

A GH1 6-phospho-β-galactosidase from G. stearothermophilus T1 (Gan1D) has been purified and crystallized in the triclinic space group P1. A full diffraction data set has been measured for the wild-type enzyme to a maximal resolution of 1.33 Å to be further used for a detailed three-dimensional structural analysis of the protein.
Keywords: Geobacillus stearothermophilus, galactan utilization, glycoside hydrolases, galactosidases, GH1, P1, high resolution
Abstract
Geobacillus stearothermophilus T1 is a Gram-positive thermophilic soil bacterium that contains an extensive system for the utilization of plant cell-wall polysaccharides, including xylan, arabinan and galactan. The bacterium uses a number of extracellular enzymes that break down the high-molecular-weight polysaccharides into short oligosaccharides, which enter the cell and are further hydrolyzed into sugar monomers by dedicated intracellular glycoside hydrolases. The interest in the biochemical characterization and structural analysis of these proteins originates mainly from the wide range of their potential biotechnological applications. Studying the different hemicellulolytic utilization systems in G. stearothermophilus T1, a new galactan-utilization gene cluster was recently identified, which encodes a number of proteins, one of which is a GH1 putative 6-phospho-β-galactosidase (Gan1D). Gan1D has recently been cloned, overexpressed, purified and crystallized as part of its comprehensive structure–function study. The best crystals obtained for this enzyme belonged to the triclinic space group P1, with average crystallographic unit-cell parameters of a = 67.0, b = 78.1, c = 92.1 Å, α = 102.4, β = 93.5, γ = 91.7°. A full diffraction data set to 1.33 Å resolution has been collected for the wild-type enzyme, as measured from flash-cooled crystals at 100 K, using synchrotron radiation. These data are currently being used for the detailed three-dimensional crystal structure analysis of Gan1D.
1. Introduction
6-Phospho-β-galactosidases (EC 3.2.1.85) are enzymes that hydrolyze the β-glycosidic bond between a terminal nonreducing galactose-6-phosphate and other organic molecules. These enzymes are usually associated with the phosphotransferase system (PTS transport system), an alternative intake system involved in the transport and phosphorylation of numerous monosaccharides, disaccharides, amino sugars, polyols and a series of other sugar derivatives. These unique PTS transport systems, also referred to as phosphoenolpyruvate–carbohydrate phosphotransferase systems (PEP-PTS), are only found in bacteria (Postma et al., 1993 ▶; Robillard & Broos, 1999 ▶; Deutscher et al., 2006 ▶). According to the CAZy database, all of the 6-phospho-β-galactosidases reported to date belong to family 1 of the glycoside hydrolases (GH1; Henrissat & Davies, 1997 ▶) and seem to act via a retaining mechanism. Based on sequence analysis, 6-phospho-β-galactosidases are identified to be involved in lactose utilization in Staphylococcus aureus, Lactococcus lactis, Lactobacillus casei and Streptococcus mutans (de Vos & Gasson, 1989 ▶; Breidt & Stewart, 1987 ▶; Porter & Chassy, 1988 ▶; Honeyman & Curtiss, 1993 ▶). In these bacteria, lactose is transported into the cell and phosphorylated via a phosphotransferase system and further hydrolyzed inside the cell by a 6-phospho-β-galactosidase, resulting in glucose and galactose 6-phosphate (Breidt et al., 1987 ▶; de Vos et al., 1990 ▶; Alpert & Chassy, 1990 ▶; Rosey & Stewart, 1992 ▶). Only a small number of these enzymes have been biochemically characterized, most of them in lactic bacteria such as Lactococcus lactis, Lactobacillus gasseri and Streptococcus lactis (McKay et al., 1970 ▶; Suzuki et al., 1996a ▶,b ▶; Honda et al., 2012 ▶). Of these enzymes, only the 6-phospho-β-galactosidase from L. lactis has been subjected to full crystallographic analysis (Wiesmann et al., 1995 ▶, 1997 ▶).
