

Mass spectrometry was used to aid in the identification and structure determination of serendipitously purified glycerol dehydrogenase from the bacterial genus Serratia.
Keywords: glycerol dehydrogenase, Serratia
Abstract
The 1.90 Å resolution X-ray crystal structure of glycerol dehydrogenase derived from contaminating bacteria present during routine Escherichia coli protein expression is presented. This off-target enzyme showed intrinsic affinity for Ni2+-Sepharose, migrated at the expected molecular mass for the target protein during gel filtration and was crystallized before it was realised that contamination had occurred. In this study, it is shown that liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) can efficiently identify the protein composition of crystals in a crystallization experiment as part of a structure-determination pipeline for an unknown protein. The high-resolution X-ray data enabled sequencing directly from the electron-density maps, allowing the source of contamination to be placed within the Serratia genus. Incorporating additional protein-identity checks, such as tandem LC-MS/MS, earlier in the protein expression, purification and crystallization workflow may have prevented the unintentional structure determination of this metabolic enzyme, which represents the first enterobacterial glycerol dehydrogenase reported to date.
1. Introduction
Structure determination of unidentified proteins can be a challenging endeavor. Often, a search of unit-cell parameters in the PDB will allow the identification of commonly crystallized proteins derived from the host expression organism (i.e. Escherichia coli). More advanced searches have enabled the identification of unidentified protein crystals by performing molecular replacement on up to 100 000 protein domains (Stokes-Rees & Sliz, 2010 ▶). Often, these methods may not be feasible owing to the computing power needed for brute-force molecular replacement. Experimental phasing techniques do not require a known sequence for phase determination, but in both cases knowing the sequence of the protein contained in the crystals can be of immense help for efficient structure solution.
When structure solution does not progress as smoothly as planned, it is often tempting, and wise, to double-check the identity of the protein contained within the crystals that were exposed to X-rays. There are multiple examples of the purification and crystallization of contaminants (Bolanos-Garcia & Davies, 2006 ▶; Psakis et al., 2009 ▶; Kiser et al., 2007 ▶; Tiwari et al., 2010 ▶), and successful identification of the protein is often the key to successful structure solution.
In this study, we crystallized an off-target protein derived from an unknown source of contamination that diffracted to 1.9 Å resolution. The protein was directly collected from hanging-drop vapour-diffusion experiments and subjected to sequencing by mass spectrometry. This enabled the structure determination of the metabolic enzyme glycerol dehydrogenase (GDH) from the bacterial contaminate with a peptide sequence that most closely matches GDH within the genus Serratia.
2. Materials and methods
2.1. Protein production and purification
A human target protein (predicted molecular weight of ∼40 kDa) was cloned into the pLIC_His pET vector kindly provided by John Sondek (UNC-Chapel Hill, North Carolina, USA), which contains an N-terminal 6×His tag followed by a TEV protease cleavage site preceding the target gene. The sequence of the resulting plasmid was confirmed, showing insertion of the human target gene in frame with the N-terminal tag and TEV cleavage site. In-house Terrific broth (TB) medium was prepared as a 10× concentrated stock, autoclaved and diluted in filtered water. A flask containing 100 ml TB supplemented with 100 µg ml−1 ampicillin was inoculated with a single colony containing E. coli BL21(DE3)pLysS cells transformed with the expression plasmid. This flask was maintained at 37°C with vigorous agitation overnight. Flasks containing 1.3 l TB supplemented with 100 µg ml−1 ampicillin were inoculated with 1%(v/v) of this overnight starter culture and were maintained at 37°C with vigorous agitation. At mid-log phase, IPTG was added to a final concentration of 1 mM and the temperature was lowered to 23°C for 24 h. Cells were harvested by centrifugation and formed a visibly red-tinted pellet. The bacteria were lysed by sonication and cleared by centrifugation. Cleared lysates were passed over an Ni2+-affinity column in a buffer consisting of 150 mM NaCl, 20 mM Tris pH 7.4, 5% glycerol, 20 mM imidazole. Trapped protein was eluted using a stepped protocol with the same buffer with the imidazole concentration increased to 250 mM (Fig. 1 ▶ a). The 50 and 100% peaks consisted primarily of a protein that migrated near the 37 kDa standard on SDS–PAGE analysis (Fig. 1 ▶ b). These peaks were pooled and further purified by size-exclusion chromatography (Figs. 1 ▶ c and 1 ▶ d). The protein eluted at a volume suggesting a mass of ∼160 kDa, which was consistent with the expected mass of a tetramer of the target protein. The final protein yield was ∼1 mg per litre of culture.
