

Abstract
Pathologies that result in early afterdepolarizations (EADs) are a known trigger for tachyarrhythmias, but the conditions that cause surrounding tissue to conduct or suppress EADs are poorly understood. Here we introduce a cell culture model of EAD propagation consisting of monolayers of cultured neonatal rat ventricular myocytes treated with anthopleurin-A (AP-A). AP-A-treated monolayers display a cycle length dependent prolongation of action potential duration (245 ms untreated, vs. 610 ms at 1 Hz and 1200 ms at 0.5 Hz for AP-A-treated monolayers). In contrast, isolated single cells treated with AP-A develop prominent irregular oscillations with a frequency of 2.5 Hz, and a variable prolongation of the action potential duration of up to several seconds. To investigate whether electrotonic interactions between coupled cells modulates EAD formation, cell connectivity was reduced by RNA silencing gap junction Cx43. In contrast to well-connected monolayers, gap junction silenced monolayers display bradycardia-dependent plateau oscillations consistent with EADs. Further, simulations of a cell displaying EADs electrically connected to a cell with normal action potentials show a coupling strength-dependent suppression of EADs consistent with the experimental results. These results suggest that electrotonic effects may play a critical role in EAD-mediated arrhythmogenesis.
Key points
Early afterdepolarizations (EADs) are a known trigger for arrhythmias, but the effect of surrounding tissue on EADs is poorly understood.
Neurotoxin anthopleurin-A (AP-A) increases action potential duration and gives rise to EADs in isolated myocytes. We investigate the effect of AP-A on connected networks of cultured cardiac cells.
We show that EADs are markedly suppressed in well-coupled neonatal rat ventricular monolayers treated with AP-A, but reappear when gap junction connectivity is blocked.
The ability of cell coupling to electrotonically damp EADs is confirmed in a two-cell simulation where connectivity is systematically varied.
Taken together, these results suggest that cell–cell coupling can act to suppress EADs in normal cardiac tissue. Results also suggest that EADs may emerge and propagate in poorly coupled tissue.
Introduction
Many studies suggest that early afterdepolarizations (EADs) arise from Purkinje fibres under pathological conditions that result in prolongation of the action potential duration (APD) and conduct to overlying myocardium to initiate the triggered tachyarrhythmia usually called Torsade de pointes (El-Sherif et al. 1988, 1990; Schimizu et al. 1991; Boutjdir et al. 1994; Gilmore & Moise, 1996). The high membrane resistance intrinsic to Purkinje myocytes (Cordeiro et al. 1998) promotes EAD formation subsequent to a small increase in net inward current (Boutjdir et al. 1994). For example, in a canine model of acquired long Q-T syndrome, El-Sherif et al. (1988) showed that the first ectopic beat of tachycardias induced by anthopleurin-A (APA) resulted from EADs. In that study, EADs developed in Purkinje fibres but not ventricular fibres subsequent to differential APD prolongation.
Purkinje and ventricular myocytes may either facilitate or suppress EAD formation at the Purkinje–ventricular interface. In dogs with inherited sudden death, Gilmore and Moise (1996) identified the site of EAD initiation as the middle of a false tendon far from Purkinje–ventricular junctions. They suggested that electrotonic interactions probably suppressed EAD formation at the Purkinje–ventricular junction because these Purkinje action potentials were shortened by coupling to ventricular cells. In contrast, Li et al. (1992, 1994) found that electrotonic interactions at the Purkinje–ventricular junction were instrumental in inducing triggered activity. EDTA preferentially prolonged APD in Purkinje fibres, which yielded phase 3 EADs that triggered ventricular activation.
Although much has been learned about cardiac electrotonic interactions from multicellular preparations, fundamental principles governing electrophysiological function may be best studied in simplified cardiac models. In recent years, cultured cardiac cell monolayers have become a contemporary experimental preparation for the study of basic mechanisms that underlie normal and pathological electrophysiology at the tissue level (Himel et al. 2012). We investigated the initiation and propagation characteristics of EADs in the neonatal rat ventricular myocyte (NRVM) monolayer superfused with AP-A. The drug results in predictable bradycardia-dependent prolongation of APD and induction of plateau-level EADs in isolated rat ventricular myocytes (Boutjdir et al. 1994). However, our preliminary observations showed that although AP-A resulted in both dose- and cycle length (CL)-dependent prolongation of APD in the NRVM monolayer, no EADs developed on the prolonged plateau. Our findings suggested that electrotonic interactions between myocytes in the monolayer could suppress the formation of EADs. The present study was planned to investigate this phenomenon further.