Geobacillus stearothermophilus is a thermophilic, Gram-positive, soil bacterium which possesses an extensive system for the utilization of plant cell-wall polysaccharides, including xylan, arabinan and galactan (Shulami et al., 1999 ▶, 2011 ▶; Tabachnikov & Shoham, 2013 ▶). The bacterium secretes a small number of extracellular enzymes that cleave high-molecular-weight polysaccharides into shorter decorated oligosaccharides. The resulting oligosaccharides are transported into the cell via specialized ABC transporters (Rees et al., 2009 ▶) and are further hydrolyzed into sugar monomers by a battery of specific intracellular glycoside hydrolases. The most studied and the best characterized of these complex utilization systems is that of xylan, where the bacterium secretes an extracellular xylanase (Gat et al., 1994 ▶; Teplitsky et al., 1997 ▶, 2004 ▶; Bar et al., 2004 ▶) that partially degrades xylan to short decorated xylooligosaccharides, which are then transported into the cell via dedicated ABC transporters (Shulami et al., 2007 ▶). Inside the cell, the decorated xylooligosaccharides are hydrolyzed by several side-chain-cleaving enzymes, including arabinofuranosidases (Shallom, Belakhov, Solomon, Gilead-Gropper et al., 2002 ▶; Shallom, Belakhov, Solomon, Shoham et al., 2002 ▶; Hövel et al., 2003 ▶), an α-glucuronidase (Teplitsky et al., 1999 ▶; Golan et al., 2004 ▶; Zaide et al., 2001 ▶; Shallom et al., 2004 ▶), acetylesterases (Alalouf et al., 2011 ▶; Lansky, Alalouf et al., 2013 ▶; Lansky et al., 2014 ▶) and finally by an intracellular xylanase (Teplitsky et al., 2000 ▶; Solomon et al., 2007 ▶) and several xylosidases (Bravman et al., 2003 ▶; Shallom et al., 2005 ▶; Brüx et al., 2006 ▶; Ben-David et al., 2007 ▶).
In the framework of a wider study of the hemicellulolytic utilization systems in G. stearothermophilus T1, we have characterized a galactan-utilization gene cluster in this bacterium (Tabachnikov & Shoham, 2013 ▶). This 9.4 kb cluster includes several transcriptional units, which encode, among other proteins, an extracellular galactanase (Gan53A), an intracellular galactosidase (Gan42B) (Solomon et al., 2013 ▶) and a specific ABC sugar transporter (GanEFG) (Tabachnikov & Shoham, 2013 ▶). It is therefore suggested that Gan53A cleaves the high-molecular-weight polymer galactan outside of the cell into galactooligosaccharides; these oligosaccharides are then transported via the ABC sugar transport system into the cell, where they are further hydrolyzed to galactose by Gan42B. In addition to this system, we recently identified another 12.5 kb gene cluster that, based on real-time RT-PCR analysis, is induced by galactan and galactose, suggesting that this cluster is also involved in the utilization of galactan (unpublished results). The newly discovered cluster includes five transcriptional units, which encode a putative GH1 6-phospho-β-galactosidase (Gan1D), a transcriptional regulator (GanR2), a GH4 6-phospho-β-glucosidase (Gan4C), a three-component regulatory system (GanPST) and another dedicated ABC sugar transport system (GanE2F2G2). Relatively little is known about these five protein components, except that the ABC transporter seems to recognize galactose and that the sugar-binding lipoprotein of the three-component regulatory system, GanP, has been shown to bind both galactose and glucose (unpublished results).
The current study is part of a larger project geared to characterize in detail the proteins involved in this new transport system, both alone and as an integrated cellular system. In the present report, we describe the purification, crystallization and preliminary crystallographic characterization of one of these proteins, Gan1D, as the first step toward its three-dimensional structural determination and the corresponding structure–function analysis. As a key player in the biochemical hydrolysis of polymeric β-1,4-galactosaccharides into galactose monomers, Gan1D and related enzymes play an important part of the hemicellulolytic utilization system of many microorganisms which use plant biomass for growth. The interest in the biochemical characterization and structural analysis of these enzymes stems therefore not only from basic scientific interest, but also from their numerous potential biotechnological applications.
2. Experimental
2.1. Cloning, expression and purification of Gan1D
The gan1D gene was amplified via PCR fused to a six-His-tag extrapeptide at its N-terminus and was cloned into pET-9d (Novagen, Madison, Wisconsin, USA). The gene was amplified from chromosomal DNA of G. stearothermophilus T1 using PCR, based on the genomic sequence (GenBank accession No. KF840174, base pairs 236–1672). The PCR primers were Gan1D N-ter, 5′-TATAGTCATG ATACATCACCACCACCACCATGAGCATCGTCATCTTAAAC-3′, which included a His6 tag and a BspHI restriction site (TCATGA), and Gan1D C-ter, 5′-TATACGGATCCCTACAGCTCGGCACCGTTC-3′, which included a BamHI restriction site (GGATCC). The PCR product was digested with the appropriate restriction enzymes and cloned into pET-9d vector linearized with NcoI and BamHI to give pET-9d-gan1D. Expression of gan1D was carried out by growing Escherichia coli BL21 (λDE3) cells (Stratagene, La Jolla, California, USA) harbouring pET-9d-gan1D in 2 l shake flasks containing 0.5 l Terrific broth medium supplemented with 25 µg ml−1 kanamycin, which were shaken at 230 rev min−1 at 310 K. Following overnight growth (final OD600 nm of about 13), cells from 1 l culture were harvested, resuspended in 40 ml buffer (20 mM imidazole, 20 mM phosphate buffer, 500 mM NaCl pH 7.0), disrupted by two passages through an Avestin Emulsiflex C3 Homogenizer (Avestin) and centrifuged (14 000 rev min−1 for 30 min) to obtain soluble extract. The His-tagged fused protein was isolated using a 5 ml HisTrap column (GE Healthcare Life Sciences) mounted on an ÄKTA Avant-25 chromatography system (GE Healthcare Life Sciences) according to the manufacturer’s instructions. The final protein sample was more than 95% pure, based on SDS–PAGE, with a total yield of approximately 50 mg per litre of overnight culture. Gel-permeation chromatography on a Superose 12 column (GE Healthcare Life Sciences) indicated that the native protein has a molecular weight of about 110 kDa.