Figure 1.

(a) Ni2+-affinity chromatogram showing steps of 5, 50 and 100% elution buffer containing 250 mM imidazole and (b) SDS–PAGE analysis of the resulting peaks. (c) Size-exclusion chromatography and (d) SDS–PAGE analysis of the pooled 50% and 100% peaks.
2.2. Crystallization
The purified protein was concentrated to 6 mg ml−1 and dialyzed against 150 mM ammonium acetate, 50 mM sodium chloride, 20 mM Tris pH 7.4, 5% glycerol. This protein solution was used to set up low-volume (0.2 µl protein solution and 0.2 µl crystallant) sitting-drop crystallization trials with a Phoenix robot (Art Robbins Instruments, Sunnyvale, California, USA) using commercially available screens. Initial crystals were discovered in well G4 of The PEGs Suite (Qiagen, Germantown, Maryland, USA). Larger crystals were obtained by mixing 1 µl protein solution with 1 µl crystallant consisting of 5–10% PEG 3350, 0.2 M calcium acetate, 4% glycerol in a hanging-drop vapour-diffusion experiment. These crystals exhibited a prolate spheroid-like morphology that lacked defined edges and faces (Fig. 2 ▶ a). Manipulation of these fragile crystals proved to be difficult and the diffraction limit varied between crystals. Crystal robustness and diffraction reproducibility were improved by chemical cross-linking with glutaraldehyde using 2 µl 25% glutaraldehyde, which was exposed to the crystals by vapor diffusion for 1 h (Lusty, 1999 ▶). The diffraction of these crystals was limited to ∼3 Å and they were highly sensitive to radiation damage. To improve the crystal morphology and diffraction limits, we screened crystallization additives. The addition of 4% 2,2,2-trifluoroethanol significantly altered the crystal morphology, generating crystals with a cuboid morphology with clearly defined edges and faces (Fig. 2 ▶ b). This crystal form withstood manipulation and initially diffracted to ∼1.6 Å resolution. Crystals were cryoprotected with crystallant solution plus 20% glycerol before flash-cooling in liquid nitrogen.
Figure 2.

Crystals of Serratia glycerol dehydrogenase before (a) and after (b) optimization with 2,2,2-trifluoroethanol, measuring approximately 75 × 75 × 200 and 50 × 50 × 150 µm, respectively.
2.3. Diffraction data collection
150° of data were collected in 0.5° oscillation frames from a single crystal. Data were collected at 100 K using a MAR Mosaic 300 mm CCD on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Diffraction data to 1.9 Å resolution were integrated and scaled using HKL-2000 (HKL Research; Table 1 ▶).
Table 1. Crystallographic statistics.
Values in parentheses are for the outer shell.
| PDB code | 4mca |
| Data collection | |
| Space group | I422 |
| Unit-cell parameters (, ) | a = b = 117.5, c = 259.9, = = = 90 |
| Beamline | APS beamline 22-ID, SER-CAT |
| Wavelength () | 1.0 |
| Resolution range () | 501.90 (1.971.90) |
| No. of unique reflections | 71425 (3706) |
| Completeness (%) | 91.6 (52.3)† |
| Multiplicity | 8.7 (4.0) |
| I/(I) | 20.2 (3.0) |
| R merge | 0.1 (0.41) |
| Refinement | |
| Refinement software | REFMAC v.5.7.0032 |
| Resolution range () | 39.61.90 (1.941.90) |
| Completeness (%) | 91.5 (52.3) |
| No. of reflections, working set | 65855 (2473) |
| No. of reflections, test set | 3341 (110) |
| Final R cryst | 0.177 (0.258) |
| Final R free | 0.215 (0.298) |
| No. of non-H atoms | |
| Protein | 5397 |
| Ion | 6 |
| Ligand | 24 |
| Water | 365 |
| Total | 5792 |
| R.m.s. deviations | |
| Bonds () | 0.020 |
| Angles () | 1.863 |
| Overall average B factor (2) | 30.0 |
| Ramachandran plot analysis, residues in (%) | |
| Most favored regions | 98 |
| Additionally allowed regions | 2 |
| Disallowed regions | 0 |
The data collected were anisotropic, resulting in low completeness in the two highest resolution shells. These data were included in spite of the low completeness owing to an I/(I) of 3 in the highest shell, a multiplicity of 4 and an R merge of 0.41.