Methods
The investigation conforms to the Guide for the Care and use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and was approved by the Institutional Animal Care and Use Committee of the VA NY Harbor Healthcare System.
The neonatal rat ventricular myocyte monolayer anthopleurin-A model
NRVM were obtained using a standard enzymatic digestion protocol. Briefly, 2–3 day old neonatal rat pups were killed by decapitation and hearts were excised by application of gentle pressure to the upper chest. Ventricles were isolated and placed in ice-cold phosphate-buffered saline solution containing 0.1% heparin. Ventricles were minced into approximately 1 mm3 cubes, which were then subjected to successive 8 min digestions in warmed (37°C) trypsin (0.2%) solution. Digestion solution containing individual cardiomyocytes was then removed and deactivated by addition of foetal bovine serum. Myofibroblasts were reduced through a 2 h preplating step. Cells were plated on to 35 mm cell culture-treated dishes, and then incubated for 3–7 days in Dulbecco's modified essential cell culture media (Life Technologies, Carlsbad, CA, USA) supplemented with 5% foetal bovine serum and 50 U mL−1 penicillin. The NRVM monolayer was stained by immersion into oxygenated Tyrode's solution (in mmol l−1: 136 NaCl, 5.4 KCl, 1.8 CaCl2, 0.33 NaH2PO4, 1 MgCl2, 10 Hepes and 10 glucose; pH 7.3) containing the fluorescent calcium dye Rhod-2AM (5 μmol l−1 for 30 min, Molecular Probes, Eugene, OR, USA) and/or the voltage dye RH-237 (5 μmol l−1 for 5 min) at 37°C. In a series of later experiments, specifically in the Cx43 silenced monolayer, voltage recordings were obtained using ANNINE-6plus (5 μmol l−1 for 5 min; courtesy of Dr Marlon Hinner, Johann-Straub-Weg 8, Munich, Germany). The dye belongs to a new class of fast response voltage-sensitive dyes with improved voltage discrimination (Fromherz et al. 2008). Both intracellular Ca2+ transients (FCai) and membrane voltage (FV) were mapped at a resolution of 80 × 80 pixels and a temporal resolution of 500 Hz using a CCD Camera (Cardio CCD-SMQ; Redshirt Imaging, Decatur, GA, USA). Our macroscope incorporated an optical cube to filter and reflect excitation light toward the object, and to pass and filter emission before reaching the CCD sensor. All filters were contained within a single optical cube, and could be changed rapidly (<2 min) and repeatedly without the need to refocus the optics of the system. This allowed multiple parameters (Vm and Cai) to be examined in a single dual-stained culture within a short period. Fluorescence values were recorded with a 14-bit resolution. The circular mapping field of view was 15 mm in diameter, giving a spatial resolution of 187.5 × 187.5 μm2 pixel−1.
A stock solution of AP-A (Sigma-Aldrich, St Louis, MO, USA) was prepared by dissolving 100 μg of AP-A into 5 ml of Tyrode's solution, giving a stock solution of 4 μm. For perfusion during experiments, 100 μl aliquots of the stock solution were dissolved in 100 ml of Tyrode's solution, resulting in a final concentration of 4 nm.
For the monolayer to be selected for study, it was first examined to confirm the presence of an interconnected confluent network that was either silent or had an intrinsic rhythm <0.5 Hz. S1 stimulation was applied at 1.0 Hz at the start of each experiment to assess overall wavefront propagation. Following control perfusion with normal Tyrode's solution, the monolayer was perfused with AP-A solution at a flow rate of 1–3 ml min−1. The preparation was paced at a CL of 0.25–1.0 Hz with 10 ms duration 2× threshold stimulus using bipolar electrodes placed at the lower edge of the preparation. Pacing was performed with a Grass stimulator triggered by a computer-controlled programmable pacing sequence.
Single cell recordings in isolated neonatal rat ventricular myocytes and monolayers
To investigate the absence of detectable plateau level oscillations consistent with EADs in the NRVM monolayer AP-A model, we recorded FCai from isolated single cardiomyocytes following AP-A perfusion and compared it with FCai of single cardiomyocytes in an interconnected confluent monolayer. Single cell recordings were obtained from 1 to 2 day cultures that were plated at a low density (5–10 × 104 cells plate−1). Our experience is that these cultures contain isolated cells, which had no neighbours, as well as cell pairs and small islands of four to 20 cells.