2.2. Biochemical experiments
The activity of Gan1D towards various substrates has been qualitatively determined in a number of preliminary experiments using a continuous colorimetric assay. The reactions were performed at 313 K in 100 mm citric acid–Na2HPO4 buffer pH 7.6, 0.5 M NaCl, 1 mg ml−1 BSA and 10 µg ml−1 Gan1D. Substrate concentrations were in the range 0–35 mM. The reactions were initialized by adding 50 µl of appropriately diluted (within the range 0.1–10 K m) pre-warmed substrate to 150 µl pre-warmed enzyme solution in a 96-well plate. The absorbance at 405 nm was measured at 30 s intervals for 20 min using a Synergy HT Multi-Mode Microplate Reader (Bio-Tek Instruments, Winooski, Vermont, USA).
2.3. Crystallization experiments
Crystallization experiments were set up immediately after the last purification step of the recombinant wild-type Gan1D protein. The purified protein was concentrated to approximately 4 mg ml−1 using Centricon centrifugal concentrators (Millipore, Massachusetts, USA) and this protein solution was used for the crystallization experiments. All initial crystallization experiments were performed by the hanging-drop vapour-diffusion method, using an extensive series of different factorial screens (Jancarik & Kim, 1991 ▶). In general, these initial conditions were based on commercially available sets of ‘ready-to-use’ screening solutions. Once positive results had been obtained (i.e. crystals or microcrystals), further refinement experiments of these crystallization conditions were performed with specially prepared solutions, optimizing parameters such as pH, ionic strength, protein concentration, temperature, precipitating additives, protein drop volume and drop-to-reservoir ratio (Almog et al., 1993 ▶, 1994 ▶; Teplitsky et al., 1997 ▶, 1999 ▶, 2000 ▶; Bar et al., 2004 ▶; Lansky, Alalouf et al., 2013 ▶; Lansky, Salama et al., 2013 ▶; Solomon et al., 2013 ▶).
The final Gan1D protein crystallization drops were prepared by mixing the concentrated protein solution (2.0–3.0 µl, ∼4 mg ml−1) with an equal amount of each of the specific screen solutions to give a final drop volume of 4.0–6.0 µl. Each of these protein drops was suspended over 0.5–1.0 ml reservoir solution in 4 × 6 VDX crystallization plates (Hampton Research, California, USA) for a period of about 1–8 d at a constant temperature of 293 K. One of these initial screening experiments resulted in reasonable crystals (in terms of size and shape) and fine refinement of this condition resulted in two different crystal forms. As explained below, although the two crystal forms showed reasonable X-ray diffraction properties, only one of them was found to be suitable for further crystallographic analysis at high resolution.
Most of the X-ray diffraction data measurements were performed at the European Synchrotron Research Facility (ESRF), Grenoble, France. Processing, reduction, indexing, integration and scaling of the synchrotron diffraction data were conducted using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶). Some of the crystals were further analyzed at the Technion Center for Structural Biology (TCSB), Technion, Haifa, Israel, using our in-house X-ray source (see below). Processing, reduction, indexing, integration and scaling of the TCSB diffraction data were conducted using the programs iMosflm (Battye et al., 2011 ▶) and SCALA (Evans, 2006 ▶).
3. Results and discussion
3.1. Preliminary characterization of Gan1D
The gan1D gene is part of a 12.5 kb cluster that is upregulated when G. stearothermophilus T1 grows on galactose as the sole carbon source. The gene encodes a 478-amino-acid protein with a calculated molecular mass of 55 204 Da. Based on BLAST analysis, Gan1D shows more than 35% sequence homology to a number of GH1 6-phospho-β-galactosidases, including those from L. lactis (37% sequence identity), S. aureus (38%), S. mutans (37%) and L. casei (36%), and the GH1 6-phospho-β-glucosidases from Lactobacillus plantarum (39% sequence identity) and S. mutans (37%) (de Vos & Gasson, 1989 ▶; Breidt & Stewart, 1987 ▶; Porter & Chassy, 1988 ▶; Honeyman & Curtiss, 1993 ▶; Michalska et al., 2013 ▶). The 6-phospho-β-galactosidases and 6-phospho-β-glucosidases show relatively high sequence similarity to each other. However, using the reported crystal structures of the GH1 6-phospho-β-galactosidase from L. lactis and the GH1 6-phospho-β-glucosidases from L. plantarum and S. mutans, it should be possible, in principle, to predict the preferred activity of GH1 enzymes of this type based only on their three-dimensional structure (Wiesmann et al., 1997 ▶; Michalska et al., 2013 ▶). In the 6-phospho-β-galactosidase from L. lactis the key residue Trp429 is localized in the phosphate-binding pocket, at a hydrogen-bonding distance from the axial position of the O4 hydroxyl group of a bound galactose (Wiesmann et al., 1997 ▶). In the 6-phospho-β-glucosidases this Trp residue is replaced by an Ala (Ala431 in L. plantarum), which clashes with the axial O4 hydroxyl group of galactose 6-phosphate but not with the equatorial O4 hydroxyl group of glucose 6-phosphate (Michalska et al., 2013 ▶). Since Gan1D contains the Trp433 residue in this position (based on sequence alignment), it is suggested that the enzyme is likely to be a 6-phospho-β-galactosidase.