2.4. Structure determination
Molecular replacement using search models generated from homologous proteins of our target protein was unsuccessful. A search for similar unit-cell parameters in the PDB also failed to provide a molecular-replacement search model. Since we could not identify an appropriate model for molecular replacement, we performed extensive heavy-atom and halide screening to facilitate de novo phasing. Osmium chloride preserved crystal quality and provided a suitable signal for SAD experiments. This allowed us to calculate the initial phases, which resulted in an interpretable map with visible density for side chains. Attempts to build the target human protein sequence into this map were unsuccessful, suggesting that the target protein was not present in these crystals.
To expedite the identification of the crystallized protein, a drop from the hanging-drop experiment was analysed by SDS–PAGE and the single visible band was excised and subjected to tryptic digestion. The resulting peptides were analysed by reverse-phase liquid chromatography coupled with LC-MS/MS on an LTQ Orbitrap Hybrid Mass Spectrometer (Thermo Scientific) as described previously (Xu et al., 2009 ▶). A search for peptides matching a human sequence database confirmed that the target protein was not present. To our surprise, a subsequent search of an E. coli sequence database also returned no clear matches. We then expanded our search to include all known protein sequences, which provided peptide matches to glycerol dehydrogenase from the bacterial genus Serratia (Fig. 3 ▶).
Figure 3.

Peptides identified by LC-MS/MS depicted as red letters within the sequences of glycerol dehydrogenase from (a) S. proteamaculans and (b) S. odorifera.
After protein identification, structure solution was readily achieved using PDB entry 1jpu (the crystal structure of glycerol dehydrogenase; Ruzheinikov et al., 2001 ▶) as a search model for molecular replacement using Phaser (McCoy et al., 2007 ▶). Subsequent cycles of model building with Coot (Emsley et al., 2010 ▶) were alternated with crystallographic refinement using REFMAC v.5.7.0032 (Murshudov et al., 1997 ▶, 2011 ▶). The final model has no geometric outliers and a MolProbity score of 1.27, placing it in the 99th percentile among structures of comparable resolution.
2.5. Miscellaneous
Figures were prepared with Geneious (Biomatters), PyMOL (Schrödinger) and LigPlot+ (Laskowski & Swindells, 2011 ▶). Model validation was performed with MolProbity (Chen et al., 2010 ▶). Coordinates and structure factors have been deposited in the PDB (http://www.pdb.org) as entry 4mca.
3. Results and discussion
Like many laboratories interested in protein structure–function relationships, we set out to express, purify and structurally characterize a human protein of interest. We cloned the target gene into a popular expression vector containing a hexahistidine tag for convenient purification. Initial purification utilizing Ni2+-affinity chromatography resulted in a protein that was near the expected molecular weight as analysed by SDS–PAGE and formed what appeared to be the expected multimer when analysed by size-exclusion chromatography. After purification, initial crystals were identified with limited screening efforts and both the morphology and diffraction limit of the initial crystals were vastly improved by additive screening.
Initial failed attempts at molecular replacement were attributed to potentially poor search models owing to a lack of closely related structures in the PDB. Our efforts then pivoted to de novo phasing techniques. Initial phases were calculated from a SAD data set utilizing the signal from bound osmium ions, and the resulting map suggested that the sequence of our target protein was not present. Mass-spectrometric analysis of a crystallization drop ruled out that we had crystallized our target human protein: rather, it identified the protein as a probable glycerol dehydrogenase from the bacterial genus Serratia.