Cells were observed using light microscopy (Nikon Eclipse Ti, Nikon Inc., Melville, NY, USA). The cells were field stimulated (Myopacer EP, IonOptix Corp., Milton, MA, USA) with platinum electrodes at 0.5 Hz (square pulses, 5 ms in duration; 2× threshold pacing voltage, 10–40 V) to verify contractile activity. Cells were illuminated from beneath the stage using a filtered (480 ± 15 nm) xenon arc lamp excitation source (Optosource; Cairn Research, Faversham, Kent, UK). Cells were loaded with fluo-4 and emitted fluorescence was filtered (535 ± 20 nm) and measured using a photomultiplier tube (IonOptix Corp.) and recordings were obtained at a rate of 500 Hz.
Cells were paced normally at 0.5 Hz for approximately 5 min. Normal Tyrode's solution was then removed and replaced with Tyrode's solution containing AP-A (4 nm, Sigma-Aldrich Corp.). Cells were paced at rates of 0.25–1 Hz and recordings were continuously obtained as AP-A solution was perfused. For single cell recordings, the perfusion temperature was maintained at 37°C.
Fluorescence values were processed initially using IonWizard® (IonOptix Corp.). The background fluorescence was subtracted from the raw fluorescence values, followed by division by the baseline fluorescence (F0). Signals were then filtered (13 point moving average) and plotted in MATLAB® (The Mathworks, Natick, MA, USA). Data values are given as means ± standard deviation.
Cx43 silencing
The sequence of the siRNA specifically targeting the rat Cx43 gene was either purchased from Ambion (Silencer Pre-designed siRNA) or designed through siRNA Target Finder (Ambion, Austin, TX, USA). The oligonucleotides were selected by BLAST homology search among cDNA sequences of target genes from various species, including mouse, rat and human. The oligos were synthesized (Integrated DNA Technologies, Coralville, IA, USA) with the following format: sense oligo: 50T-(19nt)-TTCAAGAGA)-(91nt)-TTTTTTC (sense siRNA–loop–antisense siRNA–stop). The designed sequences were also matched with Cx43 siRNA described in an earlier report (Zlochiver et al. 2008). Myocte culture was transfected with custom-designed rat-specific Stealth RNAi construct directed against Cx43 (sequence: GGCUUGCUGAGAACCUACAUCAUCA) (Invitrogen, Carlsbad, CA, USA). The sequence GAUACACUGCGUUUCGCUUCGAGUA was used as a negative control. RNAi transfection was performed using lipofectamine RNAiMAX (Invitrogen) in serum-free M199 medium without antibiotics. Serum was restored after 4 h. RNAi uptake in myocytes was assessed using BLOCK-iT Control Fluorescent Oligo (Invitrogen), and transfection efficiency was 80–85%. (see Supplementary Fig. 1).
Mathematical model
Simulations were conducted on a pair of electrically coupled cells, each based on the Pasek, Simurda, Orchard and Christe ventricular cardiomyocyte model (Pasek et al. 2008). As there are presently no biophysically realistic models of neonatal rat myocytes in the literature, we chose to screen the CellML Model Repository (http://models.cellml.org/) for cardiac electrophysiological models that display prolonged trains of EADs similar to those observed experimentally, using published parameters but with a voltage shift in the Na+ inactivation gate (h) curve, which is consistent with the effects of AP-A on Na+ conductance (Sheets & Hanck 1995). The Pasek et al. (2008) ventricular myocyte model was found to display sustained EADs with an 11 mV shift in the h curve. Two models were electrically coupled with conductance and the Na+ inactivation curve shift for each cell as control parameters. Simulations were carried out using custom software based on the Sundials CVODE ordinary differential equation solver (Hindmarsh et al. 2005). The code is available on request.
Statistical analysis
Values were listed as means ± standard deviation. The two-tailed Student t test assessing unequal variance was used to test for difference between groups. P < 0.05 was considered significant.
