata et al. 2007) and VOH (Takewa et al. 2009) might be the first comparative assessment of myocardial oxygen supply–demand mismatch in POH and VOH. Our evaluation of neurohormonal factors (Fig. 4) was limited; both the renin–angiotensin system (Schultz et al. 2002) and the adrenergic system (Plante et al. 2008) have known complex roles in cardiac fibrosis. Finally, we previously studied myocardial oxygen consumption at equal PVA (Sakata et al. 2007; Takewa et al. 2009) but did not address metabolic differences such as mitochondrial function. Morphological abnormalities of the intramyocardial vasculature were a notable factor of ischaemia propensity in POH in our study; they may be a consequence of pressure overload, but they are observed in HCM as well (Maron et al. 2009).
Conclusion
Smaller amounts of replacement fibrosis in VOH compared to POH are most likely due to a smaller propensity for ischaemia in VOH. Also, reactive fibrotic pathways are attenuated in VOH, with reduced neurohormonal activation in VOH versus POH as a possible source. This paradigm is validated by the pattern of resistin expression in LV tissue and the interaction of resistin with the profibrotic factors.
Acknowledgments
None.
Glossary
- AR
aortic regurgitation
- AS
aortic stenosis
- AT1R
angiotensin II receptor type 1
- CLVH
compensated/concentric left ventricular hypertrophy
- CTGF
connective tissue growth factor
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- LOX
lysyl-oxidase
- LV
left ventricle
- LVEDP
LV end-diastolic pressure
- LVH
left ventricular hypertrophy
- MI
myocardial infarction
- MMP
matrix metalloprotease
- MOI
multiplicity of infection
- MR
mitral regurgitation
- POH
pressure overload LVH
- PVA
pressure–volume area
- RV
right ventricle
- TGFβ1
transforming growth factor β1
- TIMP
tissue inhibitor(s) of metalloprotease(s)
- VOH
volume overload LVH
Additional information
Competing interests
None declared.
Author contributions
Conception and design of the experiments: E.R.C., S.K., R.J.H. and D.L. Collection, analysis and interpretation of data: E.R.C., S.K., S.Z., L.McC., J.C., L.B., K-R.P. and D.L. Drafting the manuscript or revising it critically for important intellectual content: E.R.C., R.J.H. and D.L. All authors approved the final version of the manuscript. E.R.C and D.L. are the guarantors of this work and assume full responsibility for the integrity of the experimental data and analysis.
Funding
This work was supported by Leducq Foundation through the Caerus network (R.J.H.) and by NIH (D.L., R.J.H.) and Federal Contract No. HHSN268201000045C (R.J.H.) NIH-T32-HL007824 (E.R.C.).
References
- Arias T, Chen J, Fayad ZA, Fuster V, Hajjar RJ, Chemaly ER. Comparison of echocardiographic measurements of left ventricular volumes to full volume magnetic resonance imaging in normal and diseased rats. J Am Soc Echocardiogr. 2013;26:910–918. doi: 10.1016/j.echo.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache RJ, Dai XZ. Myocardial oxygen consumption during exercise in the presence of left ventricular hypertrophy secondary to supravalvular aortic stenosis. J Am Coll Cardiol. 1990;15:1157–1164. doi: 10.1016/0735-1097(90)90258-q. [DOI] [PubMed] [Google Scholar]
- Beeri R, Yosefy C, Guerrero JL, Nesta F, Abedat S, Chaput M, del Monte F, Handschumacher MD, Stroud R, Sullivan S, Pugatsch T, Gilon D, Vlahakes GJ, Spinale FG, Hajjar RJ, Levine RA. Mitral regurgitation augments post-myocardial infarction remodeling failure of hypertrophic compensation. J Am Coll Cardiol. 2008;51:476–486. doi: 10.1016/j.jacc.2007.07.093. [DOI] [PubMed] [Google Scholar]
- Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. doi: 10.1161/01.cir.89.1.151. [DOI] [PubMed] [Google Scholar]
- Borer JS, Herrold EM, Carter JN, Catanzaro DF, Supino PG. Cellular and molecular basis of remodeling in valvular heart diseases. Heart Fail Clin. 2006;2:415–424. doi: 10.1016/j.hfc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Borer JS, Truter S, Herrold EM, Falcone DJ, Pena M, Carter JN, Dumlao TF, Lee JA, Supino PG. Myocardial fibrosis in chronic aortic regurgitation: molecular and cellular responses to volume overload. Circulation. 2002;105:1837–1842. doi: 10.1161/01.cir.0000014419.71706.85. [DOI] [PubMed] [Google Scholar]
- Brower GL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, Janicki JS. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg. 2006;30:604–610. doi: 10.1016/j.ejcts.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- Carabello BA. Volume overload. Heart Fail Clin. 2012;8:33–42. doi: 10.1016/j.hfc.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Carabello BA, Nakano K, Ishihara K, Kanazawa S, Biederman RW, Spann JF., Jr Coronary blood flow in dogs with contractile dysfunction due to experimental volume overload. Circulation. 1991;83:1063–1075. doi: 10.1161/01.cir.83.3.1063. [DOI] [PubMed] [Google Scholar]
- Chemaly ER, Chaanine AH, Sakata S, Hajjar RJ. Stroke volume-to-wall stress ratio as a load-adjusted and stiffness-adjusted indicator of ventricular systolic performance in chronic loading. J Appl Physiol. 2012;113:1267–1284. doi: 10.1152/japplphysiol.00785.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemaly ER, Hadri L, Zhang S, Kim M, Kohlbrenner E, Sheng J, Liang L, Chen J, K-Raman P, Hajjar RJ, Lebeche D. Long-term in vivo resistin overexpression induces myocardial dysfunction and remodeling in rats. J Mol Cell Cardiol. 2011;51:144–155. doi: 10.1016/j.yjmcc.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chemaly ER, Liang LF, LaRocca TJ, Yaniz-Galende E, Hajjar RJ. A new model of congestive heart failure in rats. Am J Physiol Heart Circ Physiol. 2011;301:H994–H1003. doi: 10.1152/ajpheart.00245.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HN, Sonnenblick EH, Braunwald E. Myocardial oxygen consumption associated with external work: the Fenn effect. Am J Physiol. 1969;217:291–296. doi: 10.1152/ajplegacy.1969.217.1.291. [DOI] [PubMed] [Google Scholar]
- Dorn GW., 2nd The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009–1086. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- Eckhouse SR, Spinale FG. Changes in the myocardial interstitium and contribution to the progression of heart failure. Heart Fail Clin. 2012;8:7–20. doi: 10.1016/j.hfc.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez-Sarano M, Avierinos JF, Messika-Zeitoun D, Detaint D, Capps M, Nkomo V, Scott C, Schaff HV, Tajik AJ. Quantitative determinants of the outcome of asymptomatic mitral regurgitation. N Engl J Med. 2005;352:875–883. doi: 10.1056/NEJMoa041451. [DOI] [PubMed] [Google Scholar]
- Faris RF, Flather M, Purcell H, Poole-Wilson PA, Coats AJ. Diuretics for heart failure. Cochrane Database Syst Rev. 2012;2:CD003838. doi: 10.1002/14651858.CD003838.pub3. [DOI] [PubMed] [Google Scholar]