Identification of the protein contained in the crystals as a probable member of the glycerol dehydrogenase family from Serratia allowed the identification of homologous crystal structures in the PDB suitable for molecular-replacement search models. The final model contains two glycerol dehydrogenase monomers in the asymmetric unit, with each monomer containing two glycerols, two zinc ions and a sodium ion (Fig. 4 ▶). While the protein does not contain more than two consecutive histidine residues in its primary sequence, it displays 11 surface-exposed histidines, with His31, His59, His60 and His83 forming a cluster on the surface of the protein. These histidine residues are within van der Waals contact distance, and His31 and Cys85 coordinate a zinc ion at this site. We hypothesize that this zinc cluster may have contributed to the affinity of this glycerol dehydrogenase for the Ni2+ medium. The probable sequence of the protein, as determined by interpretation of electron density, has >95% sequence identity to S. proteamaculans, S. odorifera, S. plymuthica, S. marcescens and S. liquefaciens GDH, but is not identical to any of them (Fig. 5 ▶ a). At only three positions does the electron density suggest an amino acid other than one found in close homologs: Leu33, Val154 and Val319 (Figs. 5 ▶ b, 5 ▶ c and 5 ▶ d, respectively). The next closest match identified in the nonredundant NCBI protein database is GDH from Yersinia intermedia, which shows significantly less identity at 78%. Thus, the protein that we have purified is most likely either from one of the known Serratia species with naturally occurring variations or from an unsequenced species within the genus.
Figure 4.

Overall structure of Serratia glycerol dehydrogenase represented by a ribbon diagram with α-helices colored blue and β-strands colored yellow. Glycerol is depicted as sticks (C, green; O, red), bound zinc is depicted as a black sphere and bound sodium is depicted as a gray sphere.
Figure 5.

(a) Sequence of 4mca compared by alignment with the five closest glycerol dehydrogenase sequence matches from various Serratia species. (b, c, d) 2F o − F c electron density contoured at 1σ (blue) and F o − F c contoured at 2σ (green) and −2σ (red) at amino acids (Leu33, Val154 and Val319, respectively) where electron density was used to interpret a sequence that differed from those of close homologs.
Glycerol dehydrogenase is the enzyme responsible for the oxidation of glycerol to dihydroxyacetone. This permits its entrance into the glycolytic pathway (Forage & Lin, 1982 ▶; May & Sloan, 1981 ▶). Thus, many organisms express glycerol dehydrogenase under anaerobic conditions to utilize glycerol as an energy source (Lin, 1976 ▶; May & Sloan, 1981 ▶). This oxidation requires the concurrent reduction of NAD+ to NADH (May & Sloan, 1981 ▶) along with the presence of an active-site zinc responsible for coordinating glycerol in the active site of the enzyme (Ruzheinikov et al., 2001 ▶; Spencer et al., 1989 ▶). It is plausible that in our culture conditions glycerol dehydrogenase expression was increased to take advantage of the 10% glycerol supplementation in TB medium.
Overall, the structure is highly similar to the previously published structures of glycerol dehydrogenase, with an r.m.s.d. of 1.1 Å versus PDB entry 1jqa (Bacillus stearothermophilus glycerol dehydrogenase complex with glycerol; Ruzheinikov et al., 2001 ▶; Fig. 6 ▶ a) and 0.96 Å versus PDB entry 1kq3 [crystal structure of a glycerol dehydrogenase (Tm0423) from Thermotoga maritima at 1.5 Å resolution; Brinen et al., 2003 ▶; Fig. 6 ▶ b] as calculated by matchmaker in the Chimera software package (Pettersen et al., 2004 ▶). The structure maintains a two-domain architecture separated by a deep cleft that has been observed to be the NAD-binding site in other crystal structures (Ruzheinikov et al., 2001 ▶). Modeling of a NAD molecule by superposition of PDB entry 1jq5 (Ruzheinikov et al., 2001 ▶) onto the Serratia structure predicts that binding is conserved at this site, maintaining all of the predicted hydrogen-bonding and hydrophobic interactions (Fig. 7 ▶). The active site is highly conserved and contains a zinc ion coordinated by two histidines and an aspartate. This zinc is responsible for coordinating the bound glycerol molecule (Fig. 8 ▶).
Figure 6.
