

Polysaccharide deacetylases are bacterial enzymes that catalyze the deacetylation of acetylated membrane sugars on Gram-positive bacteria, allowing them to evade host immune systems. Here, the first X-ray crystal structure of BA0150, a putative polysaccharide deacetylase from B. anthracis, is reported to 2.0 Å resolution.
Keywords: polysaccharide deacetylases, carbohydrate esterases, Bacillus anthracis
Abstract
Polysaccharide deacetylases are bacterial enzymes that catalyze the deacetylation of acetylated sugars on the membranes of Gram-positive bacteria, allowing them to be unrecognized by host immune systems. Inhibition of these enzymes would disrupt such pathogenic defensive mechanisms and therefore offers a promising route for the development of novel antibiotic therapeutics. Here, the first X-ray crystal structure of BA0150, a putative polysaccharide deacetylase from Bacillus anthracis, is reported to 2.0 Å resolution. The overall structure maintains the conserved (α/β)8 fold that is characteristic of this family of enzymes. The lack of a catalytic metal ion and a distinctive metal-binding site, however, suggest that this enzyme is not a functional polysaccharide deacetylase.
1. Introduction
Polysaccharide deacetylases (PDs) are family 4 carbohydrate esterases (CE-4) found in bacteria, and include acetyl xylan esterases, chitin deacetylases, chitooligosaccharide deacetylases and peptidoglycan deacetylases (Caufrier et al., 2003 ▶; Davies et al., 2005 ▶). These enzymes catalyze the N- or O-deacetylation of acetylated sugars on the membranes of Gram-positive bacteria. In altering their membrane recognition, the bacteria become invisible to mammalian hydrolases, such as lysozyme, and defend themselves against host immune systems (Boneca, 2005 ▶). These enzymes are so critical for bacterial virulence that the genomes of Bacillus anthracis and its close homolog B. cereus both encode at least ten PDs (Psylinakis et al., 2005 ▶). Deletion of the peptidoglycan N-acetylglucosamine deacetylase A gene (pgdA) resulted in the bacteria being more susceptible to lysozyme (Vollmer & Tomasz, 2000 ▶; Boneca et al., 2007 ▶), and deletion of pgdA in Streptococcus pneumoniae specifically caused the pathogen to become less infective in a mouse model (Vollmer & Tomasz, 2002 ▶). Inhibition of PDs will similarly thwart such bacterial defensive mechanisms, and offers a promising route for the development of novel antibiotic therapeutics and/or prophylatic drugs. This information is especially crucial as we consider the threat of bioterrorism and potentially weaponizable bacteria in the present day.
In order to be effective against a wide range of pathogenic bacteria, an inhibitor should target conserved enzyme features, such as folds, surfaces and catalytic metal ions. Although the CE-4 enzymes share relatively low sequence identities, the available crystal structures reveal a conserved (α/β)8-barrel fold, termed the NodB homology domain for its likeness to NodB proteins (Kafetzopoulos et al., 1993 ▶; Blair & van Aalten, 2004 ▶; Blair et al., 2005 ▶; Taylor et al., 2006 ▶; Oberbarnscheidt et al., 2007 ▶; Deng et al., 2009 ▶; Fadouloglou et al., 2013 ▶; PDB entry 1ny1, Northeast Structural Genomics Consortium, unpublished work; PDB entry 3rzx, Midwest Center for Structural Genomics, unpublished work). In addition, a divalent metal ion (typically zinc) that is required for enzyme activity is found in the active site coordinated by a largely conserved Asp–His–His motif (Blair et al., 2005 ▶). The metal coordination sphere is completed by exogenous anions from the crystallization conditions (acetate, cacodylate, phosphate etc.) in the known structures. The proposed general acid/base mechanism of deacetylation is thought to be similar to that of other zinc-dependent deacetylases (Blair et al., 2005 ▶; Hernick & Fierke, 2005 ▶).
Here, we report the first X-ray crystal structure of BA0150, a putative PD from B. anthracis, to 2.0 Å resolution. The overall structure of BA0150 is similar to other known structures and reveals the conserved (α/β)8 fold that is characteristic of this family of enzymes. Notably, however, BA0150 lacks the conserved Asp–His–His metal-binding motif and a catalytic metal ion, suggesting that it is not a functional PD.