- Frankel DS, Vasan RS, D’Agostino RB, Sr, Benjamin EJ, Levy D, Wang TJ, Meigs JB. Resistin, adiponectin, and risk of heart failure: the Framingham offspring study. J Am Coll Cardiol. 2009;53:754–762. doi: 10.1016/j.jacc.2008.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster V, Danielson MA, Robb RA, Broadbent JC, Brown AL, Jr, Elveback LR. Quantitation of left ventricular myocardial fiber hypertrophy and interstitial tissue in human hearts with chronically increased volume and pressure overload. Circulation. 1977;55:504–508. doi: 10.1161/01.cir.55.3.504. [DOI] [PubMed] [Google Scholar]
- Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, Larson MG, Levy D. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol. 2012;60:1249–1256. doi: 10.1016/j.jacc.2012.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson KR, Guggilam A, Cismowski MJ, Galantowicz ML, West TA, Stewart JA, Jr, Zhang X, Lord KC, Lucchesi PA. Temporal pattern of left ventricular structural and functional remodeling following reversal of volume overload heart failure. J Appl Physiol. 2011;111:1778–1788. doi: 10.1152/japplphysiol.00691.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson KR, Stewart JA, Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol. 2010;48:564–569. doi: 10.1016/j.yjmcc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandalam V, Basu R, Moore L, Fan D, Wang X, Jaworski DM, Oudit GY, Kassiri Z. Lack of tissue inhibitor of metalloproteinases 2 leads to exacerbated left ventricular dysfunction and adverse extracellular matrix remodeling in response to biomechanical stress. Circulation. 2011;124:2094–2105. doi: 10.1161/CIRCULATIONAHA.111.030338. [DOI] [PubMed] [Google Scholar]
- Kang S, Chemaly ER, Hajjar RJ, Lebeche D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J Biol Chem. 2011;286:18465–18473. doi: 10.1074/jbc.M110.200022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim YJ, Lee SP, Kim HK, Seo JW, Sohn DW, Oh BH, Park YB. Survival, exercise capacity, and left ventricular remodeling in a rat model of chronic mitral regurgitation: serial echocardiography and pressure-volume analysis. Korean Circ J. 2011;41:603–611. doi: 10.4070/kcj.2011.41.10.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Oh JK, Sakata S, Liang I, Park W, Hajjar RJ, Lebeche D. Role of resistin in cardiac contractility and hypertrophy. J Mol Cell Cardiol. 2008;45:270–280. doi: 10.1016/j.yjmcc.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortekaas KA, Hoogslag GE, de Boer RA, Dokter MM, Versteegh MI, Braun J, Marsan NA, Verwey HF, Delgado V, Schalij MJ, Klautz RJ. Galectin-3 and left ventricular reverse remodelling after surgical mitral valve repair. Eur J Heart Fail. 2013;15:1011–1018. doi: 10.1093/eurjhf/hft056. [DOI] [PubMed] [Google Scholar]
- Krysiak R, Sierant M, Marek B, Okopien B. The effect of perindopril and enalapril on plasma resistin levels in normotensive patients with coronary heart disease. Endokrynol Pol. 2010;61:683–690. [PubMed] [Google Scholar]
- Leask A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- Lopes MM, Ribeiro GC, Tornatore TF, Clemente CF, Teixeira VP, Franchini KG. Increased expression and phosphorylation of focal adhesion kinase correlates with dysfunction in the volume-overloaded human heart. Clin Sci (Lond) 2007;113:195–204. doi: 10.1042/CS20070036. [DOI] [PubMed] [Google Scholar]
- Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, Udelson JE, Camici PG. The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:866–875. doi: 10.1016/j.jacc.2009.04.072. [DOI] [PubMed] [Google Scholar]