The gan1D gene was readily overexpressed in the E. coli pET system, and gel-permeation chromatography of the purified protein suggested that the native protein is a dimer in solution, with a molecular mass of about 110 kDa per dimer. As noted above, relatively little is presently known about Gan1D and its catalytic properties. In a series of preliminary experiments, however, Gan1D exhibited residual activity towards 4-nitrophenyl β-d-galactopyranoside and 2-nitrophenyl β-d-glucopyranoside, and significantly higher activity towards 4-nitrophenyl β-d-glucose 6-phosphate and 4-nitrophenyl β-d-galactose 6-phosphate. These experiments support our initial assignment of Gan1D as a 6-phospho-β-galactosidase, which was based only on its sequence homology and its observed genetic regulation.
3.2. Crystal forms of wild-type Gan1D
The preliminary screening experiments were based on several commercial screens, including Crystal Screen and Crystal Screen 2 (Hampton Research, California, USA), and Wizard Classic Screens 1–4 (Emerald BioSystems, Bainbridge Island, Washington, USA). As noted above, those initial crystallization screens indicated only one condition that produced useful crystals. This was solution No. 39 of the Wizard Classic 4 Screen, consisting of 20%(w/v) polyethylene glycol 8000 (PEG 8K), 3%(v/v) (±)-2-methyl-2,4-pentanediol (MPD), 0.1 M imidazole buffer pH 6.5. This initial condition was further refined, resulting in two slightly different crystallization conditions that differed from each other only by the total content of the PEG component and the exact concentration of the protein. In condition 1, the initial protein concentration was around 3.9–4.3 mg ml−1 and the reservoir solution (and half of the protein crystallization drop) consisted of 16–18%(w/v) PEG 8K, 3%(v/v) MPD, 0.1 M imidazole buffer pH 6.48. In condition 2, the initial protein concentration was around 4.3–4.5 mg ml−1 and the reservoir solution contained the same general components but with 19–22% rather than 16–18% PEG 8K. Surprisingly, this relatively small difference in the PEG component led to two different crystal forms. The crystals obtained from condition 1 usually appeared as small elongated flat prisms (EFP habit), varying in the shape of the flat face from elongated triangles to elongated rectangles (Figs. 1 ▶ a, 1 ▶ b and 1 ▶ c). The typical dimensions of the EFP crystals were in the range 0.05–0.12 × 0.02–0.07 mm for the flat face, with a thickness of around 0.01–0.03 mm (Figs. 1 ▶ a and 1 ▶ b). These crystals appeared usually in clusters of three to eight crystals, all originating from a common crystallization core. In some of the drops it was possible to isolate single, nonclustered crystals of this form, some of which were used for the detailed crystallographic characterization and data collection described below. The crystals obtained from condition 2 usually appeared as larger rectangular plates (RP habit), with typical dimensions in the range 0.4–0.7 × 0.2–0.4 × 0.01–0.05 mm (Fig. 1 ▶ d). Although larger in overall size, the RP crystals usually appeared in multiple layers (Fig. 1 ▶ d), making it difficult to isolate a suitable single monolayered crystal for crystallographic analysis.
Figure 1.

Crystals of the two habits obtained for Gan1D. (a–c) Typical crystals of the EFP habit. It is noted that crystals of the EFP habit vary in shape between elongated rectangular bars (a, top left), almost perfect square boxes (b, top centre), flat rectangular plates (a, right; b, bottom) and various polyhedral prisms (a, b, c). They also vary in size from typically around 0.05 × 0.05 × 0.01 mm (a, b) to sometimes as large as 0.12 × 0.07 × 0.03 mm (c). Larger crystals of the type presented in (c) were used for the measurement of the full diffraction data set described here at 1.33 Å resolution (Fig. 2 ▶). (d) A typical crystal of the RP habit, with dimensions of around 0.7 × 0.4 × 0.05 mm.