2. Materials and methods
2.1. Protein expression and purification
The BA0150 plasmid was a gift from Professor Vassilis Bouriotis (University of Crete); the construct encodes BA0150 (∼28 500 kDa) with a C-terminal His tag. BA0150 was recombinantly expressed in Escherichia coli BL21(DE3)pLysS cells. The cells were grown in LB medium supplemented with kanamycin (50 µg l−1) at 37°C for ∼3 h, at which point the cells were induced by the addition of isopropyl β-d-1-thiogalactopyranoside (0.3 mM final concentration) and grown for ∼6 h at 37°C. The cells were pelleted by centrifugation, resuspended in 25 ml buffer A (50 mM Tris pH 7.6, 200 mM NaCl, 10% glycerol) and lysed by sonication on ice. The cell debris was pelleted by centrifugation and the cell-free extract was purified using affinity chromatography (Talon Cobalt resin; buffer A, as above; buffer B, 50 mM Tris base pH 7.6, 200 mM NaCl, 10% glycerol, 500 mM imidazole) and size-exclusion chromatography (HiLoad 26/60 Superdex, GE Healthcare) in 25 mM Tris base pH 8.0, 300 mM NaCl, 10% glycerol. The final yield was >200 mg l−1 and the final purity of BA0150 was >95% as determined by SDS–PAGE. The concentration of BA0150 was determined from its absorbance using a calculated extinction coefficient (∊280 = 37 410 M −1 cm−1; Gill & von Hippel, 1989 ▶).
2.2. Crystallization and data collection
Clusters of rectangular crystals were obtained in 1–2 d using the hanging-drop vapor-diffusion method with the following conditions: 2 µl protein solution (24–28 mg ml−1 BA0150 in 25 mM Tris base pH 8.0, 300 mM NaCl, 10% glycerol) were mixed with 2 µl precipitant solution (0.1 M Tris base pH 8.0, 0.2 M NaCl, 30–34% PEG 3350) and equilibrated against a 500 µl reservoir of precipitant solution. Single crystals were harvested and flash-cooled using the precipitant buffer supplemented with 20% glycerol. Crystals diffracted X-rays to 2.0 Å resolution on the Advanced Photon Source beamline NE-CAT 24-ID-C (Argonne National Laboratory) with a Pilatus 6MF detector. Diffraction data were indexed and scaled using XDS as implemented in the Rapid Automated Processing of X-ray Data package (https://github.com/RAPD/RAPD). The crystals belonged to space group P212121, with unit-cell parameters a = 47.14, b = 75.79, c = 124.54 Å.
2.3. Energy-dispersive X-ray spectroscopy
Elemental analysis of cryogenically protected BA0150 crystals was performed on NE-CAT 24-ID-C using an Amptek X-123SDD silicon drift detector. X-ray excitation of the crystals occurred at 12 662 eV. Fluorescence emission amplitudes were detected from 6000 to 13 000 eV. Only Compton scattering from the excitation was observed.
2.4. Structure determination and refinement
The structure was determined using an HHpred alignment and Rosetta as implemented in PHENIX (Terwilliger et al., 2012 ▶) on a TeamHPC 128-core computing cluster. The Matthews coefficient predicted a single molecule in the asymmetric unit, but successful molecular replacement using a carbohydrate esterase from B. anthracis (30% sequence identity; PDB entry 2j13; Oberbarnscheidt et al., 2007 ▶) and phenix.mr_rosetta required the explicit statement of two molecules in the asymmetric unit. This resulted in a solvent content of 37%. Phenix.mr_rosetta produced a model with an R work of 0.24 and an R free of 0.29. The model was refined using iterative cycles of refinement in PHENIX (Adams et al., 2002 ▶) and manual model rebuilding in Coot (Emsley & Cowtan, 2004 ▶). Coot was used to define secondary-structural elements in PyMOL (v.0.99rc6; Schrödinger). Disordered segments in the final model include Met1–Pro46 and Ser247–Gln254 in both monomers A and B. Data-collection and refinement statistics are given in Table 1 ▶. The atomic coordinates and structure factors of BA0150 have been deposited in the Protein Data Bank (http://www.rcsb.org) with accession code 4m1b.