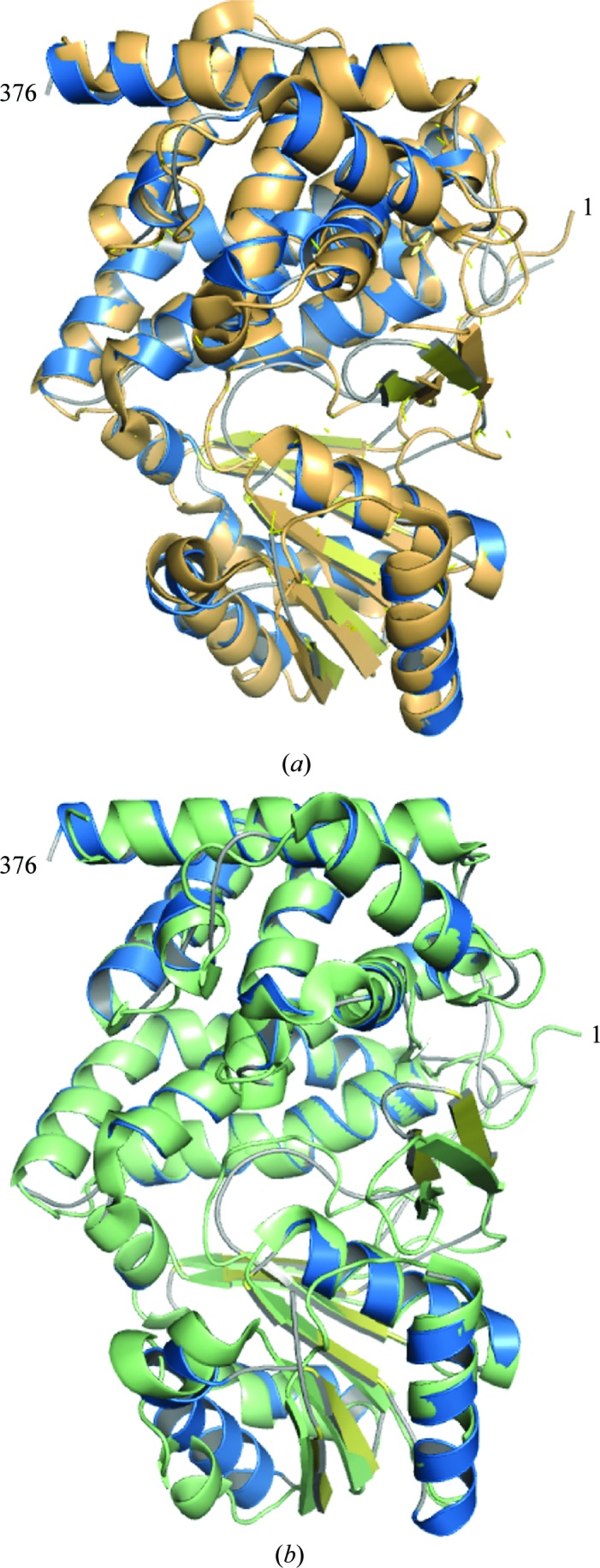
Superposition of Serratia glycerol dehydrogenase (blue) with (a) B. stearothermophilus glycerol dehydrogenase (copper) with an r.m.s.d. of 1.14 Å and (b) T. maritima glycerol dehydrogenase (green) with an r.m.s.d. of 0.96 Å as calculated by matchmaker in the Chimera software package (Pettersen et al., 2004 ▶).
Figure 7.

Molecular surface of Serratia glycerol dehydrogenase (blue) with bound glycerol depicted as sticks (C, green; O, red). NAD+, depicted as sticks (C, copper; O, red; P, magenta; N, blue), is placed from a superposition of B. stearothermophilus glycerol dehydrogenase (PDB entry 1jq5), which shares a sequence identity of 46%.
Figure 8.

(a) Active site of Serratia glycerol dehydrogenase (blue ribbon, with close side chains colored C, blue; O, red; N, blue) with bound glycerol depicted as sticks (C, green; O, red) and bound zinc depicted as a black sphere. (b) Active site of Serratia glycerol dehydrogenase represented in two dimensions with noncovalent interactions highlighted by either green dashes or red combs. This figure was generated by LigPlot+ (Laskowski & Swindells, 2011 ▶).
The genus Serratia contains bacteria that are Gram-negative, rod-shaped and facultatively anaerobic (Mahlen, 2011 ▶). Serratia can often be visually identified, as some strains have a characteristic red pigment, and is often found as a red biofilm in bathrooms and in nature (Mahlen, 2011 ▶). It is considered to be an opportunistic pathogen and is known to infect the respiratory and urinary tracts. Infections are most often the result of S. marcescens (Mahlen, 2011 ▶).