Results
The neonatal rat ventricular myocyte anthopleurin-A monolayer
Figure 1A illustrates control recordings of voltage (FV) and Ca2+ (FCai) fluorescence from a 4-day-old NRVM monolayer paced at 1 Hz. The recordings have a triangular configuration and lack a prominent plateau. The duration of FV at 85% repolarization (FVD85) was 245 ms compared to the duration of FCai at 85% relaxation (FCaiD85) of 285 ms. Figure 1B was obtained following 1 min of perfusion with AP-A, and showed plateau extension and lengthening of both FVD85 and FCaiD85 to 610 and 620 ms respectively. In Fig. 1C, the preparation was paced at a slower rate of 0.5 Hz, which resulted in CL-dependent prolongation of both FVD85 and FCaiD85 to 1200 and 1220 ms, respectively. There were no oscillations observed on the plateau of either FV or FCai recordings consistent with EADs in any of the cells in the preparation.
Figure 1. Recordings of voltage (Fv) and Ca2+ (FCai) fluorescence.
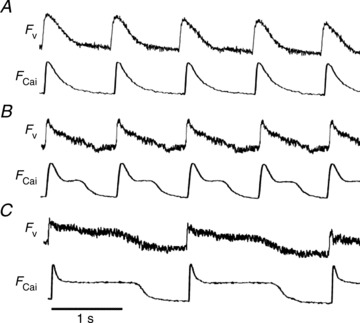
A, perfused with normal Tyrode, paced at 1 Hz. B, perfused with anthopleurin-A (4 nm), paced at 1 Hz. C, perfused with anthopleurin-A (4 nm), paced at 0.5 Hz.
The prolongation of both FvD85 and FCaiD85 were associated with significant dispersion of signal duration. An overall dispersion in the recording field of up to 200 ms was seen and spatial dispersion of 20–40 ms between pixels less than 1 mm apart was not uncommon. (Please see Supplementary Fig. 2.)
Single cell recordings
Figure 2A illustrates FCai recordings from an isolated NRVM. Figure 2A-I, left, shows a control recording during stimulation at 0.5 Hz. Panel I, right, and panel II illustrate changes in FCaiD85 following perfusion of AP-A (4 nm). The drug resulted in gradual prolongation of FCai followed by the development of prominent irregular oscillations at the plateau level, with an approximate frequency of 2.5 Hz, and resulting in variable prolongation of FCaiD85 of up to several seconds.
Figure 2. The effect of AP-A on isolated and connected neonatal rat ventricular myocytes.
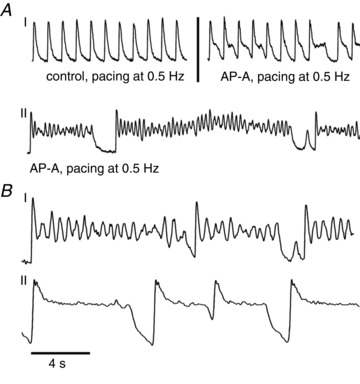
A, panel I (left) perfused with normal Tyrode, paced at 0.5 Hz; (right) AP-A 4 nm perfusion, paced at 0.5 Hz; panel II) AP-A 4 nm perfusion, paced at 0.5 Hz. B, comparison between isolated cardiomyocyte (I) and confluent monolayer (II), paced at 0.25 Hz. AP-A, anthopleurin-A.
Figure 2B was obtained from different experiments and compares effects of AP-A on FCai oscillations from an isolated cardiomyocyte (panel I) and a cardiomyocyte from an interconnected confluent monolayer (panel II), both stimulated at 0.25 Hz. The recordings show that prominent FCai oscillations in panel I were markedly suppressed in panel II. To estimate the relative amplitude of Ca2+ oscillations occurring during the plateau phase, we calculated the amplitude of the largest plateau level FCai oscillation in relation to the overall FCai signal amplitude. In panel I, for example, the largest FCai oscillation amplitude was 52% of the overall FCai signal amplitude. In panel II, the largest FCai amplitude was only 5% of the overall FCai amplitude, illustrating an approximate 10-fold reduction of FCai oscillation amplitudes. We analysed 16 isolated myocytes from eight different preparations and 14 myocytes from seven different confluent monolayers. Oscillation amplitudes from confluent monolayer preparations were significantly depressed vs. amplitudes from isolated myocytes (5.5 ± 1.8%vs. 47.2 ± 5.9%, P < 0.0001).