3.3. Crystallographic characterization of the RP crystal habit
The X-ray diffraction pattern of the RP crystals indicated that they belong to a C-centred monoclinic crystal system (space group C2), with average unit-cell parameters of a = 108.0, b = 68.9, c = 152.9 Å, β = 100.7°. Using a synchrotron setup (ESRF beamline BM14) the best crystals of this form displayed detectable X-ray diffraction to a resolution limit of around 1.8 Å, but with smeared spots and relatively large overall mosaicity values. In comparison (at the time), the EFP crystals proved to be more suitable for crystallographic structural analysis (see below), as based on the reproducibility of the crystallization experiments, handling and cooling of the resulting crystals and the quality of the diffraction pattern obtained. At this point, it was therefore decided to continue further crystallographic studies (at the ESRF) mainly with this type of crystals, as further described in §§3.4 and 3.5.
At a later point (after the full characterization of the EFP crystals), we decided to further characterize the RP crystals for use as potential ‘back-up’ crystals for future enzyme–substrate structural studies, if needed. These crystallographic analyses were performed at the TCSB, using an FR-X rotating-anode (Rigaku, Japan) X-ray source (λ = 1.54 Å) coupled to an R-AXIS HTC (Rigaku, Japan) detector and an Oxford Cryosystem crystal-cooling system (100 K). Several crystals of this form (from different crystallization batches) have been examined, some of which proved to be quite reasonable for crystallographic analysis. One such RP Gan1D crystal (Fig. 1 ▶ d) was used for a full X-ray diffraction data measurement to 1.92 Å resolution. This oscillation data set included 555 frames, measured with a Δϕ of 0.3°, 2 min exposure time per frame and a crystal-to-detector distance of 140 mm. The raw diffraction images were processed with iMosflm (Battye et al., 2011 ▶) and scaled with SCALA (Evans, 2006 ▶). A total of 264 862 reflections were measured in the 40.00–1.92 Å resolution range and resulted in 86 922 independent reflections, with 99.7% overall completeness to 1.92 Å resolution and 98.5% completeness for the highest resolution shell (2.03–1.92 Å). The overall diffraction multiplicity of the processed data set was 3.2, the overall mosaicity was 0.69°, the mean I/σ(I) was 6.0 and the final R merge for the whole data was 10.4%. These parameters indicate that the data collected from the RP crystal represent a reasonable diffraction data set that could potentially be used as a basis for crystal structure analysis (Table 1 ▶). These parameters confirm, however, that the EFP crystals are preferable over the RP crystals for high-resolution structural studies of the Gan1D protein, as demonstrated in detail below.
Table 1. Representative parameters for the crystallographic data of Gan1D.
Values in parentheses are for the outer resolution shell.
| Protein crystal | Gan1D (EFP habit) | Gan1D (RP habit) |
|---|---|---|
| X-ray system | ESRF BM14 | Technion TCSB |
| Wavelength (Å) | 0.979 | 1.54 |
| Space group | P1 | C2 |
| Resolution range (Å) | 40.0–1.33 (1.35–1.33) | 40.0–1.92 (2.03–1.92) |
| Unit-cell parameters | ||
| a (Å) | 67.0 | 108.0 |
| b (Å) | 78.1 | 68.9 |
| c (Å) | 92.1 | 152.9 |
| α (°) | 102.4 | 90 |
| β (°) | 93.5 | 100.7 |
| γ (°) | 91.7 | 90 |
| No. of reflections | ||
| Total | 2986976 | 264862 |
| Unique | 429175 | 86922 |
| Multiplicity | 3.9 (3.8) | 3.2 (3.1) |
| 〈I〉/〈σ(I)〉 | 11.9 (2.4) | 6.0 (2.3) |
| Mosaicity (°) | 0.32 | 0.69 |
| Completeness (%) | 95.5 (92.3) | 99.7 (98.5) |
| R merge † (%) | 5.5 (59.6) | 10.4 (32.3) |
R
merge = 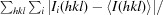
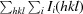 , where Ii(hkl) is the intensity of observation i of reflection hkl.
, where Ii(hkl) is the intensity of observation i of reflection hkl.
3.4. Crystallographic characterization of the EFP crystal habit
After refinement of the initial conditions, it was determined that the best conditions for obtaining EFP Gan1D crystals are those of the general hanging-drop procedure described above using a protein concentration of 4.3 µg µl−1 and a reservoir solution consisting of 17%(w/v) PEG 8K, 3% MPD, 0.1 M imidazole buffer pH 6.48. In these conditions, a larger portion of the resulting crystals appeared as single crystals or as small clusters of relatively thick crystals (Fig. 1 ▶ c) that were rather easy to break into single crystals.