Table 1. Data-collection and structure-refinement statistics for BA0150.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Resolution range () | 48.112.00 (2.112.00) |
| Space group | P212121 |
| Unit-cell parameters () | a = 47.14, b = 75.79, c = 124.54 |
| R merge † | 0.081 (0.548) |
| R meas ‡ | 0.096 (0.683) |
| Completeness (%) | 99.2 (95.9) |
| Multiplicity | 5.9 (3.7) |
| I/(I) | 16.4 (2.4) |
| No. of reflections | 181499 |
| No. of unique reflections | 30744 |
| Mosaicity () | 0.19 |
| Refinement | |
| R work/R free § | 0.17/0.21 |
| No. of atoms | |
| Protein | 3156 |
| Polyethylene glycol | 10 |
| Water | 295 |
| B factors (2) | |
| Overall | 28.8 |
| Protein | 28.0 |
| Polyethylene glycol | 44.5 |
| Water | 36.5 |
| R.m.s.d.s | |
| Bond lengths () | 0.007 |
| Bond angles () | 1.040 |
| Ramachandran statistics (%) | |
| Preferred | 98 |
| Allowed | 2 |
| Outliers | 0 |
R
merge = 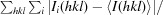
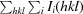 , where Ii(hkl) is the ith observation of reflection hkl and I(hkl) is the weighted average intensity for all observations iofreflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and I(hkl) is the weighted average intensity for all observations iofreflection hkl.
R
meas = 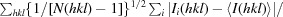 , where R
meas is a merging R factor independent of data redundancy.
, where R
meas is a merging R factor independent of data redundancy.
R
work = 
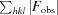 , where F
obs and F
calc are the observed and calculated structure factors, respectively, and the statistic is calculated for all reflections except for the test set. R
free is calculated accordingly for reflections excluded from refinement (the test set).
, where F
obs and F
calc are the observed and calculated structure factors, respectively, and the statistic is calculated for all reflections except for the test set. R
free is calculated accordingly for reflections excluded from refinement (the test set).
3. Results and discussion
Here, we report the first X-ray crystal structure of BA0150, a putative PD from B. anthracis, to 2.0 Å resolution. The structure was determined using an HHpred alignment and phenix.mr_rosetta (Terwilliger et al., 2012 ▶) with two molecules in the asymmetric unit, resulting in a solvent content of 37%. The two independent monomers of BA0150 in the P212121 unit cell are structurally quite similar, with r.m.s. deviations of 0.86 Å for 200 Cα atoms and 1.38 Å for all atoms (Maiti et al., 2004 ▶). A polyethylene glycol molecule was modeled into density in monomer B (not shown). The structure of monomer A is shown in Fig. 1 ▶. The conserved (α/β)8-barrel homology domain is clearly visible, and the overall structure aligns well with other known PD structures from Gram-positive bacteria.
Figure 1.
Stereoview of monomer A of BA0150. The monomer is colored in ‘chainbows’ to highlight the secondary structure of the NodB domain.
Interestingly, however, the crystal structure does not contain a catalytic metal ion. While most PDs have a metal ion (typically zinc) that is required for enzyme activity, three independent data collections have shown that BA0150 reproducibly crystallizes as an apoenzyme under the reported conditions. Energy-dispersive X-ray spectroscopy confirmed the complete absence of any metals in the protein crystals. Sequence alignments show that BA0150 does not contain the conserved Asp–His–His metal-binding triad that is found in many CE-4 enzymes (Blair et al., 2005 ▶). Fig. 2 ▶ is a partial sequence alignment of the structurally characterized PDs from Gram-positive bacteria that contain a metal ion in the active site. Rather than the Asp–His–His triad, BA0150 contains Ile, Met and Tyr residues in the metal-binding sites, respectively, none of which have a strong affinity for metal binding (Fig. 3 ▶). Furthermore, the Ile residue points away from the putative active site and makes long-range hydrophobic interactions with Ala72, Trp95 and Ala211. Crystallization trials using a zinc salt (50–200 mM ZnCl2) in place of NaCl largely resulted in protein precipitation, and zinc-soaking experiments with pre-formed crystals (0.5–5 mM ZnCl2, ∼2 h) failed to produce any zinc anomalous signal or zinc difference density in the active site. The inability to bind metal suggests that BA0150 is not a metalloenzyme and, in fact, is not a functional PD. While the catalytic metal ion is missing, the putative active site has nearby Asp (Asp64) and His (His210) residues, which are largely conserved among the PDs, and suggests that the enzyme may have some hydrolytic activity. Studies to measure activity and determine specific functionality are in progress.
Figure 2.

Partial sequence alignments of structurally characterized PDs from Gram-positive bacteria that contain metal ions in the active sites. Largely conserved residues are shown in bold and underlined. The largely conserved Asp–His–His metal-binding residues are shown in bold.
Figure 3.

Active-site alignment of BA0150 with a carbohydrate esterase from B. anthracis (PDB entry 2j13). The active-site residues in BA0150 are Ile65, Met116 and Tyr120 (C, green; N, blue; O, red; S, yellow). The active-site residues in 2j13 are His103 and His107, with metal coordination completed by acetate and cacodylate anions (C, cyan; N, blue; O, red; As, purple). The catalytic zinc ion of 2j13 is shown as a gray sphere.