Cx43 silencing
In contrast to control, monolayers in which Cx43 was silenced and that were stimulated at long CLs showed oscillations on prolonged plateaus of FV and FCai consistent with EADs. Figure 3 is a representative example from one of the monolayers in which Cx43 was silenced following 2 min of perfusion with AP-A. The preparation was stimulated from a site at the bottom of the monolayer and representative pixels of FCai are shown. Figure 3 shows a recording during pacing at 1 Hz (top trace). FCaiD85 was 675 ms and showed no oscillations, Fig. 3B, bottom three traces shows a simultaneous recording from three different pixels, X, Y and Z, during pacing at 0.25 Hz. There is a marked prolongation of FCaiD85 and development of irregular oscillations at the plateau level at an approximate frequency of 3 Hz. Recordings at the two pixels close to the stimulating electrode (X and Y) show the presence of a certain degree of synchronous propagation at a limited spatial scale despite of the high degree of uncoupling. On the other hand, recordings at sites distal from the stimulating electrode (Z) showed evidence of both propagation delay and intermittent failure of propagation.
Figure 3. Cx43 silenced monolayer perfused with anthopleurin-A.
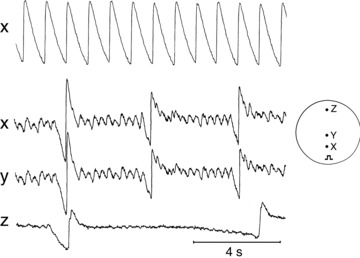
Top trace shows FCai recorded from a 3-day monolayer paced at 1 Hz. Bottom three traces show FCai recorded from a monolayer paced at 0.25 Hz. Bottom three traces correspond to traces obtained from different locations relative to the pacing site as shown in the inset.
Figure 4 is a representative example from one of the monolayers in which Cx43 was silenced following 3 min of perfusion with AP-A. Figure 4A shows recordings from two pixels within 1 mm in the optical field during pacing at 1 Hz. FvD85 was 615 ms and showed no oscillations. Figure 4B shows recordings from the same two pixels during pacing at 0.25 Hz. There is a marked prolongation of FvD85 and development of irregular oscillations at the plateau level consistent with EADs at an approximate frequency of 2 Hz.
Figure 4. Cx43 silenced monolayer perfused with anthopleurin-A.
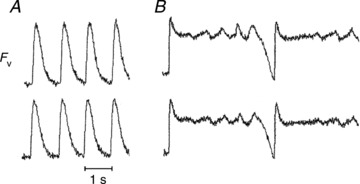
A, Fv recorded from a monolayer paced at 1 Hz. B, Fv recorded from a monolayer paced at 0.25 Hz. The two traces correspond to traces obtained from two locations within 1 mm in the optical field.
Mathematical model
The Pasek et al. (2008) model displays sustained EADs if the Na+ inactivation gate (h) curve is shifted by 11 mV or more (hs > 11 mV, Fig. 5A). To investigate whether a quiescent cell can modulate EAD activity, we simulated the effects of coupling a cell with hs > 11 mV with one with normal dynamics (hs= 0), for a range of conductances (G) and values of hs. Here, we classify EADs as damped if the plateau remains elevated (>50 mV) but have oscillations with less than 5 mV amplitude for periods of 2000 ms or more (Fig. 5B). There is a large region in the (hs, G) parameter space that displays damped oscillations similar to those seen experimentally (Fig. 5C). The effect of Cx43 silencing would be to shift the dynamics outside of this region, recovering the uncoupled (single cell) EAD behaviour.
Figure 5. Simulation results from the two-cell Pasek et al. (2008) model.
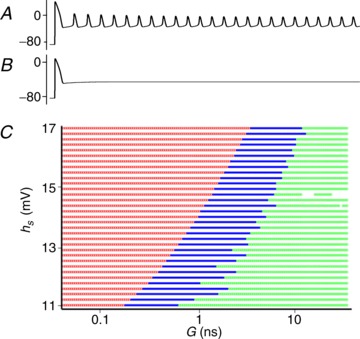
A, shift in the Na+ inactivation gate (h) curve (hs > 11 mV) results in sustained EADs. B, EADs are suppressed when two cells are coupled for some values of conductance (G) and hs. C, two-cell simulations where a quiescent cell (hs= 0 mV) is coupled to a EAD expressing cell (hs > 11 mV) for 6000 different values of G and hs. The results are classified as blocked (no EADs – green), suppressed (elevated baseline with no oscillations – blue), and expressed EADs (red). EADs, early afterdepolarizations.