Before the pathogenicity of Serratia was appreciated, its red pigment was utilized as a tracer agent by the US government in biological warfare and medical tests (Mahlen, 2011 ▶). It is now known that Serratia bacteria often contain widely ranging resistance to a variety of antibacterial agents (Stock et al., 2003 ▶). This antibiotic resistance presumably allowed the Serratia to escape ampicillin and chloramphenicol selection during protein expression. During routine cleaning and maintenance of our laboratory, we identified a small (∼2 mm diameter) pink biofilm growing inside the in-house filtered-water spigot that we hypothesize was the source of contamination in this experiment. The spigot was immediately sterilized and replaced, preventing further investigation into the source of this contamination. It is apparent, however, that this Gram-negative facultatively anaerobic bacteria was able to out-compete, or at least co-exist, with E. coli in antibiotic-containing culture.
While it is possible to determine the structure of crystals without a priori knowledge of their sequence, this remains a difficult endeavor. In this study, we used LC-MS/MS to unambiguously identify the protein we crystallized as a Serratia GDH without a reported DNA sequence. This highlights the importance of quality-control measures, such as protein identification by mass spectrometry, as part of a protein-production protocol. In our case, attempting to express a potentially toxic human protein in E. coli may have allowed the Gram-negative facultatively anaerobic Serratia bacteria to thrive rather than being outcompeted by a rapidly growing E. coli population.
Supplementary Material
PDB reference: glycerol dehydrogenase, 4mca
Acknowledgments
We thank N. T. Seyfried in the Center for Neurodegenerative Diseases’ Proteomics Core at Emory University for his help in acquiring the MS data for protein identification. The research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award No. RO1DK095750 to EAO and by AHA predoctoral grant 12PRE12060583 and an Emory–National Institute of Environmental Health Sciences Graduate and Postdoctoral Training in Toxicology grant (T32ES012870) to PMM. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
References
- Bolanos-Garcia, V. M. & Davies, O. R. (2006). Biochim. Biophys. Acta, 1760, 1304–1313. [DOI] [PubMed]
- Brinen, L. S. et al. (2003). Proteins, 50, 371–374. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Forage, R. G. & Lin, E. C. C. (1982). J. Bacteriol. 151, 591–599. [DOI] [PMC free article] [PubMed]
- Kiser, P. D., Lodowski, D. T. & Palczewski, K. (2007). Acta Cryst. F63, 457–461. [DOI] [PMC free article] [PubMed]
- Laskowski, R. A. & Swindells, M. B. (2011). J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed]
- Lin, E. C. C. (1976). Annu. Rev. Microbiol. 30, 535–578. [DOI] [PubMed]
- Lusty, C. J. (1999). J. Appl. Cryst. 32, 106–112.
- Mahlen, S. D. (2011). Clin. Microbiol. Rev. 24, 755–791. [DOI] [PMC free article] [PubMed]
- May, J. W. & Sloan, J. (1981). Microbiology, 123, 183–185.
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed]
- Psakis, G., Polaczek, J. & Essen, L.-O. (2009). J. Struct. Biol. 166, 107–111. [DOI] [PubMed]
- Ruzheinikov, S. N., Burke, J., Sedelnikova, S., Baker, P. J., Taylor, R., Bullough, P. A., Muir, N. M., Gore, M. G. & Rice, D. W. (2001). Structure, 9, 789–802. [DOI] [PubMed]
- Spencer, P., Bown, K. J., Scawen, M. D., Atkinson, T. & Gore, M. G. (1989). Biochim. Biophys. Acta, 994, 270–279. [DOI] [PubMed]
- Stock, I., Burak, S., Sherwood, K. J., Gruger, T. & Wiedemann, B. (2003). J. Antimicrob. Chemother. 51, 865–885. [DOI] [PubMed]
- Stokes-Rees, I. & Sliz, P. (2010). Proc. Natl Acad. Sci. USA, 107, 21476–21481. [DOI] [PMC free article] [PubMed]
- Tiwari, N., Woods, L., Haley, R., Kight, A., Goforth, R., Clark, K., Ataai, M., Henry, R. & Beitle, R. (2010). Protein Expr. Purif. 70, 191–195. [DOI] [PubMed]
- Xu, P., Duong, D. M. & Peng, J. (2009). J. Proteome Res. 8, 3944–3950. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: glycerol dehydrogenase, 4mca