4. Conclusions
In summary, we report the first X-ray crystal structure of BA0150. The overall structure reveals that the protein maintains the conserved (α/β)8 barrel (NodB domain) that is characteristic of the CE-4 family of enzymes. The enzyme reproducibly crystallized without a catalytic metal, however; the lack of the conserved Asp-His-His triad indicates that this enzyme is not a metalloenzyme and is not a functional PD. The largely conserved Asp and His residues near the putative active site, however, suggest that BA0150 may have some hydrolytic activity. Studies to determine specific functionality are ongoing.
Supplementary Material
PDB reference: BA0150, 4m1b
Acknowledgments
The work of DK and VB was co-financed by EU (ERDF) and Greek funds through the ‘THALIS’ program. This work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) of the National Institutes of Health. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. KEC and RJS would like to thank Ithaca College for funding.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Blair, D. E., Schüttelkopf, A. W., MacRae, J. I. & van Aalten, D. M. F. (2005). Proc. Natl Acad. Sci. USA, 102, 15429–15434. [DOI] [PMC free article] [PubMed]
- Blair, D. E. & van Aalten, D. M. F. (2004). FEBS Lett. 570, 13–19. [DOI] [PubMed]
- Boneca, I. G. (2005). Curr. Opin. Microbiol. 8, 46–53. [DOI] [PubMed]
- Boneca, I. G. et al. (2007). Proc. Natl Acad. Sci. USA, 104, 997–1002.
- Caufrier, F., Martinou, A., Dupont, C. & Bouriotis, V. (2003). Carbohydr. Res. 338, 687–692. [DOI] [PubMed]
- Davies, G. J., Gloster, T. M. & Henrissat, B. (2005). Curr. Opin. Struct. Biol. 15, 637–645. [DOI] [PubMed]
- Deng, D. M., Urch, J. E., ten Cate, J. M., Rao, V. A., van Aalten, D. M. F. & Crielaard, W. (2009). J. Bacteriol. 191, 394–402. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fadouloglou, V. E., Kapanidou, M., Agiomirgianaki, A., Arnaouteli, S., Bouriotis, V., Glykos, N. M. & Kokkinidis, M. (2013). Acta Cryst. D69, 276–283. [DOI] [PubMed]
- Gill, S. C. & von Hippel, P. H. (1989). Anal. Biochem. 182, 319–326. [DOI] [PubMed]
- Hernick, M. & Fierke, C. A. (2005). Arch. Biochem. Biophys. 433, 71–84. [DOI] [PubMed]
- Kafetzopoulos, D., Thireos, G., Vournakis, J. N. & Bouriotis, V. (1993). Proc. Natl Acad. Sci. USA, 90, 8005–8008. [DOI] [PMC free article] [PubMed]
- Maiti, R., Van Domselaas, G. H., Zhang, H. & Wishart, D. S. (2004). Nucleic Acids Res. 32, W590–W594. [DOI] [PMC free article] [PubMed]
- Oberbarnscheidt, L., Taylor, E. J., Davies, G. J. & Gloster, T. (2007). Proteins, 66, 250–252. [DOI] [PubMed]
- Psylinakis, E., Boneca, I. G., Mavromatis, K., Deli, A., Hayhurst, E., Foster, S. J., Vårum, K. M. & Bouriotis, V. (2005). J. Biol. Chem. 280, 30856–30863. [DOI] [PubMed]
- Taylor, E. J., Gloster, T. M., Turkenburg, J. P., Vincent, F., Brzozowski, A. M., Dupont, C., Shareck, F., Centeno, M. S. J., Prates, J. A. M., Puchart, V., Ferreira, L. M. A., Fontes, C. M. G. A., Biely, P. & Davies, G. J. (2006). J. Biol. Chem. 281, 10968–10975. [DOI] [PubMed]
- Terwilliger, T. C., DiMaio, F., Read, R. J., Baker, D., Bunkóczi, G., Adams, P. D., Grosse-Kunstleve, R. W., Afonine, P. V. & Echols, N. (2012). J. Struct. Funct. Genomics, 13, 81–90. [DOI] [PMC free article] [PubMed]
- Vollmer, W. & Tomasz, A. (2000). J. Biol. Chem. 275, 20496–20501. [DOI] [PubMed]
- Vollmer, W. & Tomasz, A. (2002). Infect. Immun. 70, 7176–7178. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: BA0150, 4m1b