Several such EFP Gan1D crystals were used for a detailed crystallographic characterization and measurement of X-ray diffraction data under cryogenic conditions. These experiments were performed using X-ray synchrotron radiation (λ = 0.979 Å) and a CCD area detector (MAR 225, MAR Research, USA) on the BM14 beamline of the ESRF. The crystal-cooling procedure used for these experiments included a short soak of the selected crystal (for about 20–60 s) in a cryoprotecting solution consisting of 30%(w/v) PEG 4K, 14%(w/v) PEG 8K, 3%(v/v) MPD, 0.1 M imidazole buffer pH 6.48. Each of these pre-soaked crystals was then submitted to a flash-cooling procedure by immediate immersion in liquid nitrogen followed by quick transfer into the centre of a cold nitrogen-gas stream (100 K; Oxford Cryosystem), which flowed around the crystal throughout the X-ray data collection.
The observed diffraction pattern of some of these crystals exceeded the 1.4 Å resolution limit and indicated that the crystals belonged to the triclinic crystal system (space group P1), with average crystallographic unit-cell parameters of a = 67.0, b = 78.1, c = 92.1 Å, α = 102.4, β = 93.5, γ = 91.7°. Different crystals of this crystallization batch gave similar unit-cell parameters, with an overall deviation from these average values of less than 0.2%.
3.5. X-ray diffraction data for Gan1D
One such Gan1D crystal was used for a full X-ray diffraction data measurement to 1.33 Å resolution (Fig. 2 ▶). This oscillation data set included 360 frames, measured with a Δϕ of 1°, 5 s exposure and a crystal-to-detector distance of 114.3 mm on the BM14 beamline at the ESRF, as described above (λ = 0.979 Å, 100 K). The raw CCD diffraction images were processed with the HKL-2000 software package (Otwinowski & Minor, 1997 ▶). A total of 2 986 976 accepted reflections [F > 0σ(F)] were measured in the 40.00–1.33 Å resolution range and resulted in 429 175 independent reflections with 95.5% overall completeness to 1.33 Å resolution and 92.3% completeness for the highest resolution shell (1.35–1.33 Å). The overall diffraction multiplicity of the processed data set was 3.9, the overall mosaicity value was 0.32°, the average I/σ(I) was 11.9 and the final R merge for the whole data set was 5.5% (59.6% for the highest resolution shell). These parameters confirm that the data collected represent an almost complete diffraction data set of relatively high quality (Table 1 ▶).
Figure 2.

X-ray diffraction pattern of the Gan1D crystals (EFP habit), obtained using a synchrotron source (BM14, ESRF). The outer circle corresponds to a 1.33 Å resolution limit. The insets represent magnified views of the sections indicated by the corresponding squares (top, medium resolution; bottom, high resolution).
A rough calculation of the specific ratio of volume/protein (V M) was performed in order to estimate the number of protein monomers per crystallographic asymmetric unit. The volume of the Gan1D crystallographic unit cell, as determined from the mean value of the unit-cell parameters at 100 K, is 4.83 × 105 Å3. Assuming that the V M and the overall water content values here are within the normal range observed for soluble proteins (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶), there should be either three or four molecules of Gan1D monomers (478 amino-acid residues, molecular mass 55 204 Da per monomer) in the crystallographic asymmetric unit. With three molecules in the P1 asymmetric unit (and the unit cell), the calculated V M is 2.92 Å3 Da−1 and the corresponding solvent content in the crystals is 57.9%. With four molecules in the asymmetric unit, the calculated V M is 2.19 Å3 Da−1 and the corresponding solvent content in the crystals is 43.8%. Both of these two possibilities seem to be reasonable considering their calculated V M values and water content. However, according to the gel-filtration results, Gan1D appears to be a dimer in solution (see above), making the second possibility the more likely solution, featuring two homodimers of Gan1D in the P1 unit cell. The presence of two protein dimers per unit cell was also supported by self-rotation calculations.
This full 1.33 Å resolution data set should now be used for the detailed three-dimensional structural analysis of Gan1D. A search in the Protein Data Bank (PDB) indicated two relevant three-dimensional protein structures that share more than 40% sequence identity with Gan1D. One of these proteins is a β-glucosidase from Streptomyces sp. (PDB entry 1gnx, 1.68 Å resolution, 42% sequence identity; A. Guasch, M. Vallmitjana, R. Perez, E. Querol, J. A. Perez-Pons & M. Coll, unpublished work). The second protein is a β-glucosidase from Bacillus circulans sp. alkalophilus (PDB entry 1qox, 2.7 Å resolution, 41% sequence identity; Hakulinen et al., 2000 ▶). Also relevant is the structure of the 6-phospho-β-galactosidase from L. lactis, for which a full crystallographic analysis has been performed at 2.3 Å resolution (PDB entries 1pbg and 4pbg; 37% sequence identity; Wiesmann et al., 1995 ▶, 1997 ▶). These three structures could be used now as suitable reference models for molecular-replacement calculations, which are expected to solve the phase problem and provide good starting models for the full structural analysis of the Gan1D protein. Such analyses are currently in progress in our laboratory.