Discussion
Early afterdepolarization suppression
There are two plausible explanations of the failure to observe prominent plateau-level oscillations that could be easily induced in isolated rat cardiomyocytes exposed to AP-A. One possible explanation is a limitation of the resolution of the optical recordings in the monolayer due to averaging effects caused by fluorescence arriving at a single pixel from multiple cells, each having random EAD patterns. The second, more interesting, possibility is that asynchronous oscillations from cells in a confluent monolayer could be suppressed by electrotonic interactions between well-coupled cells. To investigate this possibility, we recorded FCai from isolated single cells and from single cells within a confluent monolayer. We demonstrated that plateau level prominent oscillations in isolated single cardiomyocytes were markedly suppressed in electrotonically coupled myocytes.
We initially tried to suppress Cx43 using pharmacological means (18-α glycyrrhetinic acid) but we quickly abandoned this approach. Available agents typically have effects other than decreasing gap junction conductance, in particular on the fast Na+ current, which counteracts the effects of AP-A in inducing bradycardia-dependent prolongation of the plateau. On the other hand, silencing Cx43 clearly showed the development of bradycardia-dependent plateau oscillations consistent with EADs. This evidence strongly suggests that cellular coupling in the monolayer can explain the failure to demonstrate EADs despite marked prolongation of the plateau.
To confirm further the experimental observation, we conducted simulations using two gap junction coupled cells. The simulations confirmed that quiescent cells can damp oscillations in neighbouring cells that display EADs (Fig. 5). Furthermore, cell–cell conductances where oscillations are damped were within physiologically expected values (0.05–35 nS, see Burt & Spray 1998). The models were based on the assumption that the experimental system consists of both cells that display EADs in isolation and a quiescent cell population. In this simulation, we used two identical cell models with different shifts in the Na+ inactivation gate curve. However, in principle this approach would work if we modelled myocytes coupled to passive fibroblasts, which are a known population of cells in monolayer preparations (Gaudesius et al. 2003).
The model has several limitations. First, the single cell myocyte model was chosen based on its ability to accommodate biophysically realistic Na+ inactivation gate curve shifts and the presence of sustained EADs given minimal changes in published parameters. The mechanism is based on known effects of AP-A on the Na+ current, which are different from those modelled in previous EAD studies (Luo & Rudy 1994; Sato et al. 2009). However, the single cell myocyte model extended in this study is not a neonatal rat model, which complicates comparison to the experimental NVRM system. Developing and validating a new neonatal cell model with sustained EADs is beyond the scope of this study. Still, we propose that the mechanism for EAD suppression seen here is due to electrotonic effects, and therefore any biophysically realistic cardiac cell system will probably display similar dynamics. Second, we anticipate that a two-dimensional sheet of cells, where cells are coupled to neighbours on all sides, may display more complex behaviours than the two-cell system simulated here. However, spatial effects will probably not prevent suppression of EADs observed in the two-cell model, and we propose that the two-cell system is sufficient to demonstrate the behaviours observed experimentally.
The effects of anthopleurin-A on membrane currents and the genesis of early afterdepolarizations
Single channel recordings have shown that AP-A and the closely related sea anemone toxin ATX-II induce burst behaviour of the fast Na+ channel as a result of a marked delay of the open-to-inactivation transition (El-Sherif et al. 1992). The persistence of inward Na+ current results in prolongation of APD, which in turn promotes Ca2+ overload. The latter can trigger spontaneous Ca2+ release from the sarcoplasmic reticulum, enhance the turnover rate of the Na+/Ca2+ exchanger and its depolarization current, INCX, which may reactivate the L-type calcium current (ICa-L) (January & Riddle, 1989; Sipido et al. 1995). The classic hypothesis of EAD genesis proposes that they arise from reactivation of ICa-L. However, a recent study suggests that reactivation ICa-L plays a prominent role in EAD genesis under oxidative stress, while spontaneous Ca2+ waves are a prominent cause for EADs under Ca2+ overload conditions (Zhao et al. 2012). In support of this mechanism are data from the Salama laboratory (Choi et al. 2002) as well as ours (Lakireddy et al. 2006), which showed that Ca2+ oscillations on the plateau preceded voltage depolarizations characteristic of EADs.