- Mullens W, Abrahams Z, Francis GS, Sokos G, Starling RC, Young JB, Taylor DO, Tang WH. Usefulness of isosorbide dinitrate and hydralazine as add-on therapy in patients discharged for advanced decompensated heart failure. Am J Cardiol. 2009;103:1113–1119. doi: 10.1016/j.amjcard.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M, Harada M, Kishimoto I, Kuwahara K, Kawakami R, Nakagawa Y, Yasuno S, Usami S, Kinoshita H, Adachi Y, Fukamizu A, Saito Y, Nakao K. Genetic disruption of angiotensin II type 1a receptor improves long-term survival of mice with chronic severe aortic regurgitation. Circ J. 2007;71:1310–1316. doi: 10.1253/circj.71.1310. [DOI] [PubMed] [Google Scholar]
- O’Keefe JH, Patil HR, Lavie CJ, Magalski A, Vogel RA, McCullough PA. Potential adverse cardiovascular effects from excessive endurance exercise. Mayo Clin Proc. 2012;87:587–595. doi: 10.1016/j.mayocp.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JT, Hallak H, Johnson L, Li H, O’Brien PM, Sliskovic DR, Bocan TM, Coker ML, Etoh T, Spinale FG. Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation. 2001;103:2303–2309. doi: 10.1161/01.cir.103.18.2303. [DOI] [PubMed] [Google Scholar]
- Piper C, Schultheiss HP, Akdemir D, Rudolf J, Horstkotte D, Pauschinger M. Remodeling of the cardiac extracellular matrix differs between volume- and pressure-overloaded ventricles and is specific for each heart valve lesion. J Heart Valve Dis. 2003;12:592–600. [PubMed] [Google Scholar]
- Plante E, Lachance D, Champetier S, Drolet MC, Roussel E, Arsenault M, Couet J. Benefits of long-term β-blockade in experimental chronic aortic regurgitation. Am J Physiol Heart Circ Physiol. 2008;294:H1888–H1895. doi: 10.1152/ajpheart.01286.2007. [DOI] [PubMed] [Google Scholar]
- Plante E, Lachance D, Gaudreau M, Drolet MC, Roussel E, Arsenault M, Couet J. Effectiveness of β-blockade in experimental chronic aortic regurgitation. Circulation. 2004;110:1477–1483. doi: 10.1161/01.CIR.0000141733.55236.9D. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Wachhorst SP, Mao L, Ross J., Jr ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am J Physiol Heart Circ Physiol. 1994;266:H2468–H2475. doi: 10.1152/ajpheart.1994.266.6.H2468. [DOI] [PubMed] [Google Scholar]
- Rosenhek R, Binder T, Porenta G, Lang I, Christ G, Schemper M, Maurer G, Baumgartner H. Predictors of outcome in severe, asymptomatic aortic stenosis. N Engl J Med. 2000;343:611–617. doi: 10.1056/NEJM200008313430903. [DOI] [PubMed] [Google Scholar]
- Rosenhek R, Rader F, Klaar U, Gabriel H, Krejc M, Kalbeck D, Schemper M, Maurer G, Baumgartner H. Outcome of watchful waiting in asymptomatic severe mitral regurgitation. Circulation. 2006;113:2238–2244. doi: 10.1161/CIRCULATIONAHA.105.599175. [DOI] [PubMed] [Google Scholar]
- Rudolph A, Abdel-Aty H, Bohl S, Boyé P, Zagrosek A, Dietz R, Schulz-Menger J. Noninvasive detection of fibrosis applying contrast-enhanced cardiac magnetic resonance in different forms of left ventricular hypertrophy: relation to remodeling. J Am Coll Cardiol. 2009;53:284–291. doi: 10.1016/j.jacc.2008.08.064. [DOI] [PubMed] [Google Scholar]
- Ruzicka M, Yuan B, Harmsen E, Leenen FH. The renin-angiotensin system and volume overload-induced cardiac hypertrophy in rats. Effects of angiotensin converting enzyme inhibitor versus angiotensin II receptor blocker. Circulation. 1993;87:921–930. doi: 10.1161/01.cir.87.3.921. [DOI] [PubMed] [Google Scholar]
- Ryan TD, Rothstein EC, Aban I, Tallaj JA, Husain A, Lucchesi PA, Dell’Italia LJ. Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Coll Cardiol. 2007;49:811–821. doi: 10.1016/j.jacc.2006.06.083. [DOI] [PubMed] [Google Scholar]
- Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, Tsuji T, Takewa Y, del Monte F, Peluso R, Zsebo K, Jeong D, Park WJ, Kawase Y, Hajjar RJ. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42:852–861. doi: 10.1016/j.yjmcc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D, Su X, Wei CC, Bishop SP, Powell P, Hankes GH, Dillon AR, Rynders P, Spinale FG, Walcott G, Ideker R, Dell’Italia LJ. Downregulation of ANG II receptor is associated with compensated pressure-overload hypertrophy in the young dog. Am J Physiol Heart Circ Physiol. 2002;282:H749–H756. doi: 10.1152/ajpheart.00228.2001. [DOI] [PubMed] [Google Scholar]
- Schwartzkopff B, Frenzel H, Dieckerhoff J, Betz P, Flasshove M, Schulte HD, Mundhenke M, Motz W, Strauer BE. Morphometric investigation of human myocardium in arterial hypertension and valvular aortic stenosis. Eur Heart J. 1992;13(Suppl. D):17–23. doi: 10.1093/eurheartj/13.suppl_d.17. [DOI] [PubMed] [Google Scholar]
- Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, Andre S, Crijns HJ, Gabius HJ, Maessen J, Pinto YM. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121–3128. doi: 10.1161/01.CIR.0000147181.65298.4D. [DOI] [PubMed] [Google Scholar]
- Sparrow P, Messroghli DR, Reid S, Ridgway JP, Bainbridge G, Sivananthan MU. Myocardial T1 mapping for detection of left ventricular myocardial fibrosis in chronic aortic regurgitation: pilot study. AJR Am J Roentgenol. 2006;187:W630–635. doi: 10.2214/AJR.05.1264. [DOI] [PubMed] [Google Scholar]
- Spinale FG. Diversity of myocardial interstitial proteolytic pathways: gene deletion reveals unexpected consequences. Circulation. 2011;124:2052–2055. doi: 10.1161/CIRCULATIONAHA.111.064071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takewa Y, Chemaly ER, Takaki M, Liang LF, Jin H, Karakikes I, Morel C, Taenaka Y, Tatsumi E, Hajjar RJ. Mechanical work and energetic analysis of eccentric cardiac remodeling in a volume overload heart failure in rats. Am J Physiol Heart Circ Physiol. 2009;296:H1117–H1124. doi: 10.1152/ajpheart.01120.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toischer K, Rokita AG, Unsold B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A, Teucher N, Seidler T, Grebe C, Preuss L, Gupta SN, Schmidt K, Lehnart SE, Kruger M, Linke WA, Backs J, Regitz-Zagrosek V, Schafer K, Field LJ, Maier LS, Hasenfuss G. Differential cardiac remodeling in preload versus afterload. Circulation. 2010;122:993–1003. doi: 10.1161/CIRCULATIONAHA.110.943431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- Wei CC, Chen Y, Powell LC, Zheng J, Shi K, Bradley WE, Powell PC, Ahmad S, Ferrario CM, Dell’Italia LJ. Cardiac kallikrein-kinin system is upregulated in chronic volume overload and mediates an inflammatory induced collagen loss. PLoS One. 2012;7:e40110. doi: 10.1371/journal.pone.0040110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zendaoui A, Lachance D, Roussel E, Couet J, Arsenault M. Effects of spironolactone treatment on an experimental model of chronic aortic valve regurgitation. J Heart Valve Dis. 2012;21:478–486. [PubMed] [Google Scholar]
- Zheng J, Chen Y, Pat B, Dell’Italia LA, Tillson M, Dillon AR, Powell PC, Shi K, Shah N, Denney T, Husain A, Dell’Italia LJ. Microarray identifies extensive downregulation of noncollagen extracellular matrix and profibrotic growth factor genes in chronic isolated mitral regurgitation in the dog. Circulation. 2009;119:2086–2095. doi: 10.1161/CIRCULATIONAHA.108.826230. [DOI] [PMC free article] [PubMed] [Google Scholar]