Acknowledgments
This work was supported by the Israel Science Foundation Grants 500/10 and 152/11, the I-CORE Program of the Planning and Budgeting Committee, the Ministry of Environmental Protection and the Grand Technion Energy Program (GTEP), and comprises part of The Leona M. and Harry B. Helmsley Charitable Trust reports on Alternative Energy series of the Technion, Israel Institute of Technology and the Weizmann Institute of Science. YS acknowledges the core services and support from the Lorry I. Lokey Center for Life Sciences and Engineering and the Russell Berrie Nanotechnology Institute, Technion. We thank the staff at the European Synchrotron Research Facility (ESRF beamline BM14) for their helpful support in the X-ray synchrotron data measurement and analysis. The synchrotron experiments at the ESRF were also supported by the ESRF internal funding program. HD thanks the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 330879-MC-CHOLESTRUCTURE for financial support. YS holds the Erwin and Rosl Pollak Chair in Biotechnology at the Technion.
References
- Alalouf, O., Balazs, Y., Volkinshtein, M., Grimpel, Y., Shoham, G. & Shoham, Y. (2011). J. Biol. Chem. 286, 41993–42001. [DOI] [PMC free article] [PubMed]
- Almog, O., Greenblatt, H. M., Spungin, A., Ben-Meir, D., Blumberg, S. & Shoham, G. (1993). J. Mol. Biol. 230, 342–344. [DOI] [PubMed]
- Almog, O., Klein, D., Braun, S. & Shoham, G. (1994). J. Mol. Biol. 235, 760–762. [DOI] [PubMed]
- Alpert, C. A. & Chassy, B. M. (1990). J. Biol. Chem. 265, 22561–22568. [PubMed]
- Bar, M., Golan, G., Nechama, M., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 545–549. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Ben-David, A., Bravman, T., Balazs, Y. S., Czjzek, M., Schomburg, D., Shoham, G. & Shoham, Y. (2007). Chembiochem, 8, 2145–2151. [DOI] [PubMed]
- Bravman, T., Zolotnitsky, G., Belakhov, V., Shoham, G., Henrissat, B., Baasov, T. & Shoham, Y. (2003). Biochemistry, 42, 10528–10536. [DOI] [PubMed]
- Breidt, F. Jr, Hengstenberg, W., Finkeldei, U. & Stewart, G. C. (1987). J. Biol. Chem. 262, 16444–16449. [PubMed]
- Breidt, F. Jr & Stewart, G. C. (1987). Appl. Environ. Microbiol. 53, 969–973. [DOI] [PMC free article] [PubMed]
- Brüx, C., Ben-David, A., Shallom-Shezifi, D., Leon, M., Niefind, K., Shoham, G., Shoham, Y. & Schomburg, D. (2006). J. Mol. Biol. 359, 97–109. [DOI] [PubMed]
- Deutscher, J., Francke, C. & Postma, P. W. (2006). Microbiol. Mol. Biol. Rev. 70, 939–1031. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gat, O., Lapidot, A., Alchanati, I., Regueros, C. & Shoham, Y. (1994). Appl. Environ. Microbiol. 60, 1889–1896. [DOI] [PMC free article] [PubMed]
- Golan, G., Shallom, D., Teplitsky, A., Zaide, G., Shulami, S., Baasov, T., Stojanoff, V., Thompson, A., Shoham, Y. & Shoham, G. (2004). J. Biol. Chem. 279, 3014–3024. [DOI] [PubMed]
- Hakulinen, N., Paavilainen, S., Korpela, T. & Rouvinen, J. (2000). J. Struct. Biol. 129, 69–79. [DOI] [PubMed]
- Henrissat, B. & Davies, G. (1997). Curr. Opin. Struct. Biol. 7, 637–644. [DOI] [PubMed]
- Honda, H., Nagaoka, S., Kawai, Y., Kemperman, R., Kok, J., Yamazaki, Y., Tateno, Y., Kitazawa, H. & Saito, T. (2012). J. Gen. Appl. Microbiol. 58, 11–17. [DOI] [PubMed]
- Honeyman, A. L. & Curtiss, R. III (1993). J. Gen. Microbiol. 139, 2685–2694. [DOI] [PubMed]
- Hövel, K., Shallom, D., Niefind, K., Baasov, T., Shoham, G., Shoham, Y. & Schomburg, D. (2003). Acta Cryst. D59, 913–915. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Solomon, V., Alhassid, A., Govada, L., Chayan, N. E., Belrhali, H., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 430–434. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Solomon, V., Alhassid, A., Govada, L., Chayen, N. E., Belrhali, H., Shoham, Y. & Shoham, G. (2014). Acta Cryst. D70, .10.1107/S139900471302840X. [DOI] [PubMed]