A significant fraction of Na+ channels is expressed in the intercalated discs. Recent studies have shown that suppression of Cx43 in ventricular (Jansen et al. 2011) or atrial (Desplantez et al. 2012) tissue is associated with a decrease in peak sodium current (INa). Although this effect would have been expected to interfere to some extent with the effects of AP-A to induce bradycardia-dependent marked prolongation of the plateau, this was not observed in the present study. On the other hand, our observation of the development of significant propagation delays and block in the monolayer in which Cx43 was silenced (Fig. 3) could be explained in part by the effects on uncoupling as well as a depression in peak INa. Despite cellular uncoupling in the monolayer, there was a certain degree of synchrony of EADs at a limited spatial scale. However, none of the EADs resulted in a fully conducting action potential. It is possible that EADs at the single cell level in the monolayer may not be sufficient to depolarize a large enough region to propagate an ectopic excitation. The biophysical basis for this hypothesis is consistent with previous experimental (Fozzard & Schoenberg, 1972) and computational (Noble, 1972) studies on the liminal length hypothesis.
In summary, our data could be best interpreted to suggest that electrotonic interaction between myocytes in the monolayer could suppress EAD formation. The application of these results to the whole heart, especially to the Purkinje–myocyte interface, requires further investigation.
Acknowledgments
None.
Glossary
- AP-A
anthopleurin-A
- APD
action potential duration
- CL
cycle length
- EADs
early afterdepolarizations
- FCai
Ca2+ fluorescence
- FCaiD85
duration of FCai at 85% repolarization
- FV
voltage fluorescence
- FVD85
duration of Fv at 85% repolarization
- G
conductance
- h
Na+ inactivation gate
- hs
Na+ inactivation gate shift
- NRVM
neonatal rat ventricular myocyte
- Rj
junctional resistance
- L-type calcium current (ICa-L)
sodium calcium exchanger current (INCX), fast sodium current (INa)
Additional information
Competing interests
None.
Author contributions
Authors’ were responsible for the following. H.D.H., conception and design of the experiments; Collection, analysis and interpretation of experimental data; Drafting the article and revising it critically for important intellectual content. A.G., simulation design and rationale, authored simulation code, manuscript editing. P.J.N., simulation design and rationale, manuscript editing. R.W., collection, analysis and interpretation of experimental data. J.S., collection, analysis and interpretation of experimental data. N.L., collection of experimental data. G.B., simulation design, conducted simulations; Drafting the article and revising it critically for important intellectual content. N.E.S., Conception and design of the experiments; Collection, analysis and interpretation of experimental data; Drafting the article and revising it critically for important intellectual content.
Funding
G.B. acknowledges support from the British Heart Foundation Centre of Research Excellence, Oxford. RE/08/004. N. E. was supported in part by a Veterans Administration MERIT grant and the Narrows Institute for Biomedical Research.
Supplementary material
Supplementary Fig. 1
Supplementary Fig. 2
References
- Boutjdir M, Restivo M, Wei Y, Stergiopoulos K, El-Sherif N. Early afterdepolarizations formation in cardiac myocytes: Analysis of phase plane patterns, action potential, and membrane currents. J Cardiovasc Electrophysiol. 1994;5:609–620. doi: 10.1111/j.1540-8167.1994.tb01302.x. [DOI] [PubMed] [Google Scholar]
- Burt JM, Spray DC. Single-channel events and gating behavior of the cardiac gap junction. Proc Natl Acad Sci U S A. 1998;85:3431–3434. doi: 10.1073/pnas.85.10.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JM, Spitzer KW, Giles WR. Repolarizing K+ currents in rabbit heart Purkinje cells. J Physiol. 1998;508(pt 3):811–823. doi: 10.1111/j.1469-7793.1998.811bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplantez T, McCain ML, Beauchamp P, Rogoli G, Rothen-Rutishauser B, Parker KK, Kleber AG. Connexin43 ablation in foetal atrial myocytes decreases electrical coupling, partner connexins, and sodium current. Cardiovasc Res. 2012;94:58–65. doi: 10.1093/cvr/cvs025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sherif N, Zeiler RH, Craelius W, Gough WB, Henkin R. QTU prolongation and polymorphic ventricular tachyarrhythmias due to bradycardia-dependent early afterdepolarizations. Circ Res. 1988;63:286–305. doi: 10.1161/01.res.63.2.286. [DOI] [PubMed] [Google Scholar]
- El-Sherif N, Craelius W, Boutjdir M, Gough WB. Early afterdepolarizations and arrhythmogenesis. J Cardiovasc Electrophysiol. 1990;1:145–160. [Google Scholar]