- Lansky, S., Salama, R., Solomon, V. H., Belrhali, H., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 695–699. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McKay, L., Miller, A. III, Sandine, W. E. & Elliker, P. R. (1970). J. Bacteriol. 102, 804–809. [DOI] [PMC free article] [PubMed]
- Michalska, K., Tan, K., Li, H., Hatzos-Skintges, C., Bearden, J., Babnigg, G. & Joachimiak, A. (2013). Acta Cryst. D69, 451–463. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Porter, E. V. & Chassy, B. M. (1988). Gene, 62, 263–276. [DOI] [PubMed]
- Postma, P. W., Lengeler, J. W. & Jacobson, G. R. (1993). Microbiol. Rev. 57, 543–594. [DOI] [PMC free article] [PubMed]
- Rees, D. C., Johnson, E. & Lewinson, O. (2009). Nature Rev. Mol. Cell Biol. 10, 218–227. [DOI] [PMC free article] [PubMed]
- Robillard, G. T. & Broos, J. (1999). Biochim. Biophys. Acta, 1422, 73–104. [DOI] [PubMed]
- Rosey, E. L. & Stewart, G. C. (1992). J. Bacteriol. 174, 6159–6170. [DOI] [PMC free article] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Gilead-Gropper, S., Baasov, T., Shoham, G. & Shoham, Y. (2002). FEBS Lett. 514, 163–167. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Shoham, G., Baasov, T. & Shoham, Y. (2002). J. Biol. Chem. 277, 43667–43673. [DOI] [PubMed]
- Shallom, D., Golan, G., Shoham, G. & Shoham, Y. (2004). J. Bacteriol. 186, 6928–6937. [DOI] [PMC free article] [PubMed]
- Shallom, D., Leon, M., Bravman, T., Ben-David, A., Zaide, G., Belakhov, V., Shoham, G., Schomburg, D., Baasov, T. & Shoham, Y. (2005). Biochemistry, 44, 387–397. [DOI] [PubMed]
- Shulami, S., Gat, O., Sonenshein, A. L. & Shoham, Y. (1999). J. Bacteriol. 181, 3695–3704. [DOI] [PMC free article] [PubMed]
- Shulami, S., Raz-Pasteur, A., Tabachnikov, O., Gilead-Gropper, S., Shner, I. & Shoham, Y. (2011). J. Bacteriol. 193, 2838–2850. [DOI] [PMC free article] [PubMed]
- Shulami, S., Zaide, G., Zolotnitsky, G., Langut, Y., Feld, G., Sonenshein, A. L. & Shoham, Y. (2007). Appl. Environ. Microbiol. 73, 874–884. [DOI] [PMC free article] [PubMed]
- Solomon, H. V., Tabachnikov, O., Feinberg, H., Govada, L., Chayen, N. E., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 1114–1119. [DOI] [PMC free article] [PubMed]
- Solomon, V., Teplitsky, A., Shulami, S., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2007). Acta Cryst. D63, 845–859. [DOI] [PubMed]
- Suzuki, M., Saito, T. & Itoh, T. (1996a). Biosci. Biotechnol. Biochem. 60, 139–141. [DOI] [PubMed]
- Suzuki, M., Saito, T. & Itoh, T. (1996b). Biosci. Biotechnol. Biochem. 60, 708–710. [DOI] [PubMed]
- Tabachnikov, O. & Shoham, Y. (2013). FEBS J. 280, 950–964. [DOI] [PubMed]
- Teplitsky, A., Feinberg, H., Gilboa, R., Lapidot, A., Mechaly, A., Stojanoff, V., Capel, M., Shoham, Y. & Shoham, G. (1997). Acta Cryst. D53, 608–611. [DOI] [PubMed]
- Teplitsky, A., Mechaly, A., Stojanoff, V., Sainz, G., Golan, G., Feinberg, H., Gilboa, R., Reiland, V., Zolotnitsky, G., Shallom, D., Thompson, A., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 836–848. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Shoham, Y. & Shoham, G. (2000). Acta Cryst. D56, 181–184. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Zaide, G., Shoham, Y. & Shoham, G. (1999). Acta Cryst. D55, 869–872. [DOI] [PubMed]
- Vos, W. M. de, Boerrigter, I., van Rooyen, R. J., Reiche, B. & Hengstenberg, W. (1990). J. Biol. Chem. 265, 22554–22560. [PubMed]
- Vos, W. M. de & Gasson, M. J. (1989). J. Gen. Microbiol. 135, 1833–1846. [DOI] [PubMed]
- Wiesmann, C., Beste, G., Hengstenberg, W. & Schulz, G. E. (1995). Structure, 3, 961–968. [DOI] [PubMed]
- Wiesmann, C., Hengstenberg, W. & Schulz, G. E. (1997). J. Mol. Biol. 269, 851–860. [DOI] [PubMed]
- Zaide, G., Shallom, D., Shulami, S., Zolotnitsky, G., Golan, G., Baasov, T., Shoham, G. & Shoham, Y. (2001). Eur. J. Biochem. 268, 3006–3016. [DOI] [PubMed]