- El-Sherif N, Fozzard HA, Hanck DA. Dose-dependent modulation of the cardiac sodium channel by the sea anemone toxin ATXII. Circ Res. 1992;70:285–301. doi: 10.1161/01.res.70.2.285. [DOI] [PubMed] [Google Scholar]
- Fozzard HA, Schoenberg M. Strength-duration curves in cardiac purkinje fibres: effects of luminal length and charge distribution. J Physiol. 1972;226:593–618. doi: 10.1113/jphysiol.1972.sp009999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromherz P, Hubenar G, Kuhn B, Hinner MJ. ANNINE-6plus, a voltage-sensitive dye with good solubility, strong membrane binding and high sensitivity. Eur Biophys J. 2008;37:509–514. doi: 10.1007/s00249-007-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–428. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- Gilmore RF, Moise NS. Triggered activity as a mechanism of inherited ventricular arrhythmias in German shepherd dogs. Am J Cardiol. 1996;27:1526–1533. doi: 10.1016/0735-1097(95)00618-4. [DOI] [PubMed] [Google Scholar]
- Himel HD, Bub G, Lakireddy P, El-Sherif N. Optical imaging of arrhythmias in the cardiomyocyte monolayer. Heart Rhythm. 2012;9:2077–2082. doi: 10.1016/j.hrthm.2012.08.035. [DOI] [PubMed] [Google Scholar]
- Hindmarsh AC, Brown PN, Grant KE, Lee SL, Serban R, Shumaker DE, Woodward CS. SUNDIALS: Suite of Nonlinear and Differential/Algebraic Equation Solvers. ACM Trans Math Softw. 2005;31:363–396. [Google Scholar]
- Jansen JA, Noorman M, Musa H, Stein M, de Long S, Van der Nagel R, Hund TJ, Mohler PJ, Vos MA, van Veen TA, de Bakker JM, Delmar M, van Rijen HV. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2011;9:600–607. doi: 10.1016/j.hrthm.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- January CT, Riddle JM. Early afterdepolarization: mechanism and induction of block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- Lakireddy V, Bub G, Baweja P, Syed A, Boutjidir M, El-Sherif N. The kinetics of spontaneous calcium oscillations and arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm. 2006;3:58–66. doi: 10.1016/j.hrthm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Li Z-Y, Maldonado C, Zee-Cheng C, Hiromasa S, Kupersmith J. Purkinje fibre-papillary muscle interaction in the genesis of triggered activity in a guinea pig model. Cardiovasc Res. 1992;26:543–548. doi: 10.1093/cvr/26.5.543. [DOI] [PubMed] [Google Scholar]
- Li Z-W, Wang Y, Maldonado C, Kupersmith J. Role of junctional zone cells between Purkinje fibres and ventricular muscle in arrhythmogenesis. Cardiovasc Res. 1994;28:1277–1284. doi: 10.1093/cvr/28.8.1277. [DOI] [PubMed] [Google Scholar]
- Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circ Res. 1994;74:1097–1113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- Noble D. The relation of Rushton's ‘liminal length’ for excitation to the resting and active conductances of excitable cells. J Physiol. 1972;226:573–591. doi: 10.1113/jphysiol.1972.sp009998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasek M, Simurda J, Orchard CH, Christe G. A model of the guinea-pig ventricular cardiac myocyte incorporating a transverse-axial tubular system. Prog Biophys Mol Biol. 2008;96:258–280. doi: 10.1016/j.pbiomolbio.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Sato D, Xie L-H, Sovari AA, Tran DX, Morita N, Xie F, Karaguezian H, Garfinkel A, Weiss AN, Qu Z. Synchronization of chaotic early afterdepolarization in the genesis of cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimizu W, Ohe T, Kurita T, Takaki H, Aihura N, Kamakura S, Matsuhisa M, Shimomura K. Early afterdepolarization induced by isoprenaline in patients with congenital long QT syndrome. Circulation. 1991;84:1915–1923. doi: 10.1161/01.cir.84.5.1915. [DOI] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Voltage-dependent open state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J Gen Physiol. 1995;106:617–640. doi: 10.1085/jgp.106.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Callewaert G, Carmeliet E. Inhibition and rapid recovery of ICai during calcium release from the sarcoplasmic reticulum in guinea-pig ventricular myocytes. Circ Res. 1995;76:102–109. doi: 10.1161/01.res.76.1.102. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Wen H, Fefelova N, Allen C, Baba A, Matsuda T, Xie L-H. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes:predominant by Ca waves or Ca currents. Am J Physiol Heart Circ Physiol. 2012;302:H1636–H1644. doi: 10.1152/ajpheart.00742.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlochiver S, Muñoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblasts-to-myocytes coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–4480. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.